

Abstract
The first structure-activity relationships for a benzothiazole scaffold acting as an antagonist at GPR35 is presented. Analogues were designed based on a lead compound that was previously determined to have selective activity as a GPR35 antagonist. The synthetic route was modular in nature to independently explore the role of the middle and both ends of the scaffold. The activities of the analogues illustrate the importance of all three segments of the compound.
Keywords: GPR35, Antagonist, G protein-coupled receptor, Benzothiazole, SAR
GPR35, a G protein-coupled receptor (GPCR), was discovered and classified as an orphan GPCR in 19981 and deorphanized in 2006 by the discovery of kynurenic acid as the endogenous agonist. 2 Since its discovery, limited references on the GPR35 receptor have appeared, due in part to a scarcity of endogenous or exogenous ligands that regulate GPR35 with sufficient potency and selectivity. The deorphanization of GPR35 receptor has even been debated due to the poor potency of kynurenic acid (IC50 of 39 μM) and it has recently been proposed that GPR35 should be renamed to CXCR8 due to a reported binding to CXCL17.3 GPR35 has gained special consideration as a result of its association with many diseases including type-2 diabetes,4 nociceptive pain,5 inflammation,1 mild mental retardation syndrome,6 metabolic disorders, 7 and gastric cancer.8 Based on the association of GPR35 with these major diseases and disorders, there is a current and urgent need for compounds to regulate the GPR35 receptor and serve as research tools in molecular characterization of GPR35 receptor. Herein, we report a structure-activity relationship (SAR) study of benzothiazoles as antagonists of GPR35. This research sets the foundation for the design of future potent and selective ligands.
An image-based high-throughput, high-content primary screen using the Molecular Libraries Probe Production Initiative evaluated ~300,000 compounds.9 This screen identified a few relatively potent and selective GPR35 antagonists: benzothiazole 1 (CID1231538), rhodanine 12 (CID2286812), and pyrazole 13 (CID2745684; Fig. 1). Compound 12 displayed high antagonism activity at GPR35 (IC50 = 20.1 nM), however, this compound has a rhodanine ring. Compounds with a rhodanine ring have been recommended to be excluded from library screening results due to the potentially misleading results, as described as Pan Assay Interference Compounds (PAINS).10 Compound 13 is also a potent GPR35 antagonist (IC50 = 160 nM), but the thiosemicarbazone functionality could also react in a nonselective manner, similar to the rhodanine ring.10 Although benzothiazole 1 was not as potent as the other potential leads (IC50 = 0.55 μM), it was selected because it could be synthetically modified and had a greater opportunity for selectivity since it was inherently less reactive for covalent attachment.
Figure 1.

GPR35 antagonists.
The route to synthesize compounds 1–3 and 8–10 initiated with ring-opening of phthalic anhydride 14 using 6-amino-2-mercaptobenzothiazole 15 to form acid 16 in 96% yield or with 5-amino-2- mercaptobenzoimidazole to form compound 17, also in good yield (95%) (Scheme 1). Alkylation of thiol 16 with 4-chlorobenzyl chloride, benzyl bromide, or 4-(chloromethyl) pyridine hydrochloride successfully produced thioethers 18, 19, and 20, respectively. These different aryl groups would test the potential and type of aromatic stacking.
Scheme 1.

General synthetic route to benzothiazole analogues. Reagents and conditions: (i) THF, 30 °C, 17 h; (ii) NaOH, EtOH, 0 °C to rt, ArCH2Cl, 17 h; (iii) (CH3CO)2O, Et3N, 1,4-dioxane, rt, 17 h; (iv) R2NH, THF, 50–35 °C, 15 h. See Supplementary data for specific conditions and yields.
Formation of the phthalisoimide ring was accomplished using acetic anhydride and triethylamine.11 Phthalisoimide intermediates 22, 23, and 24 were produced in 91%, 96%, and 87% yields, respectively, and were opened using morpholine, piperidine, or pyrrolidine to produce final analogues 1, 2, 3, 8, and 9. The morpholine surrogates were chosen to evaluate if there is a requirement for hydrogen bonding in that region of the receptor. Analogue 10 was synthesized following the same protocol, except using 5-amino-2-mercaptobenzoimidazole as starting substrate. The benzimidazole group was selected for synthesis to test the role of the endocyclic sulfur. Benzimidazole 10 may also provide a positive potential surface instead of negative potential surface of the sulfur.
Oxidation of the exocyclic sulfur to a sulfoxide or sulfone could potentially provide strong and directional hydrogen bonds. Sulfoxide 4 and sulfone 5 were synthesized using different amounts of meta-chloroperoxybenzoic acid (mCPBA) to improve this interaction (Scheme 2). Due to the chiral nature of the sulfoxide, enantiomers 6 and 7 were individually synthesized following procedures similar to those used to synthesize Nexium (esomeprazole). 12 The individual sulfoxide enantiomers (6 and 7) were synthesized using titanium tetraisopropoxide and the respective enantiomers of diethyl tartrate and cumene hydroperoxide as the oxidant. The absolute configuration of sulfoxides 6 and 7 were inferred based on the close analogy to the process for esomeprazole. 12 It was decided that the confirmation of the absolute configurations would be made if either of the two enantiomers were significantly potent.
Scheme 2.

