

Abstract
DNA methylation patterns, which are critical for gene expression, are replicated by DNA methyltransferase 1 (DNMT1) and ubiquitin-like containing PHD and RING finger domains 1 (UHRF1) proteins. This replication is initiated by the recognition of hemimethylated CpG sites and further flipping of methylated cytosines (mC) by the Set and Ring Associated (SRA) domain of UHRF1. Although crystallography has shed light on the mechanism of mC flipping by SRA, tools are required to monitor in real time how SRA reads DNA and flips the modified nucleobase. To accomplish this aim, we have utilized two distinct fluorescent nucleobase surrogates, 2-thienyl-3-hydroxychromone nucleoside (3HCnt) and thienoguanosine (thG), incorporated at different positions into hemimethylated (HM) and nonmethylated (NM) DNA duplexes. Large fluorescence changes were associated with mC flipping in HM duplexes, showing the outstanding sensitivity of both nucleobase surrogates to the small structural changes accompanying base flipping. Importantly, the nucleobase surrogates marginally affected the structure of the duplex and its affinity for SRA at positions where they were responsive to base flipping, illustrating their promise as nonperturbing probes for monitoring such events. Stopped-flow studies using these two distinct tools revealed the fast kinetics of SRA binding and sliding to NM duplexes, consistent with its reader role. In contrast, the kinetics of mC flipping was found to be much slower in HM duplexes, substantially increasing the lifetime of CpG-bound UHRF1, and thus the probability of recruiting DNMT1 to faithfully duplicate the DNA methylation profile. The fluorescence-based approach using these two different fluorescent nucleoside surrogates advances the mechanistic understanding of the UHRF1/DNMT1 tandem and the development of assays for the identification of base flipping inhibitors.
Graphical Abstract
1. INTRODUCTION
The past decade has seen an explosion in our understanding of the underlying molecular mechanisms that govern gene expression, with epigenetics taking center stage. Epigenetics refer to the heritable phenotypic changes that occur without altering the DNA sequence. Major epigenetic markers include DNA methylation, post-translational modifications of histones, histone variants, and nucleosome positioning.1–12 In eukaryotes, DNA methylation is a heritable cytosine modification, mediated by DNA methyltransferases (DNMTs),13 which methylate cytosine’s C5, mainly in a symmetrical CpG context.14–16 One of the first steps of the reproduction of the DNA methylation profile involves recognition of hemimethylated (HM) CpG sites (i.e., only one DNA strand is methylated) generated after DNA replication, which is subsequently fully methylated through the action of DNMT1. The operation of DNMT1 on HM CpG sites does not, however, explain the high fidelity in the replication of DNA methylation patterns. In this context, UHRF1 (Ubiquitin-like containing PHD and RING Finger domains 1) is thought to play a key role as it guides DNMT1 to its DNA target. This guiding is a result of the preferential affinity of UHRF1 for HM DNA over NM DNA, achieved through flipping of methylated cytosines (mCs) via its SRA (Set and Ring Associated) domain.17–23
Crystallographic studies have shed light on the selective recognition of HM CpG sites and the mechanism of mC flipping,17,18,20 which is facilitated by a specific binding hemisphere of 2 Å radius in SRA, able to perfectly accommodate a methyl group. The flipped mC is stacked between Tyr466 and Tyr478 and further stabilized by H bonding with Asp469 and Thr479 (Figure 1a).17 Importantly, Gly448 plays a crucial role at the entry of the pocket. Its mutation to Asp drastically decreases the affinity of SRA for HM DNA and prevents mC flipping.17
Figure 1.

Structure of the used duplex and fluorescent nucleobase analogues. (a) Structure of the duplex. The interactions of the duplex with SRA, as determined by X-ray crystallography17 are indicated by arrows. Hydrogen bonding and van der Waals interactions are indicated by black and white arrows, respectively. Positions substituted by 3HCnt and thG are highlighted in green and blue, respectively. (b) Structure of the normal (N*) and tautomer (T*) excited-state forms of 3HCnt and ESIPT reaction. (c) Chemical structure of guanosine (G) and its surrogate thienoguanosine (thG).
Although crystallography reveals the “static” structural elements of the SRA/HM DNA complex, other techniques are required for monitoring in real time how SRA reads DNA and flips mC. Toward this end, fluorescence-based techniques are particularly attractive, since they are highly sensitive and information-rich.24 They rely, however, on fluorescent probes, which should respond sensitively and selectively to the molecular event of interest. The most widely used strategy for nucleic acids is the site-specific incorporation of fluorescent nucleobase analogues.25–33 Monitoring base flipping is, however, especially challenging due to several constraints. The highly confined SRA binding pocket limits the choices of mC substitutes.17,18,20 Moreover, substitution of a nucleobase in the vicinity to mC should not affect the binding of SRA, nor the stability or conformation of the duplexes. Incorporation of 2-aminopurine (2-AP) close to mC has recently been found to partly fulfill these criteria.34 This extensively used emissive nucleoside revealed the binding of SRA to HM and nonmethylated (NM) duplexes, but was insufficiently sensitive for detecting mC flipping.
In the present study, we have used two versatile fluorescent nucleobase analogues to label the structurally characterized 12-bp duplex.17 One is 2-thienyl-3-hydroxychromone (3HC, Figure 1b), a highly responsive nucleobase surrogate33,35 that behaves as a universal nucleobase and displays environmentally sensitive normal (N*) and tautomeric (T*) emission bands, due to an excited-state intramolecular proton transfer (ESIPT, Figure 1b).36–38 The other nucleobase analogue used is the recently developed isomorphic guanosine surrogate, thienoguanosine (thG), an ideal probe for faithfully monitoring nucleic acid–protein interactions (Figure 1c).31,39 Strategic positions close to the central CpG recognition site were labeled in either NM or HM duplexes. When 3HCnt replaces G5′ or C8, we observe a much larger change in emission quantum yield (QY) and N*/T* ratio on SRA binding to HM duplexes, as compared to NM duplexes. Similarly, when thG is placed at the G7 position we observe much larger SRA-induced changes in fluorescence intensity for HM duplexes compared to NM duplexes. These large changes vanish when an SRA mutant, unable to perform base flipping, is used. Stopped-flow studies reveal that the comparatively slow kinetics of mC flipping significantly increases the lifetime of the SRA/HM DNA complex as compared to the SRA/NM DNA counterpart. These observations advance the mechanistic understanding of UHRF1 and its role in the replication of DNA methylation patterns and provide a potential platform for developing screening assays aimed at targeting UHRF1.
