

Abstract
We report progress on predicting tumor response to platinum-based chemotherapy with a novel mass spectrometry approach. Fourteen bladder cancer patients were administered one diagnostic microdose each of [14C]carboplatin (1% of the therapeutic dose). Carboplatin-DNA adducts were quantified by accelerator mass spectrometry (AMS) in blood and tumor samples collected within 24 hours, and compared to subsequent chemotherapy response. Patients with the highest adduct levels were responders, but not all responders had high adduct levels. Four patient-derived bladder cancer xenograft mouse models were used to test the possibility that another drug in the regimen could cause a response. The mice were dosed with [14C]carboplatin or [14C]gemcitabine and the resulting drug-DNA adduct levels were compared to tumor response to chemotherapy. At least one of the drugs had to induce high drug-DNA adduct levels or create a synergistic increase in overall adducts to prompt a corresponding therapeutic response, demonstrating proof-of-principle for drug-DNA adducts as predictive biomarkers.
Keywords: diagnostic microdosing, chemotherapy resistance, bladder cancer, accelerator mass spectrometry, patient derived xenografts
Introduction
Cisplatin, carboplatin and oxaliplatin are among the most commonly prescribed chemotherapeutic drugs and are used in over half of all chemotherapy patients for various cancers, such as bladder, lung, ovarian, and colon. Except in testicular cancer, their efficacy is modest, and drug resistance is the most common cause of treatment failure (1, 2). The feasibility of predicting drug resistance by genomics analysis has been reported for bladder cancer (3, 4). This work led to improved understanding of resistance mechanisms, and identified over 700 genes involved in cellular response to platinum-based drug treatment (5). However, the genomics approach has thus far failed to develop clinically useful tests for predicting resistance.
Platinum-based drugs kill cancer cells through covalent drug-DNA adduct formation (2, 6). Accordingly, another approach of identifying drug resistance has focused on quantification of drug-DNA adducts as biomarkers (7-12). The levels of such adducts are the result of numerous factors that govern cellular responses to drug exposure including genetics, tumor microenvironment, kidney function, overall patient health and others (13). The key challenge of this approach is the need to dose patients with full-dose chemotherapy in order to recover enough analyte to accurately quantify extremely low levels of drug-DNA adducts (14). Furthermore, therapeutic dosing is a poor diagnostics strategy and puts patients at risk. To address these problems, we developed the concept of “diagnostic microdosing” in which patients are administered a nontoxic sub-therapeutic “microdose” of 14C-labeled drug(s) followed by blood and tumor biopsy sampling and analysis with accelerator mass spectrometry (AMS), as summarized in Figure 1. AMS can easily measure one 14C-labeled drug molecule bound to DNA per 108 nucleotides (15), with a limit of detection of one adduct per 1012 nucleotides (16) and can quantify radiocarbon-labeled drug or metabolites in urine, blood or DNA samples (17, 18).
Figure 1. Carboplatin microdose and therapeutic dose elimination kinetics.

A) Schematic, summarizing the strategy for evaluating the feasibility of predicting response to cytotoxic chemotherapy using diagnostic microdosing. B) Carboplatin plasma concentration over 24 hours after microdosing (grey dashed lines) and therapeutic dosing (black solid lines) as determined from plasma by LSC (N = 3 patients). C) Linear correlation of carboplatin plasma concentrations. D) Drug elimination profiles for 11 bladder cancer patients after microdosing with [14C]carboplatin. E) Linear regression analysis between radiocarbon concentration measured by LSC and platinum concentration determined by ICP-MS after therapeutic [14C]carboplatin dosing (in plasma).
Most platinum-based treatments involve either cisplatin or carboplatin. Cisplatin does not have a carbon atom in the molecule and cannot be detected by AMS. Although less potent, carboplatin forms the same drug-DNA adduct crosslink structure as cisplatin (SI Figure 1), and clinical cross-resistance is common (19). Consequently, these two drugs are sometimes used interchangeably in clinical practice (6, 19). Therefore, the microdosing approach to identifying carboplatin resistance can possibly be applied to cisplatin.
We previously used AMS to quantify [14C]carboplatin-DNA monoadduct formation in cancer cell lines exposed to microdose-relevant drug concentrations (12, 15, 20-23). The resulting adduct-levels correlated with in vitro carboplatin cytotoxicity. AMS specifically detected carboplatin-DNA monoadducts, since the 14C-labeled cyclobutane dicarboxylate (CBDCA) group is released once the diadduct is formed (SI figure 1) (22). We report here translation of these studies into a prospective Phase I clinical trial including a cohort of bladder cancer patients (SI Figure 2). Bladder cancer was chosen for this study as a proof-of-principle because platinum-based therapy is the standard neoadjuvant treatment and is potentially curative for locally advanced cancer. The diagnostic microdosing approach may also be used for many other cancer types that implement similar platinum-based based therapies.
We tested the hypothesis that microdose-induced carboplatin-DNA adducts in peripheral blood mononuclear cells (PBMC) correlate with response to platinum-based neoadjuvant chemotherapy. We found that those patients with the highest carboplatin-DNA adduct levels were responders. However, not all responders had high adduct levels. This observation led to a second hypothesis that a combination drug partner may compensate for low platinum-DNA adducts levels to enable a response. This hypothesis was tested in mice bearing patient-derived tumor xenografts that were donated by four myoinvasive bladder cancer patients. These mice were dosed with either [14C]carboplatin and/or [14C]gemcitabine as a model of gemcitabine and platinum (GC) combination therapy. Mouse tumors with high drug-DNA adducts levels responded to chemotherapy, whereas low adduct levels correlated with resistance. Collectively, these results support the feasibility of diagnostic microdosing to improve patient outcomes by personalizing chemotherapy.
Material and Methods
Chemicals
Unlabeled gemcitabine (GEMZAR®) was obtained from Eli Lilly (Indianapolis, IN, USA), and carboplatin (CARBOplatin®, 10 mg/mL) from Hospira (Lake Forest, IL, USA). 14C-labeled carboplatin (specific activity 53 mCi/mmol with the 14C-label in the cyclobutane dicarboxcylic group) was obtained from GE Healthcare (Waukesha, WI, USA) and [14C]gemcitabine (specific activity 58.8 mCi/mmol with the 14C-label on the aromatic nucleobase at the 2 position) was purchased from Moravek Biochemicals (Brea, CA, USA). [14C]carboplatin for injection was prepared under good manufacturing practice (GMP) at the GMP facility at UC Davis. The [14C]carboplatin drug substance was dissolved with sterile water for injection (WFI). The resulting solution was filter sterilized with 0.2 μm PES syringe filter into sterile glass vials and sealed with a rubber septum. Specific activity was determined by liquid scintillation counting (LSC). Mixtures of 14C-labeled and unlabeled drug were used to minimize the usage of radiocarbon and achieve the different specific activities required for microdoses and therapeutic doses. Drug solutions for the indicated experiments were prepared immediately before use.