Oxidation of the exocyclic sulfur. Reagents and conditions: (i) mCPBA (1 eq), CH2Cl2, 0 °C to rt., 3 h, 82%; (ii) mCPBA (2.5 eq), CH2Cl2, 0 °C to rt., 5 h, 83%; (iii) (−) DET, Ti(Oi-Pr)4, C6H5C(CH3)2OOH, DIPEA, toluene, 2 h, 59%; (iv) (+) DET, Ti (Oi-Pr)4, C6H5C(CH3)2OOH, DIPEA, toluene, 2 h, 53%.
Compound 11 was synthesized in a racemic fashion with this study and plans for an enantioselective synthesis could be developed based on the activity of racemic 11. Synthesis of compound 11 followed a slightly different synthetic route due to the ineffectuality of the SN2 reaction on the secondary position (Scheme 3). The synthesis began with nucleophilic addition of the commercially available 1-phenylethyl mercaptan 26 to 2-chloro-6- nitrobenzothiazole 27 to form nitrobenzothiazole 28 in 80% yield. The nitro group was then reduced to amine 29 through catalytic hydrogenation. The resulting aniline was used to open phthalic anhydride to produce acid 30 in high yield. Formation of the phthalisoimide ring (31) and opening by morpholine successfully produced analogue 11 in 52% yield over three steps.
Scheme 3.

Synthesis of compound 11. Reagents and conditions: (i) NaH, DMF, 5 h, 80%; (ii) H2, Pd/C, EtOH, 17 h; (iii) phthalic anhydride, THF, 30 °C, 17 h; (iv) (CH3- CO)2O, Et3N, 1,4-dioxane, rt, 17 h, (52% over 3 steps); (v) morpholine, THF, 35 °C, 15 h, 76%.
The activities of the synthesized analogues as antagonists at GPR35 were examined using U2OS cells permanently expressing HA-GPR35a and βarr2-GFP (UGPR35β) assay; 10 μM Zaprinast was used as the agonist as in our previous publication.9 Concentration- effect curves for agonist-mediated receptor activation were analyzed by nonlinear regression techniques using GraphPad Prism 5.0 software (GraphPad) and data were fitted to sigmoidal dose-response curves to obtain IC50 (Fig. 2, curves for inactive and less active compounds are not shown for clarity).
Figure 2.