2. EXPERIMENTAL SECTION
2.1. Materials
The SRA domain of hUHRF1 (SRA, residues 408–643) was expressed in Escherichia coli BL21-pLysS (DE3) and purified as previously described.40 The mutant G448D SRA was produced as described elsewhere.41 Unmodified oligonucleotides (DNAs) were synthesized and HPLC-purified by IBA GmbH Nucleic Acids Product Supply (Germany). 3HCnt and DNAs labeled by 3HCnt were produced as described previously.33 The sequence of the 12-bp duplex was 5′-GGGCCXGCAGGG-3′/5′-CCCTGCGGGCCC-3′ with a single CpG site that was either nonmethylated (X = C) or hemimethylated (X = 5mC). The 3HCnt was selectively introduced at different positions (5, 7, 8, 9, 6′ or 5′) within this duplex (Figure 1a).33 thG was incorporated at different positions (5′, 6′, 7, or 8′) within the duplex and kindly gifted by TriLink Biotechnologies (U.S.A.). The duplexes were prepared by mixing the complementary strands in equal molar amounts in 20 mM phosphate buffer pH 7.5 containing 50 mM NaCl and 1 mM EDTA (referred to as the buffer) and annealing them by heating to 90 °C for 5 min, followed by cooling down to room temperature. Samples were then kept on ice. EDTA was critical to avoid the formation of the anionic form of 3HCnt.42,43 Lyophilized calf thymus DNA (ctDNA) from Sigma-Aldrich was solubilized overnight at 10 °C and its molar concentration in nucleotides was determined from its absorption at 260 nm using an extinction coefficient of 6600 M−1 cm−1.
2.2. Absorption Spectroscopy
Absorption spectra were recorded on a Cary 400 spectrophotometer (Varian). Extinction coefficients at 260 nm were used to determine the concentration of single stranded sequences. Extinction coefficients for the nonlabeled sequences 5′-GGGCCCGCAGGG-3′ and 5′-CCCTGCGGGCCC-3′ were 112 500 M−1cm−1 and 97 300 M−1cm−1, respectively. The extinction coefficient for single strand DNAs labeled by 3HCnt at positions 5′ or 6′ was 97 200 M−1cm−1. For sequences labeled with 3HCnt at positions 5, 7, 8, or 9, extinction coefficients were 115 300 M−1cm−1, 113 000 M−1cm−1, 116300 M−1cm−1, and 109700 M−1cm−1 respectively. These values were corrected for the 10 000 M−1cm−1 extinction coefficient of 3HCnt at 260 nm. Extinction coefficients for single strand DNA sequences labeled with thG at positions 5′, 6′, 7, or 8′ were 87 400 M−1cm−1, 87 200 M−1cm−1, 103 000 M−1cm−1, and 88 400 M−1cm−1, respectively. The extinction coefficient for SRA or its mutants is 43 890 M−1cm−1 at 280 nm. All experiments were performed at 20 °C in a buffer containing 2.5 mM TCEP and PEG 20 000 to prevent protein adsorption on the cuvette walls.44
Melting curves of the duplexes (2 μM of each strand in the buffer) were recorded by following the temperature-dependence of the absorbance changes at 260 nm with a Cary 400 spectrophotometer (Varian) equipped with a Peltier thermostated cell holder. The optical path length of the cell was 1 cm. The temperature range was 20–80 °C, with a speed of heating of 0.5 °C/min. The melting temperatures were extracted from melting curves as described elsewhere.45
2.3. Steady-State Fluorescence Spectroscopy
Fluorescence spectra were recorded at 20 °C on a FluoroLog spectrofluorometer (Jobin Yvon) equipped with a thermostated cell compartment. Excitation wavelength was set at 374 nm for 3HCnt and 350 nm for thG. Spectra were corrected for buffer fluorescence, lamp fluctuations, and detector spectral sensitivity. QYs of the labeled duplexes in the absence or presence of the SRA protein were determined by using quinine sulfate (QY = 0.546 in 0.5 M H2SO4) as a reference.46 Measurements were performed using SRA concentrations ensuring that at least 80% of the 3HCnt/thG -labeled duplexes are bound to SRA.
Anisotropy measurements were performed on the same instrument. Excitation wavelength for 3HCnt was at 374 nm and emission was collected at 556 nm, which corresponds to the T* band emission. Excitation wavelength for thG was at 350 nm and emission was collected at 465 nm. Anisotropy values were obtained by averaging 10 measurements. The affinity of wild-type SRA and its G448D mutant to labeled DNAs was obtained by titrating a fixed amount of 3HCnt/thG -labeled duplex by the protein and monitoring the fluorescence intensity and anisotropy signals, simultaneously. The affinity constants were determined by fitting the fluorescence anisotropy changes to the following equation:
| (1) |
where r and rt are the anisotropy values at a given and a saturating SRA concentration respectively, and rd is the anisotropy in the absence of protein. R is the ratio of the QYs of the bound to free forms, Ka is the apparent affinity constant, ν is the fraction of bound SRA calculated as follows:
| (2) |
where Pt and Lt are the concentrations of SRA and duplexes, respectively, and n is the number of SRA proteins bound per duplex.24,47
2.4. Isothermal Titration Calorimetry (ITC)
Affinity of SRA for nonmodified HM DNA at 20 °C was also determined by Isothermal titration calorimetry, using a VP ITC microcalorimeter (Microcal, Northampton, MA, U.S.A.). Titration was performed by monitoring under constant stirring (310 rpm) the thermal power generated by repeated injections (interval of 4 min) of 4 μL aliquots (0.5 μL·s −1) of 40 μM HM DNA contained in the syringe into a 6 μM SRA solution contained in the 1.42 mL cell compartment of the instrument. The total heat resulting from an injection of titrant was calculated as the integral versus time of the experimental signal. A control experiment in which DNA was titrated into the buffer alone was done to determine the heat of dilution. Instrument control, data acquisition, and analysis were done with the VPViewer and Origin software provided by the manufacturer.
