

Abstract
Catalytic carbonylations of organohalides are important C–C bond formations in chemical synthesis. Carbonylations of unactivated alkyl halides remain a challenge, and currently require the use of alkyl iodides under harsh conditions and high pressures of CO. Herein, we report a palladium-catalyzed alkoxycarbonylation of secondary alkyl bromides that proceeds at low pressure (2 atm CO) under mild conditions. Preliminary mechanistic studies are consistent with a hybrid organometallic-radical process. These reactions efficiently deliver esters from unactivated alkyl bromides across a diverse range of substrates and represent the first catalytic carbonylations of alkyl bromides with carbon monoxide.
Graphical abstract

The catalytic carbonylation of organohalides is a fundamental transformation of organometallic catalysis, most notably demonstrated by the Monsanto-Cativa acetic acid synthesis.1 Carbonylations of aryl or vinyl electrophiles, or activated sp3-hybridized substrates, have also been used in diverse transformations for the synthesis of small molecules (Figure 1).2 Conversely, there are few efficient catalytic carbonylations of unactivated alkyl halides.3,4 Recent studies have demonstrated the utility of palladium catalysts in these processes, but require the use of alkyl iodides under high pressures of CO (~50 atm) using elevated temperatures or intense Xe lamp irradiation.3b,5,6 Alternatively, nickel-catalyzed carboxylations of unactivated alkyl halides have recently been reported; however, substrates are limited to primary bromides.7 The lack of simple, general protocols for carbonylations of unactivated alkyl halides significantly limits applications in chemical synthesis.
Figure 1.
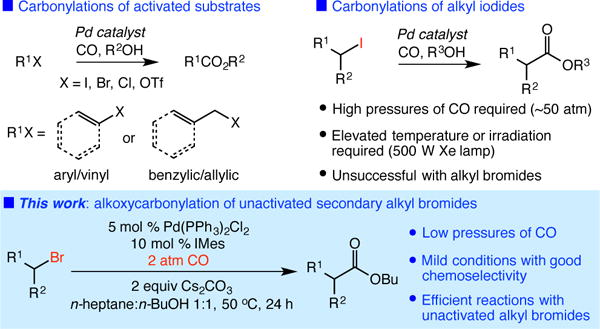
Palladium-catalyzed carbonylations of organohalides.
There are a number of challenges inherent to a catalytic carbonylation of unactivated alkyl halides with carbon monoxide. The oxidative addition of alkyl halides is expected to be more challenging in the presence of π-acidic CO, which would decrease the electron density on the metal center.8 Moreover, should a successful oxidative addition take place, undesired β-hydride elimination of an alkylmetal intermediate is prone to occur,9 especially at lower CO pressures. Herein, we report the development of an efficient catalytic alkoxycarbonylation of unactivated secondary alkyl bromides that overcomes these challenges. This palladium-catalyzed transformation enables a mild, low-pressure synthesis of diverse esters and constitutes the first examples of catalytic carbonylations of unactivated alkyl bromides with CO.
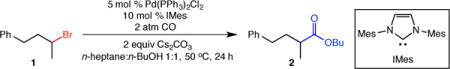
Our studies commenced with the alkoxycarbonylation of unactivated secondary alkyl bromide 1 (Table 1). We determined that a catalytic system comprised of 5 mol % Pd(PPh3)2Cl2 and 10 mol % of the N-heterocyclic carbene ligand IMes (IMes = N,N’-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene) facilitated an efficient alkoxycarbonylation of substrate 1, providing ester 2 in high yield (85%, entry 1).10 Other palladium precatalysts, such as [Pd(allyl)Cl]2 and PdCl2, were inferior to Pd(PPh3)2Cl2 (entries 2–3). Decreasing the amount of IMes ligand (5 mol %) slightly reduced the reaction yield (80% yield instead of 85% yield, entry 4). Substituting less electron-donating SIMes (SIMes = N,N’-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene) for IMes greatly reduced reaction efficiency, and the absence of IMes led to no alkoxycarbonylation (entries 5–6). Amine bases did not facilitate the reaction (entry 7), and the use of 1 atm of CO (balloon) was inferior to the optimized conditions (2 atm, entry 8). Performing the reaction in n-BuOH diminished the yield (entry 9), and no product was formed in the absence of Pd(PPh3)2Cl2 (entry 10).