Clinical Study
The study “A phase 0 clinical trial of microdosing carboplatin and molecular profiling for chemoresistance” (ClinicalTrials.gov identifier NCT01261299) is an ongoing, multisite feasibility study of the diagnostic microdosing approach. The patient population consisted of non-small cell lung cancer patients (NSCLC), stage IV with measurable lesions, and bladder transitional cell carcinoma (TCC) patients, stage II disease and above for neoadjuvant treatment, or stage III and IV metastatic disease (only data for TCC are presented here). Informed consent was obtained after the nature and possible consequences of the studies were explained. Microdoses of [14C]carboplatin were administered to patients as a diagnostic reagent, followed by standard of care full dose platinum-based chemotherapy (G/C or MVAC) and evaluation of response. PBMC and tumor tissue were collected from the patients for analysis of carboplatin-DNA monoadduct frequencies. Toxicity of the [14C]carboplatin diagnostic reagent administered as a microdose was assessed using Common Terminology Criteria for Adverse Events (CTCAE). Patient response to chemotherapy was evaluated using the RECIST for correlation to carboplatin-DNA monoadduct frequency. The carboplatin dose for human chemotherapy is calculated using the Calvert formula with an AUC of 6. Therefore, individual patients were given a microdose of [14C]carboplatin (1.0 × 107 dpm/kg of body weight) containing a total carboplatin dose at 1% of their therapeutic dose by a 2-minute bolus intravenous infusion (IV). Unlabeled carboplatin and [14C]carboplatin were mixed immediately before dosing and injected through the peripheral vein at one arm. Peripheral blood specimens were drawn into BD Vacutainer CPT™ tubes with sodium citrate (Becton Dickinson) from the other arm at specified time points before and after the administration of the microdose. PBMC were isolated within 2 hours of collection by centrifugation according to manufacturer's instruction. A proportion of total plasma was used for LSC measurement and PK determination. Tumor biopsy specimens were collected 24 hours after microdosing. Outcomes related to chemotherapy (including response and adverse events) were collected and correlated with carboplatin-DNA monoadduct data.
Animal handling and treatment
All animal experiments were performed in compliance with institutional guidelines and approved by the Animal Use and Care Administrative Advisory Committee at the University of California, Davis (CA, USA). All animals were kept under pathogen-free conditions. Female NOD scid gamma severe combined immunodeficient (NSG) mice (5-8 weeks of age, body weight: 20-25 g) were purchased from Jackson laboratories (CA, USA). Patient derived tumor xenografts (PDX) were created by subcutaneous injection at the flank of approximately 1 mm3 cancerous tissue from a patient's primary tumor directly into a NSG. PDX tumors (passage 2-4) were allowed to grow to at about 200mm3 before being assessed for drug efficacy and microdosing studies.
Animal dosing with radiolabeled drugs
When the xenografts reached approximately 200 mm3, mice were intravenously injected through the tail vein with one microdose 10 μL/g of radiolabeled carboplatin (0.375 mg/kg, 50,000 dpm/g) or gemcitabine (0.092 mg/kg, 1,000 dpm/g) or one therapeutic dose of radiolabeled carboplatin (37.5 mg/kg, 50,000 dpm/g) or gemcitabine (9.2 mg/kg, 1,000 dpm/g). Tumor tissues were collected after 4 and 24 hours and stored at −80°C until DNA isolation.
Efficacy Study
For each treatment group, 8-12 mice were used to allow statistical analysis. When tumor sizes reached approximately 200 mm3, mice were randomly assigned into different treatment groups and treated with indicated therapeutic agents (Cisplatin at 2 mg/kg IV Q7Dx3, carboplatin at 30 mg/kg IP Q7Dx4, and gemcitabine at 150 mg/kg IP Q7Dx4). Mice were monitored for tumor growth and alterations in weight. Tumor size was determined with a caliper, and tumor volumes were calculated using an ellipsoidal formula (1/2 × length × width squared). Mice were sacrificed when the tumor length reached 20 mm in any direction.
DNA isolation
DNA was extracted from minced tumor tissue or PBMC pellet with the Wizard Genomic DNA Purification Kit according to the manufacturer's instruction (Promega, Madison, WI, USA). DNA quantity and quality was determined with a NanoDrop 1000 spectrophotometer, the purity was ensured by obtaining a 260/280 nm OD ratio of approximately 1.9.
AMS
All DNA samples were submitted to Lawrence Livermore National Laboratory (LLNL) for AMS analysis of radiocarbon content using an established protocol (24). Ten micrograms of DNA per sample was converted to graphite and measured by AMS for 14C quantification as previously described.
RNA isolation, cDNA Synthesis, RT-PCR
Total RNA was isolated using Qiagen RNeasy Mini Kit and QIAshredder spin columns (Qiagen, Valencia, CA) following manufacturer instructions. Column bound RNA was pretreated with Qiagen RNase-Free DNase to remove genomic DNA before elution with RNase free water. Nucleotide concentration and purity was determined via NanoDrop 1000 spectrophotometer. RNA purity was ensured by obtaining a 260/280 nm OD ratio approximately 2.0. cDNA was synthesized using RevertAid RT Kit (Thermo Scientific, Carlsbad, CA) using 1 μg freshly purified RNA and random hexamer primer. Real-Time PCR (RT-PCR) was performed with BioRad CFX96 Real-Time System instrument in triplicates of each sample in a volume of 10 μL using a standard protocol (95°C, 2 min, 40 cycles: 95°C, 15 sec; 60°C, 30 sec; 72°C, 30 sec; melting curve 65°C - 95°C, 5 sec). Per reaction, 4 ng cDNA was mixed with 10 pmol forward and reverse primer, SYBR green and EconoTaq PLUS 2X Master Mix (Lucigen Corporation, Middleton, WI). Primer sequences (Integrated DNA Technologies, Coralville, IA) included: GAPDH: forward 5′-CGCGGGGCTCTCCAGAACATC-3′, reverse 5′-CTCCGACGCCTGCTTCACCAC-3′; RRM1: forward 5′-GCCGCCAAGAACGAGTCAT-3′, reverse 5′-AGCAGCCAAAGTATCTAGTTCCA-3′; RRM2: forward 5′-GTGGAGCGATTTAGCCAAGAA-3′, reverse 5′-CACAAGGCATCGTTTCAATGG-3′; hENT1: forward 5′-GTGCCTTCGGCTACTTTATCAC-3′, reverse 5′-GCTAATGAGGTCCAACTTGGTCT-3′; ERCC1: forward 5′-CTACGCCGAATATGCCATCTC-3′, reverse 5′-GTACGGGATTGCCCCTCTG-3′. mRNA expression level of indicated genes were normalized to the internal GAPDH expression via the 2−ΔΔCt method (25).
Whole exome sequencing
Whole-exome sequencing (WES) on the Illumina platform was previously performed for each bladder cancer PDX model (26), but with new data analysis specific to DNA repair genes. Details are in Supplemental Information and the the raw sequence data files are publicly available through the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) with Accession Number PRJNA352282.
Statistical analysis
All statistical analyses were performed using GraphPad Prism™ software (GraphPad Software Inc., CA, USA) using 1-way ANOVA with Newman-Keuls multiple comparison post hoc or a two-tailed Student's t-test. A p-value below 0.05 was considered statistically significant. All experiments were carried out at least in triplicate in order to enable statistically significant comparisons of the results. All results are expressed as the mean ± SEM unless otherwise noted. A simple correlation of the adduct levels and response is reported.
Results
Dose formulation and clinical toxicity associated with carboplatin microdosing
A clinical feasibility study (ClinicalTrials.gov identifier NCT01261299) was initiated with a starting (level 1) dose of [14C]carboplatin of 107 dpm/kg with a total carboplatin dose of 1% of the therapeutic dose (target AUC of 6). With this dose composition, the 14C-signal in DNA isolated from PBMC was approximately 10-100 times the background, which allowed accurate adduct measurement by AMS.
A total of 21 patients were accrued, including 14 bladder cancers, 6 non-small cell lung cancers and one mediastinal mass that was subsequently identified as Hodgkin's lymphoma. Of the bladder cancer patients, ten completed the entire study including microdosing, blood sampling and subsequent full dose chemotherapy. The patients’ characteristics are shown in SI Table 1. This paper only discusses the bladder cancer data. No microdose-associated adverse events were observed. Three patients received an additional dose of [14C]carboplatin at the end of their therapeutic dose infusion of unlabeled carboplatin in order to compare the therapeutic pharmacokinetics (PK) and formation of drug-DNA adducts with those of the microdose.