2.5. Stopped Flow Spectroscopy
The kinetics of SRA binding to the 3HCnt/thG-labeled duplexes was monitored using a stopped-flow apparatus (SFM-3, Bio-Logic, Claix, France). The excitation wavelength for 3HCnt/thG was 365/350 nm, and the fluorescence intensity above 530/430 nm was recorded with long-pass filters (Melles Griot, France/Wratten N° 2E, Kodak). The data recording frequency was 5 kHz for the first 200 ms and 2 kHz between 200 and 1200 ms. The dead time of the setup was 2 ms.48 The kinetic traces were recorded after fast mixing of 100 μL of each solution. The final concentration of labeled DNA was 0.3 μM, and the concentration of SRA was chosen to have 80% of DNA bound to protein. Blank experiments in which the SRA was omitted were performed under the same conditions. Dissociation experiments were performed by adding an excess of ctDNA to preformed complexes of SRA with HM or NM duplexes labeled with 3HCnt or thG. The concentration of SRA was chosen to have 50% of DNA bound to protein. The signal acquisition and experimental setup parameters were as for association measurements. Data acquisition and processing were done with the Biokine software from the instrument manufacturer.
3. RESULTS
3.1. Labeling Position Strategy and Spectroscopic Characterization of Labeled Duplexes
On the basis of the reported structure of SRA complexed to a 12 bp duplex (Figure 1a),17 the latter was labeled with either the universal nucleobase 3HCnt (Figure 1b) at positions 5, 7, 8, 5′, and 6′ or with the isomorphic guanosine surrogate thG (Figure 1c) to replace G residues at positions 5′, 6′, 7 and 8′. These sites were selected for their proximity to the methylcytosine (mC) at position 6 and their direct interaction with SRA. Position 9, not in contact with SRA, was also labeled with 3HCnt and used as a negative control.
Thermal denaturation measurements show all 3HCnt-labeled duplexes to have a minimal Tm drop (≤3 °C) (Figure S1 and Table S1 in the Supporting Information). Since the observed destabilization is lower than a single mismatch, the large surface area of 3HCnt must partially compensate for the lack of base pairing.33 The isomorphic thG-labeled duplexes show negligible ΔTm values, confirming the ability of thG to perfectly base pair with opposite C residues in duplexes (Figure S2 and Table S2), as previously documented.49
Incorporation of 3HCnt into duplexes significantly shifts its N* and T* bands, and strongly decreases their ratio in comparison to the free 3HCnt (Figure S3). Both changes are consistent with intraduplex stacking of 3HCnt and exclusion of bulk water.33 The emission QYs were rather low (0.5–2%; Table S3), likely due to the neighboring G and/or C residues, which act as efficient fluorescence quenchers.33 Importantly, the methylation of the cytosine at position 6 had negligible effect on the spectroscopic properties of 3HCnt at all incorporated positions (Table S3). Similarly, incorporation of thG into duplexes resulted in a significant decrease in QY due to the quenching behavior of adjacent G and C residues (Table S4). Noticeably, duplexes labeled with thG have similar QY at all positions (7–8.5%). In particular, the HM and NM duplexes display the same QY, indicating that the methylation of the cytosine at position 6 had negligible effect on their photophysical features.
3.2. Binding of SRA to DNA
To investigate whether incorporation of 3HCnt/thG alters the binding of SRA to its DNA target, fluorescence anisotropy changes were recorded as the labeled duplex was titrated with increasing concentrations of SRA (Figure 2a,b). Fluorescence anisotropy titrations were preferred over fluorescence intensity titrations, because limited changes in fluorescence intensity were observed mostly with NM duplexes. Assuming a 1:1 binding model, Kapp values were found to depend on the position of the label in the duplex (Figure 2c and 2d). For 3HCnt, the Kapp values for the duplexes modified at positions 8 and 5′ are close to the Kapp value obtained by ITC with the nonlabeled HM duplex (Figure S4) and the Kapp values reported in the literature,21,34 suggesting that 3HCnt at these positions minimally affects the binding of SRA. This conclusion is further strengthened by the observation that the affinity of SRA for HM duplexes labeled by 3HCnt at these positions is ~8–10 times higher than for NM duplex (Figure S5a), in line with the preferential binding of SRA to HM duplexes.17–23
Figure 2.

Binding of SRA to 3HCnt/thG-labeled HM and NM duplexes, as monitored by fluorescence anisotropy. Anisotropy titration curves for HM (red) and NM (blue) duplexes labeled by (a) 3HCnt at position 5′ and (b) thG at position 7. The concentration of duplexes was 2 μM for 3HCnt and 1 μM for thG-labeled duplexes. The lines correspond to the best fits of the experimental points to eq 1. Experimental points are expressed as means ± standard deviation for n = 2 independent experiments. Apparent affinity constants Kapp for (c) 3HCnt-labeled HM (yellow) and NM (blue) duplexes, and for (d) for thG-labeled HM (yellow) and NM (blue) duplexes. In (c) and (d), the purple lines describe the affinity of SRA to the nonlabeled HM duplex, as determined by ITC in Figure S4. Data in (c) and (d) are expressed as means ± standard deviation for n = 3 independent experiments. Experiments were done in phosphate buffer 20 mM, NaCl 50 mM, 2.5 mM TCEP, 1 mM EDTA, PEG 0.05%, pH 7.5, at T = 20 °C.
In contrast, 3HCnt at position 5 or 6′ substantially increases the affinity of the labeled HM duplex to SRA by 4- to 6-fold as compared to the nonlabeled HM duplex (Figure 2c). As numerous contacts were observed between these positions and SRA in the SRA/DNA complex,17 the complexes are likely stabilized through direct interaction between the 3HCnt and amino acid residues on the protein. Moreover, 3HCnt at positions 5, 7, and 9 was observed to suppress the preferential affinity of SRA to HM duplexes, suggesting that the structural changes induced by 3HCnt at these positions could alter the specific native interaction of SRA with mC6 observed in the crystal structure.17 Thus, the binding data suggest that to preserve the binding properties of SRA, positions 5′ and 8 are more favorable for labeling with 3HCnt.
Incorporation of the isomorphic G analogue (thG) at position 5′, 6′, and 8′ in the complementary strand results in a 2- to 3- fold decrease in the Kapp values of the HM duplexes with respect to the nonlabeled duplexes (Figure 2d). In contrast, a 2- fold increase in the Kapp value is observed for a duplex incorporating thG at position 7, next to mC6. Although these affinity differences correspond to relatively small changes in binding energy (<0.6 kcal/mol), the preferential binding of SRA to HM duplexes, with a ~ 4-fold higher affinity for NM duplexes, is observed only with thG at position 7 (Figures 2d and S5b). Modifying this position with the emissive thG appears, therefore, to be the most favorable for preserving the DNA binding properties of SRA.