Table 1.
Palladium-catalyzed alkoxycarbonylation of an unactivated secondary alkyl bromide.
| ||
|---|---|---|
|
| ||
| entry | variation from standard conditions above | % yielda |
| 1 | none | 85 |
| 2 | 2.5 mol % [Pd(allyl)Cl]2 instead of Pd(PPh3)2Cl2 | 53 |
| 3 | 5 mol % PdCl2 instead of Pd(PPh3)2Cl2 | 6 |
| 4 | 5 mol % IMes instead of 10 mol % IMes | 80 |
| 5 | 10 mol % SIMes instead of IMes | 6 |
| 6 | no IMes | <2 |
| 7 | 2 equiv Et3N instead of Cs2CO3 | <2 |
| 8 | 1 atm (balloon) CO instead of 2 atm CO | 31 |
| 9 | no n-heptane | 35 |
| 10 | no Pd(PPh3)2Cl2 | <2 |
Reactions were performed with [substrate]0 = 0.5 M. a Yields determined by 1H NMR spectroscopy of crude reaction mixture using an internal standard.
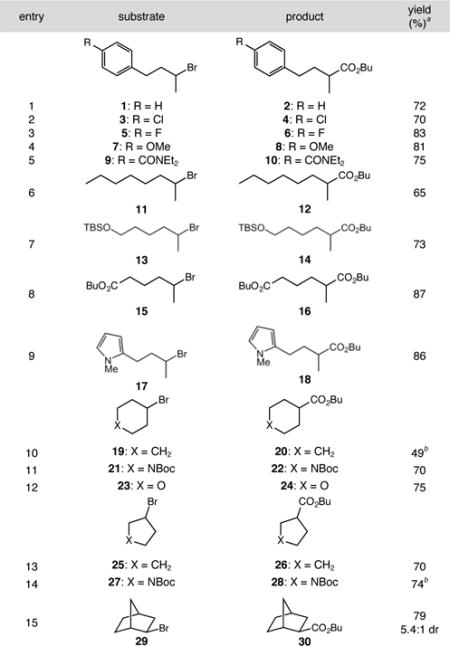
Upon identifying suitable conditions for the alkoxycarbonylation, we surveyed reactions involving a range of alkyl bromides (Table 2). A variety of aryl-substituted substrates provided esters in good yields, including one with pendant benzamide functionality (entries 1–5). Aliphatic substrate 2-bromooctane delivered butyl ester 12 in moderate yield (65%, entry 6), indicating that the aryl ring plays no role in substrate activation. Silyl protecting groups were compatible with the catalytic conditions, as demonstrated by the reaction of tert-butyldimethylsilyl ether 13 (entry 7). While pendant ester functionality was tolerated under the reaction conditions, transesterification necessitated the use of n-butyl esters (entry 8). The alkoxycarbonylation of substituted N-methylpyrrole 17 delivered ester 18 in good yield, highlighting the utility of the reaction in the presence of electron-rich aromatic systems (entry 9). Five- and six-membered carbocycles and heterocycles also reacted efficiently using our approach. Substrates examined included cyclohexyl, cyclopentyl, and tetrahydropyranyl bromides, in addition to Boc-protected piperidine and pyrrolidine substrates (entries 10–14). Lastly, exo-bromonorbornane 29 yielded primarily the exo-alkoxycarbonylation product 30 (5.4:1 dr, entry 15). Control experiments indicated that formation of the minor endo product is most likely the result of partial epimerization of the exo product under the basic reaction conditions.11
Table 2.
Low-pressure alkoxycarbonylations of secondary alkyl bromides.
|
See Table 1 for conditions. aIsolated yield. bReaction yields determined by 1H NMR spectroscopy of crude reaction mixtures using an internal standard.
We next surveyed a range of alcohols in the alkoxycarbonylation (Table 3). Importantly, these studies demonstrate the viability of the transformation when the alcohol is used in slight excess (2 equiv) rather than as reaction co-solvent. A number of primary alcohols, including those with β-branching, delivered the respective ester products in useful yields with only 2 equivalents of the alcohol as nucleophile (entries 1–5). Secondary alcohols were also effective in the alkoxycarbonylation (entries 6–8), but the reactions of isopropanol and cyclohexanol were more efficient using our standard conditions (n-heptane:alcohol 1:1).