Radiation exposure from the diagnostic microdose was not greater than 0.1 mSv. In comparison, the annual effective radiation dose equivalent from natural internal sources is 1.6 mSv per person (27), and exposure for an abdominal CT scan is 10 mSv (28). Therefore, the diagnostic [14C]carboplatin administered as a microdose appears to be safe in this patient population.
Linear correlation of carboplatin pharmacokinetics between microdose and therapeutic dose
We determined whether the microdose results in a proportionally lower drug exposure (area under the curve) compared to the therapeutic doses of carboplatin. A microdose and a full therapeutic dose containing [14C]carboplatin was administered to three patients, followed by blood sampling over 24 hours and quantitation of plasma 14C concentration by liquid scintillation counting (LSC) (Figure 1B and SI Table 2). The PK of the therapeutic and microdoses showed nearly identical elimination kinetics (SI Table 2). The clearance of carboplatin is known to occur in a triphasic manner with an initial (T1/2α), a second (T1/2β) and a terminal (T1/2γ) phase half-lives reported to be 10 to 98 minutes, 1.3 - 6.0 hours and 8.2 - 125 hours, respectively (29-32). The measured T1/2α and T1/2β half-lives of 13 - 55 minutes and 4.5 - 8.7 hours, reside well within the range of these previously reported therapeutic values. At 24 hours after dosing, over 99% of 14C labeling had been cleared from the blood, suggesting this is the optimal time for biopsy with little concern of 14C contamination. Comparison of plasma carboplatin equivalent concentration showed dose proportional linearity between microdoses and a therapeutic doses (Figure 1C).
Without prior sample fractionation, LSC only measures total 14C concentration of a given sample without differentiating the parent drug from metabolites. Therefore, the resulting PK calculations could have been confounded by the circulation of free [14C]CBDCA. To further validate the approach of determining [14C]carboplatin PK with LSC, we used inductively coupled plasma mass spectrometry (ICP-MS) to determine the platinum concentration in specimens from one patient. At the therapeutic dose a significant linear correlation was observed between ICP-MS measurements and LSC measurements (R2 = 0.724, p = 0.032, Figure 1E). However, ICP-MS was not able to detect platinum in the microdosing specimens, which supports the utility of AMS for diagnostic microdosing applications.
Correlation of microdose-induced [14C]carboplatin-DNA monoadducts in PBMC with tumor response to subsequent full dose chemotherapy
At 24 hours after intravenous microdosing, PBMC were isolated from patient blood samples followed by DNA purification. Radiocarbon content in the DNA was measured by AMS according to published protocols (24). The concentration of carboplatin-DNA monoadducts was then calculated in units of monoadducts per 108 nucleotides (Figure 2). Patients received platinum-based chemotherapy within four weeks after the microdosing procedure (typically GC or MVAC). Response was confirmed if a patient had pT1 or less tumor (partial response: PR, complete response: CR) at the time of cystectomy; or resistance if a patient still had pT2 or greater (disease progression: DP), based on standard RECIST criteria (33).
Figure 2. Correlation of microdose-induced [14C]carboplatin-DNA adduct levels to therapy response.
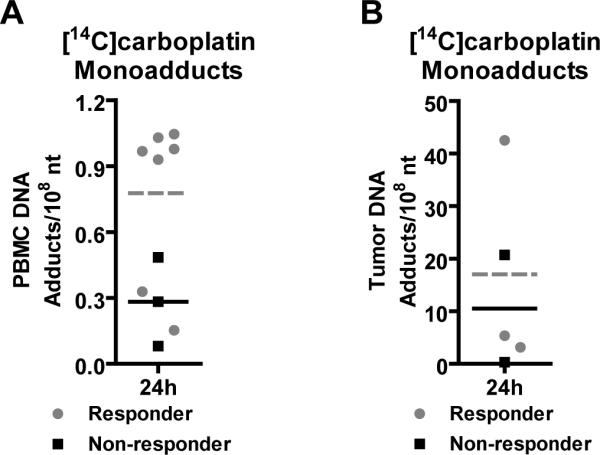
Carboplatin-DNA monoadduct levels were quantified in patient A) PBMC and B) tumor biopsy tissue 24 h after receiving one microdose of [14C]carboplatin (grey circular symbols = responder, grey dashed line = responder mean adduct level, black symbols = non-responders, black solid line = non-responder mean adduct level). Student's t-test was used to measure all p values (p<0.001 for high adduct level group compared to the low adduct level group, and p = 0.069 for differentiating responders and nonresponders). Each data point represents a value for an individual patient.
Seven patients responded to chemotherapy (grey circular symbols) whereas three patients showed disease progression (black square symbols) (Figure 2A and SI Table 2). Three patients did not receive subsequent chemotherapy: one with renal failure, and the other two with a clinical diagnosis of T2 disease because of large tumor mass, but were diagnosed with T1 disease at the biopsy during chemotherapy. Responders exhibited approximately 2-fold higher mean PBMC monoadduct levels (grey dashed line) than non-responders (black solid line) (0.741 ± 0.346 versus 0.283 ± 0.202 monoadducts/108 nt, respectively, p = 0.069). There were two distinct groups; one with high (0.941 ± 0.030 adducts per 108 nt) and the second group with low (0.266 ± 0.158 adducts per 108 nt) DNA adduct levels (p < 0.0001). All five patients in the high adduct level group responded to chemotherapy. The low adduct level group included three non-responders and two responders. Tumor adduct levels were measured for five patients. One with a very high carboplatin-DNA adduct levels responded, whereas the other tumor samples exhibited no obvious trends (Figure 2B).
Response of patient-derived bladder tumor xenografts (PDX) in mice to chemotherapy
Since two patients with low PBMC adduct levels also responded to chemotherapy, we hypothesized that tumor response in those patients with low carboplatin-DNA adducts was induced by the other components of the treatment regimen. We developed a diagnostic microdosing protocol to identify resistance to gemcitabine for use in a mouse model of human bladder cancer. We established four patient-derived tumor xenograft (PDX) mouse models from randomly selected myoinvasive bladder cancer patient volunteers using previously described protocols for nod scid gamma severe combined immune deficient (NSG) mice (26). These models were named BL0269 (TM00015), BL0293 (TM00016), BL0440 (TM00024) and BL0645 (TM01339), where the BL number is the UC Davis Comprehensive Cancer Center Biorepository number (http://www.ucdmc.ucdavis.edu/cancer/research/sharedresources/specimen.html) and the TM number is the Jackson Laboratory PDX database identifier (http://tumor.informatics.jax.org/mtbwi/pdxSearch.do). Retrospective analysis of routine quality control IHC staining showed one model, BL0645, as being CD45+, which indicates that this tumor formed a lymphocytic tumor. Formation of lymphocytic tumors from subcutaneous implantation of tumor tissue has been previously reported for PDX (34). However, we initiated this work under the assumption that the xenograft represented a tumor of origin from the bladder. The xenograft was a solid tumor with vasculature and in some regions necrosis, consistent with the other bladder tumors used in this study. Regardless of the potential molecular differences in origin, BL0645 was useful for this study as a model of synergy between cisplatin or carboplatin and gemcitabine. We included data from this model because it is not necessary for the tumor to be rigorously proven to be bladder derived in order to demonstrate correlations between drug-DNA adduct levels and treatment response.
We assessed the sensitivity of each PDX to cisplatin, carboplatin, or gemcitabine as single agent, as well as to GC combination chemotherapy (Figure 3 and SI Figure 3). Sensitivity was determined by changes in tumor volume over time (SI Figure 3), and by tumor-specific survival analysis (Figure 3). Of the four distinct tumor models, BL0269, BL0293, BL0645 were resistant to cisplatin/carboplatin chemotherapy. BL0269 and BL0645 were resistant to gemcitabine. We demonstrated that cisplatin shows similar response efficacy to carboplatin in the mouse models (SI Figure 3). Median survival of the carboplatin treatment groups is similar to the corresponding cisplatin treatment groups; for BL0269, BL0293, and BL0440 median survival was 39, 28, and 64 days, respectively, compared to cisplatin median survival of 38, 35, and 80 days post treatment initiation (SI Table 3).