3.3. Monitoring SRA–DNA Interactions by Steady- State Spectroscopy Measurements
To determine whether 3HCnt and thG could be used for monitoring SRA binding and the consequent flipping of mC at position 6, we compared the SRA-induced changes in emission spectra of the labeled HM or NM duplexes. SRA binding increases the QY of most HM and NM duplexes, but the changes were position and probe dependent (Tables S3 and S4). For 3HCnt-labeled duplexes, significant differences between HM and NM duplexes modified at positions 8 and 5′ are observed upon SRA binding (Figure 3a,b, compare red and blue spectra). Indeed, at both positions, the QY increase induced by SRA was about 2-fold higher for HM duplexes as compared to NM duplexes (Figure 4a). Concurrently, the SRA-induced changes in the N*/T* ratio are significantly different in HM and NM duplexes, only when 3HCnt is at position 5′ or 8 (Figure S6). For thG-labeled duplexes, a strong QY difference (about 2-fold) between HM and NM duplexes is only observed at position 7 (Figures 3c and 4b). As HM and NM duplexes differ only by a single methyl group at C6, the larger changes observed with HM duplexes for 3HCnt at positions 8 and 5′ and thG at position 7 are likely related to mC6 flipping.
Figure 3.
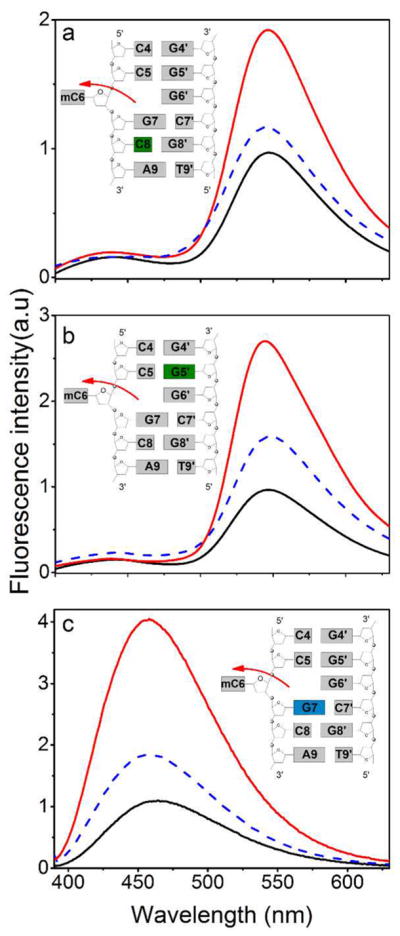
Fluorescence spectra of HM and NM duplexes labeled by (a) 3HCnt at position 8, (b) 3HCnt at position 5′ and (c) thG at position 7. Black lines: spectra of free labeled duplexes (HM and NM are superimposable), blue lines: spectra of NM duplex bound with SRA, red lines: spectra of HM duplex bound with SRA. Concentration of labeled duplexes was 1 μM, while the concentration of proteins was (a) 8.1 μM, 2.4 μM, (b) 15 μM, 2.5 μM, and (c) 1.3 μM, 2.5 μM for NM and HM DNA, respectively. The protein concentrations were chosen to ensure 80% of binding. The buffer used was the same as that given in Figure 2.
Figure 4.

Relative QY changes on binding of SRA to HM and NM duplexes labeled by (a) 3HCnt or (b) thG. QYHM/QYNM corresponds to the ratio of the QY of 3HCnt/thG in the SRA/HM-duplex complex to that in the SRA/NM-duplex. Concentration of labeled duplexes was 1 μM, while the SRA concentration was chosen to complex at least 80% of the DNA molecules (Tables S5 and S6). Data are expressed as means ± standard deviation for n = 5 independent experiments. Experiments were done in phosphate buffer 20 mM, NaCl 50 mM, TCEP 2.5 mM, 1 mM EDTA, PEG 0.05%, pH 7.5, at T = 20 °C.
Of note, the largest changes in QY and N*/T* ratios for 3HCnt are observed for positions 5, 7, and 6′ (Table S3), immediately next or opposite to position 6, confirming that 3HCnt at these positions likely interacts directly with SRA and stabilizes the SRA/duplex. This stabilization may in turn affect the proper interaction of SRA with the mC6 or C6 residues in HM and NM duplexes, respectively, as suggested by the marginal differences between the binding constants and spectroscopic properties of HM and NM duplexes (Figure 2c and Figure 4). However, only limited changes in QY and low sensitivity to mC6 flipping were observed for 3HCnt at position 9 (Table S3) and for thG at positions 5′, 6′, or 8′ (Table S4), in line with the marginal interaction of SRA with these positions, as inferred from the crystal structure (Figure 1a).17
Taken together, the binding and spectroscopy data indicate that incorporating 3HCnt into positions 8 and 5′ or incorporating thG at position 7 does not alter the binding of SRA to both HM and NM duplexes and allows monitoring the flipping of mC6. These positions were thus selected for further investigation.
3.4. Interaction of 3HCnt/thG -Labeled HM and NM Duplexes with G448D SRA Mutant
To substantiate our conclusion that the distinct responses at the selected positions of the 3HCnt- and thG-labeled HM and NM duplexes to SRA binding reflect flipping of the mC6 nucleobase, we employed a SRA mutant where the G448 residue has been replaced by a D residue. This G448 residue is located at the entrance of the mC binding pocket and substitution of this residue to a larger one was previously suggested to sterically hinder the flipping of the methylated nucleobase.17,41 Indeed, a significant decrease in the affinity of this SRA mutant to HM duplexes is seen for both probes (Figure 5a), resulting in the loss of the preferential binding to HM duplexes, in line with previous obervations.17 In sharp contrast to the wild-type SRA, the G448D SRA mutant induces only a limited increase in the QY (Figure 5b) of the 3HCnt/thG-labeled HM duplexes, comparable with that observed for binding of both wild-type and mutant SRA to NM duplexes. These data further support that 3HCnt at positions 8 and 5′ and thG at position 7 sense the SRA-induced flipping of mC6 in a similar fashion and that SRA’s preferential affinity to HM duplexes likely relies on its ability to flip the mC6 residue into its binding pocket.17,18,20
Figure 5.

Comparison of the interaction of wild-type SRA and G448D SRA with HM and NM duplexes labeled with 3HCnt at positions 8 or 5′, or thG at position 7. (a) Affinity constants of wild-type and mutant SRA to the labeled HM (red) and NM (blue) duplexes. (b) Changes in the QY of the labeled HM (red) and NM (blue) duplexes on binding to wild-type and mutant SRA. The concentration of duplexes was 1 μM. The concentration of proteins was chosen to ensure 80% of binding (Tables S5 and S6). The buffer was that same as that in Figure 2.