Concentration-response curves for GPR35 antagonists that had obtainable IC50 values.
Evaluation of the activities as antagonists at GPR35 for the analogues provide considerable information (Table 1). One of the first observations is that the parent compound is not as active as was previously determined. After some more extensive studying of this, it was determined that many of the benzothiazoles were not sufficiently stable at room temperature and decomposed within 15 h when exposed to air and light. To get reproducible data, the analogues were all frozen in DMSO until they were analyzed for their activity. The activity of the parent compound is still less than was previously determined. This could be due to the initially screened compound being partially decomposed into an unknown mixture of compounds. Fortunately, the parent compound was still moderately active when pure, which allowed for trends to be observed.
Table 1.
Biological results.
| Entry | Structure | IC50 (CI)a |
|---|---|---|
| 1 |
 1 |
4.56 (1.8–12) |
| 2 |
 2 |
>30 |
| 3 |
 3 |
>30 |
| 4 |
 4 |
13 (4.0–45) |
| 5 |
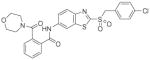 5 |
5.24 (2.2–13) |
| 6 |
 6 |
>30 |
| 7 |
 7 |
>30 |
| 8 |
 8 |
7.23 (2.3–23) |
| 9 |
 9 |
>30 |
| 10 |
 10 |
>30 |
| 11 |
 11 |
17.0 (5.6–54) |
CI = 95% Confidence Intervals.
One of the most important findings was that piperidine 2 and pyrrolidine 3 are devoid of activity as GPR35 antagonists. This indicates that there is probably a strong interaction between the oxygen of the morpholine ring and a residue in the binding site in this region. Prior modeling studies placed the morpholine ring in the region of the toggle switch,9,13 so these data can be used to further refine the model.
Oxidation of the exocyclic sulfur to sulfoxides 4, 6 and 7 and sulfone 5 decreased the activity as an antagonist. These data indicate that this area of the molecule is either not amenable to expansion or is in a non-polar environment. The electronics of the benzothiazole are also modified by having the adjacent electron-withdrawing group; this might also be detrimental. The individual enantiomers (6 and 7) appear to be less active than the racemate (4), however, the confidence intervals (CI) for the IC50 values are overlapping for sulfoxides 4, 6, and 7, which explains this otherwise unexpected result. Pyridine 9 was found to be inactive as a GPR35 antagonist, which is an indication that the basicity of the nitrogen of the pyridine ring produced an unfavorable negative potential surface. The same scenario exists when the cyclic sulfur is replaced with a nitrogen atom in benzimidazole 10.
In conclusion, we report herein an initial structure-activity relationships study for a family of benzothiazoles that act as antagonists at GPR35. A major finding was that a seemingly innocuous oxygen of the morpholine ring actually plays a major role. This and other results, including the location and type of heteroatom substitutions, could be used in concert with an updated GPR35 model, to generate more potent GPR35 antagonists.
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health Grants R21NS077347 and R01DA023204, P30DA013429. The authors thank Dr. Franklin J. Moy (UNCG) for assisting with analysis of NMR data and Dr. Brandie M. Ehrmann (UNCG) for acquisition of the high resolution mass spectrometry data at the Triad Mass Spectrometry Laboratory at the University of North Carolina at Greensboro.
Footnotes
Supplementary data (synthetic procedures and characterization of all compounds) associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmcl.2016.12.012.
References
- 1.O’Dowd BF, Nguyen T, Marchese A, et al. Genomics. 1998;47:310. doi: 10.1006/geno.1998.5095. [DOI] [PubMed] [Google Scholar]
- 2.Wang J, Simonavicius N, Wu X, et al. J Biol Chem. 2006;281:22021. doi: 10.1074/jbc.M603503200. [DOI] [PubMed] [Google Scholar]
- 3.Maravillas-Montero JL, Burkhardt AM, Hevezi PA, Carnevale CD, Smit MJ, Zlotnik A. J Immunol. 2015;194:29. doi: 10.4049/jimmunol.1401704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horikawa Y, Oda N, Cox NJ, et al. Nat Genet. 2000;26:163. doi: 10.1038/79876. [DOI] [PubMed] [Google Scholar]
- 5.Ohshiro H, Tonai-Kachi H, Ichikawa K. Biochem Biophys Res Commun. 2008;365:344. doi: 10.1016/j.bbrc.2007.10.197. [DOI] [PubMed] [Google Scholar]
- 6.Shrimpton AE, Braddock BR, Thomson LL, Stein CK, Hoo JJ. Clin Genet. 2004;66:537. doi: 10.1111/j.1399-0004.2004.00363.x. [DOI] [PubMed] [Google Scholar]
- 7.Leonard JN, Chu ZL. GPR35 and Modulators Thereof for the Treatment of Metabolic-Related Disorders. 20070077602 A1. US. 2007
- 8.Okumura S-I, Baba H, Kumada T, et al. Cancer Sci. 2004;95:131. doi: 10.1111/j.1349-7006.2004.tb03193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao P, Sharir H, Kapur A, et al. Mol Pharmacol. 2010;78:560. doi: 10.1124/mol.110.066746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baell JB, Holloway GA. J Med Chem. 2010;53:2719. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 11.de Toranzo EGD, Brieux JA. J Med Chem. 1967;10:982. doi: 10.1021/jm00317a070. [DOI] [PubMed] [Google Scholar]
- 12.Cotton H, Elebring T, Larsson M, Li L, Sörensen H, Von Unge S. Tetrahedron: Asymmetry. 2000;11:3819. [Google Scholar]
- 13.Zhao P, Lane TR, Gao HGL, et al. J Biol Chem. 2014;289:3625. doi: 10.1074/jbc.M113.508382. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.