Figure 3. Tumor specific survival after chemotherapeutic treatment of NSG PDX mice bearing four different bladder cancer xenografts.

Kaplan-Meier survival curves comparing different treatments (N ≥ 8) of A) BL0269, B) BL0293 C) BL0440 and D) BL0645. Black line = Vehicle (D5W IV Q7Dx3), blue line = cisplatin (2 mg/kg IV Q7Dx3), red line = gemcitabine (150 mg/kg IP Q7Dx4), green line = G/C combination chemotherapy (dashed lines represent treatment days).
Correlation of microdose-induced [14C]carboplatin and [14C]gemcitabine DNA-adduct levels with PDX drug sensitivity
NSG mice bearing indicated PDX were given a microdose of [14C]carboplatin (0.375 mg/kg of body weight, Figure 4A+B) or [14C]gemcitabine (0.092 mg/kg of body weight, Figure 4C+D). Each microdose was chosen to target approximately 1% of the human plasma maximum concentration of 1 μM for carboplatin and 0.3 μM for gemcitabine. Animals were sacrificed at 4h (Figure 4A+C) and 24h (Figure 4B+D) after microdose bolus IV injection and drug-DNA adduct levels were determined from purified tumor tissue DNA by AMS. Carboplatin- and gemcitabine DNA-adduct levels correlated with PDX drug sensitivity to single agent treatment in NSG mice (Figure 3+4 and SI Figure 3). The carboplatin and gemcitabine sensitive tumor BL0440 showed significant higher mean carboplatin monoadduct levels compared to the other resistant models. At 24 hours, BL0440 tumor DNA exhibited 1.5 to 2.2 fold higher mean monoadduct levels than BL0269, BL0293 and BL0645 (2.73 ± 0.66 versus 1.90 ± 0.80 (p < 0.05), 1.51 ± 0.55 (p < 0.01), and 1.27 ± 0.31 (p < 0.01) monoadducts/108 nt, respectively) (Figure 4B). There was no significant difference in carboplatin adduct levels between the three resistant PDX models.
Figure 4. Microdose-induced carboplatin- and gemcitabine DNA-adduct levels correlate with PDX drug sensitivity.

NSG mice were implanted with the indicated PDX tumors 14 to 30 days before microdosing. Animals (N ≥ 4 per group) were dosed via tail vein injection with A and B) 0.375 mg/kg carboplatin, 50,000 dpm/g or C, D and F) 0.092 mg/kg gemcitabine, 1000 dpm/g. Tumors were collected after A and C) 4h or B and D) 24h and 14C content of isolated tumor DNA was analyzed via AMS. E) Tumor volume ratio of parental PDX model BL0293 (grey line) and resistant model BL0293R (black line). BL0293 was defined as resistant after gemcitabine treatment did not affect tumor growth (8 cycles). Dashed lines represent treatment days. F) Microdose-induced gemcitabine adduct levels at 4h and 24h of BL0293 and BL0293R. One-Way ANOVA with Newman-Keuls post hoc test or Student's t-test was used to measure all p values (* p < 0.05, ** p < 0.01). Presented are means and error bars represent SEM.
Similarly, at 24h the two gemcitabine sensitive BL0293 and BL0440 PDX models display 4.1 to 6.3 fold higher gemcitabine adduct levels than the resistant PDX models BL0269 and BL0645 (10.1 ± 6.47 versus 2.44 ± 0.49 (p < 0.05) and 2.24 ± 0.81 (p < 0.05) and 14.1 ± 11.1 versus 2.44 ± 0.49 (p < 0.01) and 2.24 ± 0.81 (p < 0.05) adducts/108 nt, respectively). There is no significant difference in gemcitabine-DNA adduct levels between the two resistant PDX models. Compared to 4h, at 24h the sensitive PDX models showed a slight increase adduct levels; whereas the resistant model BL0269 showed a significant decrease in adduct frequency and BL0645 showed relatively low levels of gemcitabine-DNA adduct formation (Figure 4C+D).
We also generated a gemcitabine-resistant PDX sub-line (BL0293R) through repeated treatment of mice carrying BL0293 tumors with therapeutic doses of gemcitabine. After 4 treatment cycles, relapsed tumors were reimplanted into NSG mice and treatment was resumed for an additional 4 cycles. The combined 8 cycles of full dose gemcitabine chemotherapy resulted in BL0293R tumors that did not respond to gemcitabine treatment (Figure 4E). Progression free survival after chemotherapy decreased from 16.9 ± 1.0 days for BL0293 to 2.3 ± 0.7 days for BL0293R (p < 0.0001). A 2.5 fold decrease in gemcitabine adduct formation at 4h and 24h was detected in BL0293R compared to the parental model BL0293 (2.89 ± 1.22 versus 7.17 ± 1.97 (p < 0.001) at 4 hours, and 4.02 ± 1.23 versus 10.1 ± 6.47 (p = 0.044) adducts/108 nt at 24 hours, Figure 4F).
Synergistic GC combination chemotherapy enhanced of drug-DNA adduct levels in BL0645
We analyzed the effect of combination GC chemotherapy on drug-DNA adduct formation using BL0645 (Figure 5). This model was resistant towards each of the single agents, cisplatin and gemcitabine, but had strongly enhanced sensitivity toward gemcitabine/cisplatin (Gem/Cis) combination therapy, consistent with a synergistic effect (SI Figure 3G). BL0645 tumor bearing mice were administered a microdose of [14C]carboplatin alone (white bar) or a microdose of [14C]carboplatin in combination with a microdose of unlabeled gemcitabine (black bar). The combination of [14C]carboplatin plus unlabeled gemcitabine resulted in a significant 1.7-fold increase in carboplatin-DNA monoadduct levels (1.27 ± 0.31 versus 2.10 ± 0.60 adducts/108 nt, p = 0.024) when compared to a microdose with single agent [14C]carboplatin (Figure 5A). Similarly, BL0645 bearing mice receiving a microdose of [14C]gemcitabine in combination with a microdose of unlabeled carboplatin (white bar) showed a 1.6 fold increase in gemcitabine adduct levels (2.24 ± 0.81 versus 3.66 ± 1.13 adducts/108 nt, p = 0.051) compared to [14C]gemcitabine alone (black bar, Figure 5B). In contrast, BL0269, which is resistant to both single agents and combination therapy (Figure 3A), showed no significant increase in adduct formation upon combination treatment (Figure 5C+D) (1.90 ± 0.80 carboplatin adducts/108 nt for the combination versus 2.22 ± 0.44 carboplatin adducts/108 nt (p = 0.529) in the single agent carboplatin group, and 2.44 ± 0.49 gemcitabine adducts/108 nt for the combination versus 1.85 ± 0.49 gemcitabine adducts/108 nt (p = 0.121) for the gemcitabine group). Clearly, these observations of combination-induced increases in adduct levels the BL0645 line are consistent with the synergistic antitumor activity of the combination therapy for this tumor.
Figure 5. Pharmacodynamic evidence of in vivo for synergy between gemcitabine and carboplatin.