3.5. Kinetics of Base Flipping
To kinetically characterize the SRA-induced flipping of mC6, we comparatively investigated the interaction of the wild-type and mutant SRA proteins with the 3HCnt- and thG-labeled HM and NM duplexes by stopped-flow. For NM duplexes, a nonresolvable 3HCnt or thG fluorescence increase was observed upon mixing with SRA, indicating the reaction was essentially completed within the dead time (2 ms) of the instrument and thus, that the apparent bimolecular rate constant of the reaction is >109 M−1 s−1 (Figure 6a–c). This very fast kinetics with an apparent rate constant greater than the “diffusion-limited” one is typical of binding of proteins to nucleic acids.50,51 For HM duplexes, a similar nonresolvable component was observed, but was followed by a much slower exponential increase of the fluorescence intensity that was completed in 0.5 s (Figure 6a–c). The final fluorescence increase was fully consistent with the QY increase seen in Figure 3. This slow component, but not the initial nonresolvable component, disappeared when the HM duplexes were mixed with the G448D mutant (Figure S7), strongly suggesting that this slow component can be attributed to the kinetics of mC6 base flipping.
Figure 6.

Interaction kinetics of wild-type SRA with HM and NM duplexes labeled at position 8 and 5′ by 3HCnt and at position 7 by thG, as monitored by stopped-flow. Kinetic traces were recorded with duplexes labeled by 3HCnt at position 8 (a) or 5′ (b), or by thG at position 7 (c). The black curves correspond to HM or NM duplexes mixed with the buffer. The blue and red solid lines are the kinetic traces for the interaction of SRA with NM and HM duplexes, respectively. (d) Observed rate constants of the interaction of SRA with HM-duplexes labeled by 3HCnt at positions 8 (red) and 5′ (black) or by thG at position 7 (cyan) as a function of the SRA/DNA ratio (Solid lines show average values). For a) to c), the concentration of duplexes was 0.3 μM. The concentration of proteins was (a) 7.7 μM, 1.9 μM, (b) 14.5 μM, 2.0 μM, and (c) 2 μM, 0.7 μM for NM and HM DNA, respectively. The protein concentrations were chosen to ensure 80% of binding. The buffer was as in Figure 2.
To further corroborate this assignment, we monitored the interaction kinetics of SRA with the 3HCnt or thG-labeled HM duplexes, using SRA/DNA ratios varying from 5:1 to 25:1, and fitted the kinetic traces to single exponential functions. The observed rate constants were found to be independent of the concentration of SRA (Figure 6d), indicating that the flipping is a first-order reaction. This reaction order was fully expected as the flipping reaction can be envisioned as a conformational rearrangement that occurs after formation of the protein–DNA complex. The rate constant value associated with this slow process was found to be nearly independent of the nature and position of the probe, being about 10 s−1 for 3HCnt at both 8 and 5′ positions and 6 s−1 for thG at position 7. This confirms that both probes monitor the same process.
Dissociation experiments were next performed by adding about 80-fold excess of calf thymus DNA (ctDNA) to preformed complexes of SRA with HM or NM duplexes labeled either at the 5′ position by 3HCnt or at the 7 position by thG (Figure 7). Addition of ctDNA to the SRA/NM complexes (Figure 7, blue traces) leads to a fast fluorescence drop, indicating a fast dissociation of the complex. An identical dissociation rate constant of 150 s−1 is found for both probes. Under similar conditions, a much slower rate constant of 2.9 s−1 is observed when ctDNA is added to a complex of SRA with 3HCnt-labeled HM duplex. In case of thG labeled HM duplex, the corresponding dissociation rate constant is 3.5 s−1, in close agreement to the above value. Moreover, this rate constant is found to be only weakly dependent on ctDNA concentration (data not shown), indicating that the slower reaction rate can be considered as first order, and likely describes the flipping back of the mC6 residue into the HM duplex. Comparison of the dissociation curves obtained with HM and NM duplexes further suggests that the flipping back of the mC6 residue is the rate-limiting step and is immediately followed by the dissociation of the complex. Taken together, the kinetic data clearly confirm that 3HCnt at position 8 or 5′ and thG at position 7 can be utilized to monitor the SRA-induced flipping of the mC6 residue, facilitating, for the first time, the determination of the rate constants associated with the extrahelical flipping of the nucleobase as well as its reaccommodation within the duplex.
Figure 7.
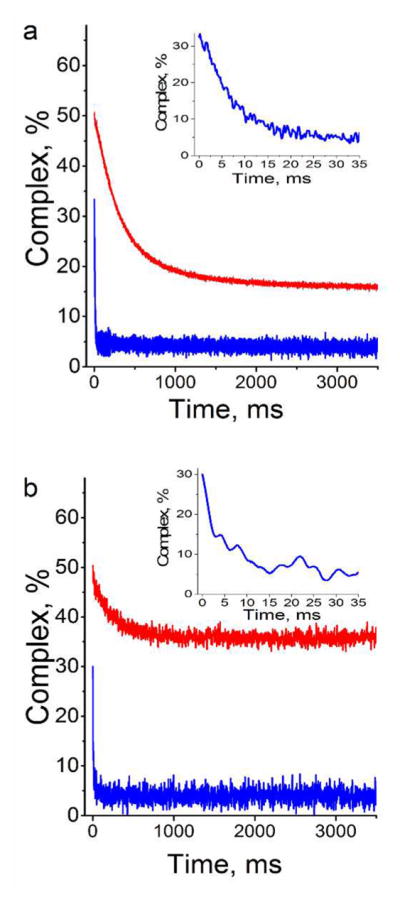
Dissociation kinetics of the complexes of SRA with HM (red) and NM (blue) duplexes labeled with (a) 3HCnt at position 5′ and (b) thG at position 7. The kinetics traces were recorded by stopped-flow after addition of an excess of ctDNA to the complexes. The concentration of HM or NM duplexes was 0.3 μM. The SRA concentration was (a) 3.6 μM, 0.6 μM and (b) 0.6 μM, 0.3 μM for NM and HM DNA, respectively. Here, the protein concentrations were chosen to ensure ~50% of binding. The concentration of ctDNA was 600 μM, as expressed in nucleotides. The measured fluorescence intensity was converted in percentage of labeled duplexes bound to SRA. Inset: Highlight of the kinetic trace recorded with the labeled NM duplex during the first 60 ms. The buffer was that same as that in Figure 2.