NSG mice were implanted with tumor model A and B) BL0645 or C and D) BL0269. Animals were dosed via tail vein injection of A and C) 0.375 mg/kg carboplatin (50,000 dpm/g), or 0.375 mg/kg carboplatin (50,000 dpm/g) plus 0.092 mg/kg gemcitabine, B, D) 0.092 mg/kg gemcitabine (1,000 dpm/g), or 0.092 mg/kg gemcitabine (1,000 dpm/g) plus 0.375 mg/kg carboplatin. Tumors were collected after 24h and isolated DNA was analyzed via AMS (N ≥ 3). Student's t-test was used to measure all p values. Presented are means and error bars represent SEM.
Similar evidence of synergy was obtained in BL0645 tumors treated with therapeutic levels of [14C]carboplatin or [14C]gemcitabine (Figure 6). A therapeutic dose of the combination [14C]carboplatin plus unlabeled gemcitabine lead to a significant increase in adduct levels (94.8 ± 16.0 versus 209.6 ± 64.7 monoadducts/108 nt, p = 0.032) compared to treatment with a therapeutic dose of [14C]carboplatin alone (Figure 6A). Likewise, combination treatment of therapeutic doses of [14C]gemcitabine with unlabeled carboplatin increases gemcitabine adduct levels (169.2 ± 9.1 versus 268.1 ± 182.8 adducts/108 nt, p = 0.421) compared to [14C]gemcitabine alone (Figure 6B), although this was not a significant difference. There was an approximately 100-fold difference in drug-DNA adduct formation when a 100-fold difference in drug concentration was administered for either drug (Figure 6C).
Figure 6. Dose linearity between the microdose and therapeutic dose in PDX xenograft model BL0645 and BL0293.

NSG mice were implanted with PDX model BL0645 14 days before microdosing. Animals were dose with A) 37.5 mg/kg carboplatin (50,000 dmp/g, red bar) or 37.5 mg/kg carboplatin (50,000 dpm/g) plus 9.2 mg/kg unlabeled gemcitabine (green bar), B) 9.2 mg/kg gemcitabine (1,000 dpm/g, red bar) or 9.2 mg/kg gemcitabine (1,000 dpm/g) plus 37.5 mg/kg unlabeled carboplatin (green bar). After 24h, DNA was isolated from collected tumor tissue and analyzed via AMS. C) Dose proportionality is shown by plotting microdose and therapeutic dose induced adduct level onto log scale. Student's t-test was used to measure all p values. Presented are means and error bars represent SEM.
Poor correlation of expression of selected genes with PDX drug sensitivity toward carboplatin and gemcitabine
We next correlated response to the expression of a selected panel of genes that are known to be involved in GC drug resistance including human equilibrative nucleoside transporter-1 (hENT1), ribonucleotide reductase (RNR, encoded by the RRM1 and RRM2 genes), and DNA repair protein, excision repair cross complementing group 1 (ERCC1). Increased expressions of these markers were previously reported to be involved in acquired resistance to gemcitabine and platinum-based chemotherapy in some cancers but not in others (35). We did not find any correlation of the expression of these genes with resistance. RT-PCR analysis of gemcitabine resistance marker RRM1 and RRM2 showed decreased mRNA expression in the gemcitabine sensitive model BL0293, and elevated levels in another gemcitabine sensitive model BL0440 (SI Figure 4A+B, SI Table 4). hENT1 expression was the highest in the sensitive model BL0440 and is lowest in the resistant model BL0269 (SI Figure 4C). ERCC1 is involved in DNA repair process and increased expression is reported to be associated with resistance to platinum based therapy (36). However, RT-PCR data shows that ERCC1 expression is highest in the carboplatin sensitive tumor model BL0440 compared to the resistant tumors BL0269, BL0293 and BL0645 (SI Figure 4D).
Analysis of Mutations in DNA Repair Genes in Bladder Cancer PDX Models
We investigated the mutation status of genes associated with DNA repair in each of the PDX models (BL0269, BL0293, BL0440, and BL0645). Targeted sequencing data (whole-exome or JAX Cancer Treatment Profile) for each of the PDX (26) were filtered for 184 known DNA repair genes, including those found to correlate with response to neoadjuvant chemotherapy (NAC), such as ATM, FANCC, RB1, BRCA1, BRCA2, ERCC1, ERCC2, and RAD51C (SI Table 5 and SI Table 6) (3, 36-39). A total of 112 somatic variants were identified in the four PDX models (SI Table 7). Each PDX tumor harbored two or more mutations in genes associated with sensitivity to cisplatin that are predicted to have moderate or high functional impact, including ATM/FANCC/RB1 (3) ERCC1/2 (36, 38), and homologous recombination deficiency (HRD) (39). While each PDX theoretically possesses one or more molecular determinant(s) of cisplatin sensitivity, it is notable that BL0440 exhibited the highest sensitivity and harbors the highest number of mutations (n=4), and in genes from 3 different prediction categories, in addition to a TP53 mutation. In contrast, BL0645 contained 3 mutations in BRCA1/2, which were within a single category (HRD), and displayed marked resistance to cisplatin.
Discussion
The main goal of the ongoing clinical study is to correlate carboplatin-DNA adducts to therapy response in patients. For the PDX mouse models with urothelial bladder carcinomas, we expanded on this theme to include gemcitabine incorporation into DNA. This work has provided novel insights into the biology of in vivo cellular responses to platinum-based treatment and delineates potential opportunities for personalized therapeutic intervention. Treatment for muscle-invasive bladder cancer has not advanced beyond platinum-based combination chemotherapy and surgery in the past 30 years (40). Standard of care guidelines recommend inclusion of platinum-based NAC, but this option is used in less than 20% of patients for a variety of reasons, including concerns over lack of response to the treatment. Therefore, the ability to predict response to platinum-based chemotherapy would be highly impactful.
Positive correlations between therapy-induced levels of platinum-based drug-DNA adducts in PBMC or other surrogate tissues, and good clinical outcome have been reported in literature for a variety of different solid tumors (7-11, 41). We evaluated the feasibility of a phenotypic approach to predicting response to platinum-based chemotherapy in bladder cancer patients by using diagnostic microdosing and AMS analysis of DNA isolated from PBMC and biopsy samples. The linear correlation between carboplatin-DNA adduct levels induced by microdose and therapeutic dose exposures is consistent with our previous cell culture studies (23). The linearity of the pharmacokinetics between these two drug doses (Fig 1C) can be explained by the fact that carboplatin is an alkylating agent that directly reacts with DNA without the need for metabolic activation or transformation (19). The PBMC carboplatin-DNA adduct data were distributed into two clusters: those patients with adduct levels above 0.7 monoadducts/108 nt all responded to subsequent platinum based therapy; and those patients with lower levels of adducts were a mixture of responders and nonresponders. With this cutoff there was a clear highly significant difference (p < 0.0001) between high and low adduct level groups of patients (Figure 2A). The ability to differentiate between responders and non-responders using the mean carboplatin-DNA adduct levels for each patient group has yet to reach statistical significance (p = 0.069). However, considering the small number of patients in the study so far, these statistics support the feasibility of diagnostic microdosing for bladder cancer. Ultimately, we envision using carboplatin-DNA adduct levels to predict response in patients by establishing one or more biomarker cutoff levels in a larger clinical study. These cutoff levels will then be used to define positive and negative predictive values for a diagnostic test.
We also showed evidence that killing of PDX bladder cancer cells in mice by gemcitabine and cisplatin/carboplatin requires a threshold level of drug-DNA damage from one or both drugs combined, and that microdose-induced drug-DNA adduct levels are predictive of whether cells can achieve that threshold during full dose therapy. One PDX was rendered resistant to gemcitabine by repeated therapeutic dosing. This tumor model of acquired resistance had microdose-induced gemcitabine-DNA adducts that were apparently below the threshold for this drug. Together, these data indicate that the diagnostic microdosing approach can be used as a phenotype-based predictive test for chemotherapy response.