4. DISCUSSION
Base flipping in nucleic acids is a fundamental phenomenon. 17,20,52–57 For the first time we were able to monitor, in diluted solution and real time, the flipping of the mC6 residue of a HM duplex promoted by the SRA domain of UHRF1. This was achieved by inserting the environment-sensitive fluorescent nucleotide surrogate 3HCnt at either position 8 or 5′ or the isomorphic guanosine surrogate thG at position 7. At these selected positions, both probes marginally perturb the binding of SRA and preserve the preferential binding to HM over NM duplex (Figure 2). Both probes sense the flipping of the methylated C residue through a significant increase in their emission QY (Figure 3). Stopped-flow studies further revealed that the kinetics of mC6 flipping is much slower than the kinetics of SRA binding to the duplex (Figure 6). Importantly, the strong spectroscopic changes as well as the slow component in the kinetic traces disappear when the HM duplex is replaced by a NM duplex or when the SRA domain is mutated and unable to execute base flipping (Figure 5, 6 and S7), clearly corroborating their relationship to the nucleobase flipping.
As the 3HCnt being incorporated in the DNA undergoes an irreversible ESIPT reaction with a fast proton transfer rate constant leading to rapid accumulation of the T* form,58 the observed spectroscopic changes with SRA likely result from a decrease of the stacking interactions of the T* form of the probe with G and C neighbors.33 These decreased stacking interactions could be rationalized by considering the 3D structure of the complex of SRA with the HM duplex.17 In this complex, the Arg491 residue of SRA is inserted into the void left by the flipped mC6 residue in DNA forming tight contacts with the C5 and G7, as well as with the G5′ and G6′ residues. Thus, when 3HCnt is at position 8, the contacts of Arg491 with G7 likely reduce its ability to dynamically quench the 3HCnt probe, explaining the increase in the emission quantum yield of emissive nucleobase surrogate. Likewise, when thG is at position 7, its interaction with Arg491 residue likely reduces its quenching by adjacent nucleobases and thus, enhances its QY. Similarly, when 3HCnt is at 5′ position, the interaction of Arg491 with G6′ likely restricts the dynamic quenching of 3HCnt by this residue.
The high sensitivity of 3HCnt and thG to flipping of the methylated C residue appears quite unique, since 2-aminopurine, an established emissive nucleobase probe, was unable to sense the UHRF1-induced conformational change in the same duplex.34 For both 3HCnt and thG, their duplex placement was found to be critical for sensing the mC6 base flipping. To shed light on these observations and explain why positions 5′ and 8 were observed to be their optimal placement for sensing mC6 flipping, NMR studies were performed on a dodecamer in which a base in the center of the duplex was substituted with 3HCnt (Figure S8A). Comparison of protons chemical shifts and nOes (nuclear Overhauser effects) with those of the corresponding unmodified dodecamer (Figure S8B,C) shows that 3HCnt strongly stacks with its neighbors, and expels the “complementary” base into the major groove, due to its increased size compared to a natural nucleobase. The perturbations imposed by 3HCnt on the neighboring residues, as monitored through proton chemical shift changes, are significant for the n+1 base pair (where n stands for the probe’s strand insertion position) and to a lesser extent for the n+2 base pair, while they are weak for the base pair in position n–1 and negligible for all other base pairs. Therefore, by transposing these conclusions to the duplex studied here, the mC6 base should be minimally affected when the 3HCnt is at position 5′ or 8. In contrast, mC6 should be expelled to the major groove when 3HCnt is at position 6′ and strongly perturbed when 3HCnt is at position 5, explaining why the base flipping process could not be observed with 3HCnt at these positions. When 3HCnt is placed at position 7, the pairing of mC6 with G6′ should only be minimally perturbed. As shown, however, from the very limited change in the melting temperature for all 3HCnt-labeled duplexes, the 3HCnt likely better stacks with its flanking bases as compared to a natural base. As a result, SRA might be unable to flip mC6 when 3HCnt is at position 7. Furthermore, insertion of 3HCnt at positions 5, 6′, and 7 was found to be associated with the largest changes in QY and lead to the highest affinity with SRA. This suggests that 3HCnt at these positions may form multiple contacts with SRA, which may further hinder the proper interaction of mC6 with the protein. Finally, when 3HCnt is at position 9, it is simply too far from mC6 to sense its flipping.
By using two distinct fluorescent nucleoside analogues, 3HCnt at positions 8 or 5′ and thG at position 7, we were able to monitor the kinetics of interaction of SRA with its DNA target and reveal, for the first time, the kinetics associated with base flipping. The two probes yielded very similar kinetic parameters, enhancing the confidence in the obtained data. The interaction of SRA with the DNA duplexes was found to be faster than a “diffusion-limited” reaction, suggesting that it may proceed through a two-step “bind-and-slide” mechanism classically described for protein/DNA interactions (Scheme 1).50,51,59
Scheme 1. Proposed Mechanism for the Interaction of SRA with DNA Duplexesa.

aSRA interacts with the duplexes through a fast two-step “bind-andslide” mechanism. At CpG recognition sites, SRA flips the methylated cytosines with rate-limiting kinetics that stabilize the binding of SRA to hemi-methylated CpG sites and allows recruiting DNMT1.