The literature covering platinum-DNA damage and DNA repair capacity arguably supports the feasibility of PBMC as a surrogate tissue for predicting tumor response to chemotherapy. In one study, platinum-DNA adduct formation in PBMCs was found to be more predictive for tumor response to platinum-based therapy than previous platinum-based therapy, stage of disease, histological type and tumor grading (42). A significantly better disease-free survival was reported for cisplatin-treated head and neck carcinoma patients with higher adduct levels (9). However, not all investigators found a relationship between surrogate tissue adduct levels and tumor response (43, 44). In spite of several clinical investigations, the utility of platinum-DNA adducts as predictive biomarkers remains unresolved due to a lack of large studies with a common design with good measurement sensitivity and precision.
Another approach has focused on correlating DNA repair capacity of PBMCs to clinical outcomes. In a large clinical study, Wang et al measured DNA repair capacity in cultured peripheral T-lymphocytes from 591 NSCLC patients that were treated with platinum-based chemotherapy (45). They found a statistically significant overall inverse correlation between DNA repair capacity and survival, especially in early stage tumors. In another study, the ratio of γH2AX, an indicator of DNA double-strand breaks, and the DNA repair protein MRE11 in PBMC significantly correlated to cisplatin and PARP inhibitor therapy response in 15 ovarian cancer patients (46). These reports are consistent with our 24 hour PBMC data, which is enough time for DNA repair to modulate the adduct levels.
The clinical microdosing study was performed with one chemotherapeutic drug, but patients were treated with combination chemotherapy. One potential pitfall is that chemoresistance to one drug may be compensated for by the partner drug in the combination therapy. This phenomenon potentially explains the results for two of our responsive patients who had low microdose-induced carboplatin-DNA adducts. To address this issue, we used gemcitabine microdosing in PDX bearing mice, which has only previously been reported in cell culture (47). Our study indeed showed that the levels of gemcitabine incorporated into DNA correlated with the response of PDXs to chemotherapy.
Another important finding of this study is that high levels of drug-DNA adducts from either component of a two drug combination is sufficient to inhibit tumor growth. This observation supports the hypothesis that a threshold level of drug-DNA damage from one or both drugs is required to achieve a response to therapy. In bladder cancer, the first-line chemotherapy is usually GC (gemcitabine and cisplatin/carboplatin) or MVAC (methotrexate, vinblastine doxorobucin/Adriamycin and cisplatin). Many physicians assume that all of the drug components in these regimens contribute to efficacy. In contrast to this assumption, even in a limited set of PDXs, some tumors responded to only one of the drugs, whereas only one model needed both drugs synergistically in order to respond. Clearly, the diagnostic microdosing approach can dissect the influence of individual combination chemotherapy components for drugs that interact with DNA. This capability may be clinically useful considering the heterogeneity of tumor response to a given drug combination.
We undertook a molecular analysis in four PDX tumor models of a few of the major mechanistic pathways involved in platinum-based and gemcitabine drug resistance. For example, the upregulation of ERCC1 expression has been reported to correlate significantly with cisplatin resistance for some cancers (36, 48, 49). Despite this, ERCC1 has been shown to have unacceptable sensitivity and specificity as a biomarker and has thus failed to gain wide-spread acceptance as standard of care for therapeutic guidance. In this study, we saw no correlation between expression level of ERCC1 and PDX tumor response toward chemotherapy (Figure 3, SI Figures 3 and 4). Gemcitabine kills cells mainly through its incorporation into DNA and termination of DNA replication (50). Several mechanisms have been implicated in mediating gemcitabine resistance, including loss of membrane import transporters, downregulation of dCK, increased formation of inactive metabolites (dFdU), upregulation of RNR or downstream inhibition of caspase executioners that control apoptosis signaling (51, 52). Interestingly, DNA repair is not associated with cellular response to gemcitabine-DNA adducts (53), even though the in vitro cytotoxicity directly correlates with the level of drug incorporation into DNA (50, 54). Analysis of three genes commonly found to be implicated in resistance toward gemcitabine based chemotherapy (RRM1, RRM2 and hENT1) showed no correlation between expression level and PDX tumor response toward chemotherapy (Figure 3, SI Figures 3 and 4). Interestingly, somatic mutations in DNA repair genes including RB1, FANCC, ATM and others did not obviously confer sensitivity to cisplatin, carboplatin or gemcitabine. These preliminary findings highlight the difficulty of finding generally applicable markers of chemotherapy resistance.
In our study, we measured drug-DNA adduct levels from PBMC and tumor samples as putative predictive biomarkers of therapy response. The formation and DNA repair of these adducts are governed by a myriad of factors including but not limited to hundreds of genes, gene-gene interactions, proteins, stroma, drug PK, organ function and even overall patient health. Therefore, drug-DNA adduct levels as pharmacodynamic endpoints are potentially more comprehensive and useful than genomic analysis of limited sets of markers for predicting drug response, at least for some drugs. We previously showed that platinum-DNA adduct levels and chemoresistance in isogenic cell cultures can be modulated by ERCC1 and RRM1 mRNA levels (23). Here we extended this analysis to include a limited number of bladder cancer PDX which showed no correlation of these genomic markers with therapeutic outcome.
The diagnostic microdosing approach is a platform technology. Even though this study focused on bladder cancer, the strategy can be applied to many other cancer and drug types as long as the target-drug complex can be measured by AMS. For example, our ongoing trial is also accruing non-small cell lung cancer patients. In addition, we have another trial with [14C]oxaliplatin in colorectal cancer (clincaltrials.gov identifier#: NCT02569723).
Human PK (AUC) and drug-DNA adduct formation of [14C]carboplatin in PBMC are dose proportional between a microdose and a therapeutic dose. Dose proportionality of drug-DNA adduct formation was also observed in the PDX mouse model. Microdose-induced drug-DNA adduct levels correlated with tumor response in the PDX model (inhibition of tumor growth and survival) and bladder cancer patients (tumor response). This applies to the use of [14C]carboplatin for prediction of cisplatin and carboplatin response, and also applies to the use of [14C]gemcitabine diagnostic microdosing to predict response to the nucleotide analogue gemcitabine (in mice). Consistent with our previous findings in cell culture, high levels of in vivo microdose-induced drug-DNA adduct levels correlated with tumor response.
Supplementary Material
Acknowledgments
The authors are grateful to Ryan R. Davis (UCDCCC Genomics Shared Resource, Department of Pathology and Laboratory Medicine) for his expert technical assistance for the analysis of the WES data. The results here are also based in part upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. We are grateful to all of the patients and families who contributed to this study, as well as to Susan and Gerry Knapp for their financial support.
Funding: Work was funded by NIH grants CA93373 (R. de Vere White), SBIR contracts to AMD Phase I HHSN261201000133C (P.T. Henderson), Phase II HHSN261201200048C (P.T. Henderson), LLNL grants LDRD 08-LW-100 (P.T. Henderson and M. Malfatti), NIH/NIGMS P41 RR13461 (K.W. Turteltaub), American Cancer Society (C.-X. Pan), the Knapp Family Fund (P.T. Henderson), and VA Career Development Award-2 (C.-X. Pan). Work performed (partially) at the Research Resource for Biomedical AMS, which is operated at LLNL under the auspices of the U.S. Department of Energy under contract DE-AC52-07NA27344. The Research Resource is supported by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program grant P41 RR13461. The UC Davis Comprehensive Cancer Center Genomics Shared Resource is supported by Cancer Center Support Grant P30CA093373 from the NCI.
Footnotes
Competing interests: Drs. Pan, Henderson, Zimmermann and Cimino are shareholders of Accelerated Medical Diagnostics, Inc.