In this mechanism, the protein nonspecifically binds to the duplex, and then slides to the CpG recognition site. As the sliding corresponds to a one-dimensional diffusion, this mechanism reduces the dimensionality of the search, explaining why the apparent rate constants obtained are faster than the diffusion limit. Due to the inability to kinetically resolve the binding reaction, we could not determine the values of k1, k2, k–1, and k–2. In contrast, as the flipping reaction was much slower, it could be represented as the third step in Scheme 1. The kinetic rate constants were determined to be k3 = 10 (±1) s–1 and k–3 = 2.9 (±0.3) s−1 using 3HCnt as the fluorescent probe, and k3 = 6 (±0.6) s−1 and k–3 = 3.5 (±0.4) s−1 using thG as the emissive nucleoside surrogate. From the ratio of these values, it can be deduced that the flipping step and the accompanying conformational changes stabilize the complex by about 2–3.5 folds. This stabilization is in good agreement with the 4-fold increase in affinity observed for HM over NM duplex.34 Importantly, comparison of the dissociation experiments with NM and HM duplexes (Figure 7) indicates that the k–3 value is much smaller than the k–2 and k–1 values. This suggests that the lifetime of UHRF1 bound to a CpG site in HM duplexes is much longer than in NM duplexes. This obviously increases the probability of recruiting DNMT1, in order to duplicate the DNA methylation profile. In contrast, the much shorter lifetime of SRA bound to NM duplexes is consistent with a reader role of SRA, which is able to slide rapidly along the DNA to scan for hemimethylated CpG sites.60,61
5. CONCLUSIONS
The environmentally sensitive fluorescent nucleoside analogue 3HCnt and the isomorphic guanosine surrogate thG, appear as highly suited tools for the high sensitivity monitoring SRA-induced flipping of mC and its dynamics. As the two nucleoside surrogates exhibit different photophysical properties and sensitivity to their environment, the very similar binding and kinetic data obtained with the two probes located at different positions along the duplex significantly enhance our confidence in the obtained conclusions. Hence, these two distinct and orthogonal tools could be used in parallel to further characterize the interaction of UHRF1 with its targets. Tools, such as 3HCnt and thG, will likely shed light on these events and will assist in identifying inhibitors of mC flipping by UHRF1. Such inhibitors may be useful to prevent DNA methylation pattern inheritance in cancer cells leading, for instance, to re-expression of tumor suppressor genes through the hindrance to UHRF1 functions.62,63
Supplementary Material
Acknowledgments
We thank the ANR (ANR-12-BS08-0003-02) and the FRM (DCM20111223038) for financial support and fellowships to V.K. and N.P.F.B, as well as the support and the use of resources of the French Infrastructure for Integrated Structural Biology FRISBI ANR-10-INSB-05 and of Instruct, a Landmark ESFRI project. Y.T. thanks the National Institutes of Health (grant number GM 069773) for support.
Footnotes
ORCID
Yves Mély: 0000-0001-7328-8269
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b00154.
Thermal denaturation, quantum yield, N*/T* ratio, isothermal titration calorimetry, stopped flow kinetics, and NMR data (PDF)
References
- 1.Feinberg AP, Tycko B. Nat Rev Cancer. 2004;4:143. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 2.Esteller M. N Engl J Med. 2008;358:1148. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 3.Jones PA, Baylin SB. Cell. 2007;128:683. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laird PW. Nat Rev Cancer. 2003;3:253. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- 5.Ng HH, Adrian B. Curr Opin Genet Dev. 1999;9:158. doi: 10.1016/s0959-437x(99)80024-0. [DOI] [PubMed] [Google Scholar]
- 6.Jenuwein T, Allis CD. Science. 2001;293:1074. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 7.Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. Genes Dev. 2009;23:781. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berger SL. Nature. 2007;447:407. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 9.Lorincz MC, Dickerson DR, Schmitt M, Groudine M. Nat Struct Mol Biol. 2004;11:1068. doi: 10.1038/nsmb840. [DOI] [PubMed] [Google Scholar]
- 10.Kouzarides T. Cell. 2007;128:693. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Li B, Carey M, Workman JL. Cell. 2007;128:707. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 12.Spruijt CG, Vermeulen M. Nat Struct Mol Biol. 2014;21:949. doi: 10.1038/nsmb.2910. [DOI] [PubMed] [Google Scholar]
- 13.Bestor TH, Ingram VM. Proc Natl Acad Sci U S A. 1983;80:5559. doi: 10.1073/pnas.80.18.5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bird A. Genes Dev. 2002;16:6. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 15.Jeltsch A. Nat Struct Mol Biol. 2008;15:1003. doi: 10.1038/nsmb1008-1003. [DOI] [PubMed] [Google Scholar]
- 16.Straussman R, Nejman D, Roberts D, Steinfeld I, Blum B, Benvenisty N, Simon I, Yakhini Z, Cedar H. Nat Struct Mol Biol. 2009;16:564. doi: 10.1038/nsmb.1594. [DOI] [PubMed] [Google Scholar]
- 17.Avvakumov GV, Walker JR, Xue S, Li Y, Duan S, Bronner C, Arrowsmith CH, Dhe-Paganon S. Nature. 2008;455:822. doi: 10.1038/nature07273. [DOI] [PubMed] [Google Scholar]
- 18.Hashimoto H, Horton JR, Zhang X, Bostick M, Jacobsen SE, Cheng X. Nature. 2008;455:826. doi: 10.1038/nature07280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bostick M, Kim JK, Estève PO, Clark A, Pradhan S, Jacobsen SE. Science. 2007;317:1760. doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- 20.Arita K, Ariyoshi M, Tochio H, Nakamura Y, Shirakawa M. Nature. 2008;455:818. doi: 10.1038/nature07249. [DOI] [PubMed] [Google Scholar]
- 21.Qian C, Li S, Jakoncic J, Zeng L, Walsh MJ, Zhou MM. J Biol Chem. 2008;283:34490. doi: 10.1074/jbc.C800169200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rottach A, Frauer C, Pichler G, Bonapace IM, Spada F, Leonhardt H. Nucleic Acids Res. 2010;38:1796. doi: 10.1093/nar/gkp1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frauer C, Hoffmann T, Bultmann S, Casa V, Cardoso MC, Antes I, Leonhardt H. PLoS One. 2011;6:e21306. doi: 10.1371/journal.pone.0021306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lakowicz JR. Principles of Fluorescence Spectroscopy. Springer; New York: 2006. [Google Scholar]