References
- 1.Chen T. Overcoming drug resistance by regulating nuclear receptors. Adv Drug Delivery Rev. 2010;62:1257–64. doi: 10.1016/j.addr.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–84. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 3.Plimack ER, Dunbrack RL, Brennan TA, Andrake MD, Zhou Y, Serebriiskii IG, et al. Defects in DNA Repair Genes Predict Response to Neoadjuvant Cisplatin-based Chemotherapy in Muscle-invasive Bladder Cancer Eur Urol. 2015;68:959–67. doi: 10.1016/j.eururo.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McConkey DJ, Choi W, Shen Y, Lee IL, Porten S, Matin SF, et al. A Prognostic Gene Expression Signature in the Molecular Classification of Chemotherapy-naïve Urothelial Cancer is Predictive of Clinical Outcomes from Neoadjuvant Chemotherapy: A Phase 2 Trial of Dose-dense Methotrexate, Vinblastine, Doxorubicin, and Cisplatin with Bevacizumab in Urothelial Cancer. Eur Urol. 2016;69:855–62. doi: 10.1016/j.eururo.2015.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen D-W, Pouliot LM, Hall MD, Gottesman MM. Cisplatin Resistance: A Cellular Self-Defense Mechanism Resulting from Multiple Epigenetic and Genetic Changes. Pharmacol Rev. 2012;64:706–21. doi: 10.1124/pr.111.005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ho GY, Woodward N, Coward JIG. Cisplatin versus carboplatin: comparative review of therapeutic management in solid malignancies. Crit Rev Oncol Hematol. 2016;102:37–46. doi: 10.1016/j.critrevonc.2016.03.014. [DOI] [PubMed] [Google Scholar]
- 7.Reed E, Parker RJ, Gill I, Bicher A, Dabholkar M, Vionnet JA, et al. Platinum-DNA Adduct in Leukocyte DNA of a Cohort of 49 Patients with 24 Different Types of Malignancies. Cancer Res. 1993;53:3694–9. [PubMed] [Google Scholar]
- 8.Schellens JH, Ma J, Planting AS, van der Burg ME, van Meerten E, de Boer-Dennert M, et al. Relationship between the exposure to cisplatin, DNA-adduct formation in leucocytes and tumour response in patients with solid tumours. Br J Cancer. 1996;73:1569–75. doi: 10.1038/bjc.1996.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoebers FJP, Pluim D, Verheij M, Balm AJM, Bartelink H, Schellens JHM, et al. Prediction of treatment outcome by cisplatin-DNA adduct formation in patients with stage III/IV head and neck squamous cell carcinoma, treated by concurrent cisplatin-radiation (RADPLAT). Int J Cancer. 2006;119:750–6. doi: 10.1002/ijc.21919. [DOI] [PubMed] [Google Scholar]
- 10.Pieck AC, Drescher A, Wiesmann KG, Messerschmidt J, Weber G, Strumberg D, et al. Oxaliplatin-DNA adduct formation in white blood cells of cancer patients. Br J Cancer. 2008;98:1959–65. doi: 10.1038/sj.bjc.6604387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim ES, Lee JJ, He G, Chow C-W, Fujimoto J, Kalhor N, et al. Tissue Platinum Concentration and Tumor Response in Non–Small-Cell Lung Cancer. J Clin Oncol. 2012;30:3345–52. doi: 10.1200/JCO.2011.40.8120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang S, Zhang H, Scharadin TM, Zimmermann M, Hu B, Pan AW, et al. Molecular Dissection of Induced Platinum Resistance through Functional and Gene Expression Analysis in a Cell Culture Model of Bladder Cancer. PloS One. 2016;11:e0146256. doi: 10.1371/journal.pone.0146256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wheate NJ, Walker S, Craig GE, Oun R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010;39:8113–27. doi: 10.1039/c0dt00292e. [DOI] [PubMed] [Google Scholar]
- 14.Cimino GD, Pan CX, Henderson PT. Personalized medicine for targeted and platinum-based chemotherapy of lung and bladder cancer. Bioanalysis. 2013;5:369–91. doi: 10.4155/bio.12.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hah SS, Stivers KM, de Vere White RW, Henderson PT. Kinetics of carboplatin-DNA binding in genomic DNA and bladder cancer cells as determined by accelerator mass spectrometry. Chem Res Toxicol. 2006;19:622–6. doi: 10.1021/tx060058c. [DOI] [PubMed] [Google Scholar]
- 16.Mauthe RJ, Dingley KH, Leveson SH, Freeman SPHT, Turesky RJ, Garner RC, et al. Comparison of DNA-adduct and tissue-available dose levels of MeIQx in human and rodent colon following administration of a very low dose. Int J Cancer. 1999;80:539–45. doi: 10.1002/(sici)1097-0215(19990209)80:4<539::aid-ijc10>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 17.Sugiyama Y, Yamashita S. Impact of microdosing clinical study — Why necessary and how useful? Adv Drug Delivery Rev. 2011;63:494–502. doi: 10.1016/j.addr.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 18.Sandhu P, Vogel JS, Rose MJ, Ubick EA, Brunner JE, Wallace MA, et al. Evaluation of microdosing strategies for studies in preclinical drug development: demonstration of linear pharmacokinetics in dogs of a nucleoside analog over a 50-fold dose range. Drug Metab Dispos. 2004;32:1254–9. doi: 10.1124/dmd.104.000422. [DOI] [PubMed] [Google Scholar]
- 19.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discovery. 2005;4:307–20. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 20.Hah SS, Sumbad RA, de Vere White RW, Turteltaub KW, Henderson PT. Characterization of oxaliplatin-DNA adduct formation in DNA and differentiation of cancer cell drug sensitivity at microdose concentrations. Chem Res Toxicol. 2007;20:1745–51. doi: 10.1021/tx700376a. [DOI] [PubMed] [Google Scholar]
- 21.Hah SS, Henderson PT, Turteltaub KW. Towards biomarker-dependent individualized chemotherapy: exploring cell-specific differences in oxaliplatin-DNA adduct distribution using accelerator mass spectrometry. Bioorg Med Chem Lett. 2010;20:2448–51. doi: 10.1016/j.bmcl.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang S, Zhang H, Malfatti M, de Vere White R, Lara PN, Jr., Turteltaub K, et al. Gemcitabine causes minimal modulation of carboplatin-DNA monoadduct formation and repair in bladder cancer cells. Chem Res Toxicol. 2010;23:1653–5. doi: 10.1021/tx1003547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henderson PT, Li T, He M, Zhang H, Malfatti M, Gandara D, et al. A microdosing approach for characterizing formation and repair of carboplatin-DNA monoadducts and chemoresistance. Int J Cancer. 2011;129:1425–34. doi: 10.1002/ijc.25814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ognibene TJ, Bench G, Vogel JS, Peaslee GF, Murov S. A high-throughput method for the conversion of CO2 obtained from biochemical samples to graphite in septa-sealed vials for quantification of 14C via accelerator mass spectrometry. Anal Chem. 2003;75:2192–6. doi: 10.1021/ac026334j. [DOI] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Pan C-X, Zhang H, Tepper CG, Lin T-y, Davis RR, Keck J, et al. Development and Characterization of Bladder Cancer Patient-Derived Xenografts for Molecularly Guided Targeted Therapy. PloS One. 2015;10:e0134346. doi: 10.1371/journal.pone.0134346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.ICRP The 2007 Recommendations of the International Commission on Radiological Protection. ICRP Publication 103. Ann. ICRP. 2007;37:2–4. doi: 10.1016/j.icrp.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 28.Brenner DJ, Hall EJ. Computed Tomography — An Increasing Source of Radiation Exposure. N Eng J Med. 2007;357:2277–84. doi: 10.1056/NEJMra072149. [DOI] [PubMed] [Google Scholar]
- 29.van der Vijgh WF. Clinical Pharmacokinetics of Carboplatin. Clin Pharmacokinet. 1991;21:242–61. doi: 10.2165/00003088-199121040-00002. [DOI] [PubMed] [Google Scholar]
- 30.Duffull S, Robinson B. Clinical Pharmacokinetics and Dose Optimis ation of Carboplatin. Clin Pharmacokinet. 1997;33:161–83. doi: 10.2165/00003088-199733030-00002. [DOI] [PubMed] [Google Scholar]
- 31.Sharma H, Thatcher N, Baer J, Zaki A, Smith A, McAucliffe CA, et al. Blood clearance of radioactively labelled cis-diammine 1,1-cyclobutane dicarboxylate platinum (II) (CBDCA) in cancer patients. Cancer Chemother Pharmacol. 1983;11:5–7. doi: 10.1007/BF00257407. [DOI] [PubMed] [Google Scholar]
- 32.Harland SJ, Newell DR, Siddik ZH, Chadwick R, Calvert AH, Harrap KR. Pharmacokinetics of cis -diammine-1,1-cyclobutane dicarboxylate platinum(II) in patients with normal and impaired renal function. Cancer Res. 1984;44:1693–7. [PubMed] [Google Scholar]
- 33.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–47. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 34.Bondarenko G, Ugolkov A, Rohan S, Kulesza P, Dubrovskyi O, Gursel D, et al. Patient-Derived Tumor Xenografts Are Susceptible to Formation of Human Lymphocytic Tumors. Neoplasia. 2015;17:735–41. doi: 10.1016/j.neo.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferrandina G, Mey V, Nannizzi S, Ricciardi S, Petrillo M, Ferlini C, et al. Expression of nucleoside transporters, deoxycitidine kinase, ribonucleotide reductase regulatory subunits, and gemcitabine catabolic enzymes in primary ovarian cancer. Cancer Chemother Pharmacol. 2010;65:679–86. doi: 10.1007/s00280-009-1073-y. [DOI] [PubMed] [Google Scholar]
- 36.Ozcan MF, Dizdar O, Dincer N, Balcı S, Guler G, Gok B, et al. Low ERCC1 expression is associated with prolonged survival in patients with bladder cancer receiving platinum-based neoadjuvant chemotherapy. Uro Oncol. 2013;31:1709–15. doi: 10.1016/j.urolonc.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 37.Liu D, Plimack ER, Hoffman-Censits J, et al. CLinical validation of chemotherapy response biomarker ercc2 in muscle-invasive urothelial bladder carcinoma. JAMA Oncology. 2016;2:1094–6. doi: 10.1001/jamaoncol.2016.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun J-M, Sung J-Y, Park SH, Kwon GY, Jeong BC, Seo SI, et al. ERCC1 as a biomarker for bladder cancer patients likely to benefit from adjuvant chemotherapy. BMC Cancer. 2012;12:1–8. doi: 10.1186/1471-2407-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manié E, Popova T, Battistella A, Tarabeux J, Caux-Moncoutier V, Golmard L, et al. Genomic hallmarks of homologous recombination deficiency in invasive breast carcinomas. Int J Cancer. 2016;138:891–900. doi: 10.1002/ijc.29829. [DOI] [PubMed] [Google Scholar]
- 40.Network NCC. NCCN Clinical Practice Guidelines in Oncology for Bladder Cancer. 2012:1. http://wwwnccnorg/professionals/physician_gls/f_guidelinesasp.
- 41.van de Vaart PJM, Belderbos J, de Jong D, Sneeuw KCA, Majoor D, Bartelink H, et al. DNA-adduct levels as a predictor of outcome for NSCLC patients receiving daily cisplatin and radiotherapy. Int J Cancer. 2000;89:160–6. [PubMed] [Google Scholar]
- 42.Reed E, Ostchega Y, Steinberg SM, Yuspa SH, Young RC, Ozols RF, et al. Evaluation of Platinum-DNA Adduct Levels Relative to Known Prognostic Variables in a Cohort of Ovarian Cancer Patients. Cancer Res. 1990;50:2256–60. [PubMed] [Google Scholar]
- 43.Motzer RJ, Reed E, Perera F, Tang D, Shamkhani H, Poirier MC, et al. Platinum-DNA adducts assayed in leukocytes of patients with germ cell tumors measured by atomic absorbance spectrometry and enzyme-linked immunosorbent assay. Cancer. 1994;73:2843–52. doi: 10.1002/1097-0142(19940601)73:11<2843::aid-cncr2820731130>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 44.Bonetti A, Apostoli P, Zaninelli M, Pavanel F, Colombatti M, Cetto GL, et al. Inductively coupled plasma mass spectroscopy quantitation of platinum-DNA adducts in peripheral blood leukocytes of patients receiving cisplatin- or carboplatin-based chemotherapy. Clin Cancer Res. 1996;2:1829–35. [PubMed] [Google Scholar]
- 45.Wang L-E, Yin M, Dong Q, Stewart DJ, Merriman KW, Amos CI, et al. DNA Repair Capacity in Peripheral Lymphocytes Predicts Survival of Patients With Non–Small-Cell Lung Cancer Treated With First-Line Platinum-Based Chemotherapy. J Clin Oncol. 2011;29:4121–8. doi: 10.1200/JCO.2010.34.3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee J-M, Gordon N, Trepel JB, Lee M-J, Yu M, Kohn EC. Development of a multiparameter flow cytometric assay as a potential biomarker for homologous recombination deficiency in women with high-grade serous ovarian cancer. J Transl Med. 2015;13:1–12. doi: 10.1186/s12967-015-0604-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wickremsinhe ER, Lutzke BS, Jones BR, Schultz GA, Freeman AB, Pratt SE, et al. Quantification of Gemcitabine Incorporation into Human DNA by LC/MS/MS as a Surrogate Measure for Target Engagement. Anal Chem. 2010;82:6576–83. doi: 10.1021/ac100984h. [DOI] [PubMed] [Google Scholar]
- 48.Zhao H, Zhang H, Du Y, Gu X. Prognostic significance of BRCA1, ERCC1, RRM1, and RRM2 in patients with advanced non-small cell lung cancer receiving chemotherapy. Tumour Biol. 2014;35:12679–88. doi: 10.1007/s13277-014-2592-7. [DOI] [PubMed] [Google Scholar]
- 49.Zhang GB, Chen J, Wang LR, Li J, Li MW, Xu N, et al. RRM1 and ERCC1 expression in peripheral blood versus tumor tissue in gemcitabine/carboplatin-treated advanced non-small cell lung cancer. Cancer Chemother Pharmacol. 2012;69:1277–87. doi: 10.1007/s00280-012-1834-x. [DOI] [PubMed] [Google Scholar]
- 50.Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991;51:6110–7. [PubMed] [Google Scholar]
- 51.Morgan MA, Parsels LA, Parsels JD, Mesiwala AK, Maybaum J, Lawrence TS. Role of Checkpoint Kinase 1 in Preventing Premature Mitosis in Response to Gemcitabine. Cancer Res. 2005;65:6835–42. doi: 10.1158/0008-5472.CAN-04-2246. [DOI] [PubMed] [Google Scholar]
- 52.Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol. 2006;17(suppl 5):v7–v12. doi: 10.1093/annonc/mdj941. [DOI] [PubMed] [Google Scholar]
- 53.Alli E, Sharma VB, Hartman AR, Lin PS, McPherson L, Ford JM. Enhanced sensitivity to cisplatin and gemcitabine in Brca1-deficient murine mammary epithelial cells. BMC Pharmacology. 2011;11:7. doi: 10.1186/1471-2210-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang P, Chubb S, Plunkett W. Termination of DNA synthesis by 9-beta-D-arabinofuranosyl-2-fluoroadenine. A mechanism for cytotoxicity. J Biol Chem. 1990;265:16617–25. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.