- 25.Secrist JA, Barrio JR, Leonard NJ. Science. 1972;175:646. doi: 10.1126/science.175.4022.646. [DOI] [PubMed] [Google Scholar]
- 26.Hawkins ME, Pfieiderer W, Mazumder A, Pommier YG, Balis FM. Nucleic Acids Res. 1995;23:2872. doi: 10.1093/nar/23.15.2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brauns EB, Madaras ML, Coleman RS, Murphy CJ, Berg MA. J Am Chem Soc. 1999;121:11644. [Google Scholar]
- 28.Okamoto A, Tanaka K, Fukuta T, Saito I. ChemBioChem. 2004;5:958. doi: 10.1002/cbic.200400010. [DOI] [PubMed] [Google Scholar]
- 29.Jones AC, Neely RK. Q Rev Biophys. 2015;48:244. doi: 10.1017/S0033583514000158. [DOI] [PubMed] [Google Scholar]
- 30.Wilson JN, Cho Y, Tan S, Cuppoletti A, Kool ET. ChemBioChem. 2008;9:279. doi: 10.1002/cbic.200700381. [DOI] [PubMed] [Google Scholar]
- 31.Sholokh M, Sharma R, Shin D, Das R, Zaporozhets Oa, Tor Y, Mély Y. J Am Chem Soc. 2015;137:3185. doi: 10.1021/ja513107r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ward DC, Reich E, Stryer L. J Biol Chem. 1969;244:1228. [PubMed] [Google Scholar]
- 33.Dziuba D, Postupalenko VY, Spadafora M, Klymchenko AS, Guérineau V, Mély Y, Benhida R, Burger A. J Am Chem Soc. 2012;134:10209. doi: 10.1021/ja3030388. [DOI] [PubMed] [Google Scholar]
- 34.Greiner VJ, Kovalenko L, Humbert N, Richert L, Birck C, Ruff M, Zaporozhets OA, Dhe-Paganon S, Bronner C, Mély Y. Biochemistry. 2015;54:6012. doi: 10.1021/acs.biochem.5b00419. [DOI] [PubMed] [Google Scholar]
- 35.Kuznetsova AA, Kuznetsov NA, Vorobjev YN, Barthes NPF, Michel BYBY, Burger A, Fedorova OS. PLoS One. 2014;9:e100007. doi: 10.1371/journal.pone.0100007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klymchenko AS, Demchenko AP. J Am Chem Soc. 2002;124:12372. doi: 10.1021/ja027669l. [DOI] [PubMed] [Google Scholar]
- 37.Kilin V, Glushonkov O, Herdly L, Klymchenko A, Richert L, Mely Y. Biophys J. 2015;108:2521. doi: 10.1016/j.bpj.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Demchenko AP, Tang KC, Chou PT. Chem Soc Rev. 2013;42:1379. doi: 10.1039/c2cs35195a. [DOI] [PubMed] [Google Scholar]
- 39.Sholokh M, Improta R, Mori M, Sharma R, Kenfack C, Shin D, Voltz K, Stote RH, Zaporozhets OA, Botta M, Tor Y, Mély Y. Angew Chem, Int Ed. 2016;55:7974. doi: 10.1002/anie.201601688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delagoutte B, Lallous N, Birck C, Oudet P, Samama JP. Acta Crystallogr, Sect F: Struct Biol Cryst Commun. 2008;64:922. doi: 10.1107/S1744309108027462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Achour M, Mousli M, Alhosin M, Ibrahim A, Peluso J, Muller CD, Schini-Kerth VB, Hamiche A, Dhe-Paganon S, Bronner C. Biochem Biophys Res Commun. 2013;430:208. doi: 10.1016/j.bbrc.2012.11.087. [DOI] [PubMed] [Google Scholar]
- 42.Tomin VI, Jaworski R. J Appl Spectrosc. 2014;81:360. [Google Scholar]
- 43.Tomin VI, Javorski R. Opt Spectrosc. 2007;103:952. [Google Scholar]
- 44.Vuilleumier C, Maechling-Strasser C, Gérard D, Mély Y. Anal Biochem. 1997;244:183. doi: 10.1006/abio.1996.9870. [DOI] [PubMed] [Google Scholar]
- 45.Breslauer KJ. Methods in Enzymology. In: Johnson ML, Ackers GK, editors. Energetics of Biological Macromolecules. Vol. 259. Academic Press; New Jersey: 1995. pp. 221–242. [DOI] [PubMed] [Google Scholar]
- 46.Melhuish WH. J Phys Chem. 1961;65:229. [Google Scholar]
- 47.Rochel N, Ciesielski F, Godet J, Moman E, Roessle M, Peluso-Iltis C, Moulin M, Haertlein M, Callow P, Mély Y, Svergun DI, Moras D. Nat Struct Mol Biol. 2011;18:564. doi: 10.1038/nsmb.2054. [DOI] [PubMed] [Google Scholar]
- 48.Maillot S, Carvalho A, Vola JP, Boudier C, Mély Y, Haacke S, Léonard J. Lab Chip. 2014;14:1767. doi: 10.1039/c3lc51283e. [DOI] [PubMed] [Google Scholar]
- 49.Shin D, Sinkeldam RW, Tor Y. J Am Chem Soc. 2011;133:14912. doi: 10.1021/ja206095a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Von Hippel PH, Berg OG. J Biol Chem. 1989;264:675. [PubMed] [Google Scholar]
- 51.Berg OG, Winter RB, von Hippel PH. Biochemistry. 1981;20:6929. doi: 10.1021/bi00527a028. [DOI] [PubMed] [Google Scholar]
- 52.Roberts RJ, Cheng X. Annu Rev Biochem. 1998;67:181. doi: 10.1146/annurev.biochem.67.1.181. [DOI] [PubMed] [Google Scholar]
- 53.Gerasimaite R, Merkiene E, Klimašauskas S. Nucleic Acids Res. 2011;39:3771. doi: 10.1093/nar/gkq1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoang C, Ferré-D’Amaré AR. Cell. 2001;107:929. doi: 10.1016/s0092-8674(01)00618-3. [DOI] [PubMed] [Google Scholar]
- 55.Parikh SS, Mol CD, Slupphaug G, Bharati S, Krokan HE, Tainer JA. EMBO J. 1998;17:5214. doi: 10.1093/emboj/17.17.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang X, Gérczei T, Glover LT, Correll CC. Nat Struct Biol. 2001;8:968. doi: 10.1038/nsb1101-968. [DOI] [PubMed] [Google Scholar]
- 57.Matthews MM, Thomas JM, Zheng Y, Tran K, Phelps KJ, Scott AI, Havel J, Fisher AJ, Beal Pa. Nat Struct Mol Biol. 2016;23:426. doi: 10.1038/nsmb.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dziuba D, Karpenko IA, Barthes NPF, Michel BY, Klymchenko AS, Benhida R, Demchenko AP, Mely Y, Burger A. Chem - Eur J. 2014;20:1998. doi: 10.1002/chem.201303399. [DOI] [PubMed] [Google Scholar]
- 59.Winter RB, Berg OG, von Hippel PH. Biochemistry. 1981;20:6961. doi: 10.1021/bi00527a030. [DOI] [PubMed] [Google Scholar]
- 60.Bronner C, Fuhrmann G, Chedin FL, Macaluso M, Dhe-Paganon S. Genet Epigenet. 2010;2009:29. [PMC free article] [PubMed] [Google Scholar]
- 61.Hashimoto H, Vertino PM, Cheng X. Epigenomics. 2010;2:657. doi: 10.2217/epi.10.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alhosin M, Sharif T, Mousli M, Etienne-Selloum N, Fuhrmann G, Schini-Kerth VB, Bronner C. J Exp Clin Cancer Res. 2011;30:41. doi: 10.1186/1756-9966-30-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bronner C, Krifa M, Mousli M. Biochem Pharmacol. 2013;86:1643. doi: 10.1016/j.bcp.2013.10.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.