Table 3.
Alkoxycarbonylations of an unactivated secondary alkyl bromide with diverse alcohols.
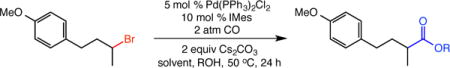
Isolated yield.
Reactions were performed with [substrate]0 = 1.0 M in PhCF3 with 2 equiv of alcohol.
Reactions were performed with [substrate]0 = 0.5 M in a mixture of n-heptane:alcohol = 1:1.
Reaction time 48 h.
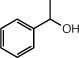
The relative stability of alkyl bromides over alkyl iodides combined with the ease of accessing alkyl bromides from their parent alcohols offers attractive opportunities for late stage C—C bond formation. As an initial demonstration of a late-stage alkoxycarbonylation, we studied the reaction of androsterone bromide 31 (eq 1). Under our optimized conditions for this substrate, the catalytic alkoxycarbonylation delivered n-butyl ester 32 in moderate yield as a mixture of diastereomers (53% isolated yield, 1.4:1 dr). The mild, catalytic transformation is successful in the presence of ketone functionality, which would present challenges in classical carboxylation using stoichiometric organometallic reagents.4e
 |
(1) |
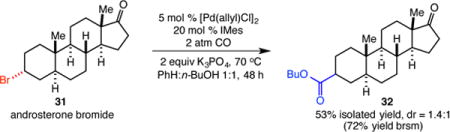
The potential for palladium to react with alkyl electrophiles via both two-electron (SN2) and single-electron pathways opens the door to a number of possible pathways for the catalytic alkoxycarbonylation.8,12 Our initial mechanistic studies commenced with reaction of alkenyl radical clock substrate 33. Under our reaction conditions, substituted cyclopentane ester 34 was the sole product, in which 5-exo cyclization preceded the carbonylation step. Importantly, the diastereoselectivity observed in the reaction is similar to reported free radical cyclizations of similar substrates (Scheme 1).3b,13 In addition, primary alkyl bromides failed to carbonylate under our conditions despite the known faster rate for SN2 oxidative addition of these substrates.11,12b These results are both consistent with the participation of radical intermediates in the carbonylation process.
Scheme 1.
Studies Probing the Reaction Mechanism
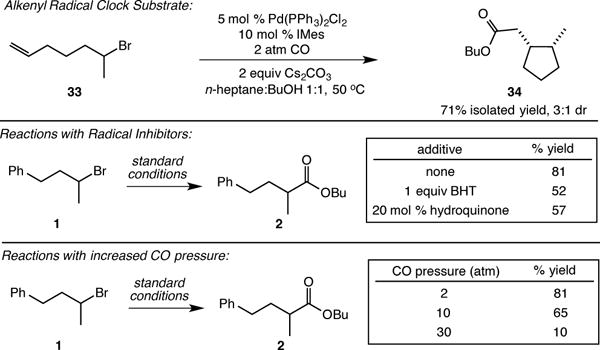
In order to uncover further details regarding the radical nature of the reaction, we studied the effect of radical inhibitors and varying pressures of carbon monoxide (Scheme 1). Reactions performed in the presence of radical inhibitors BHT and hydroquinone provided esters with only a modest decrease in yield (52% and 57% yield, respectively).14 These results are consistent with a metal-catalyzed process involving tightly associated or caged radical intermediates instead of a purely free radical carbonylation with no metal involvement.15 Furthermore, in contrast to standard, high-pressure free radical carbonylation,4a–c we found that the efficiency of the catalytic process decreased with increasing pressures of carbon monoxide. This observation is consistent with the necessity of an open coordination site on the palladium center in an inner-sphere substrate activation step, and provides evidence against an outer-sphere electron transfer.8 The high oxidation potential of alkyl bromides (~2.5 V versus SCE)16 disfavors an outer-sphere electron transfer mechanism as well. We currently favor an activation involving bromine atom abstraction by the palladium center.8 Finally, the carbonylation of an enantioenriched form of substrate 1 (96% ee) stopped at partial conversion returned the substrate with no erosion of enantiopurity, consistent with an irreversible activation step.11
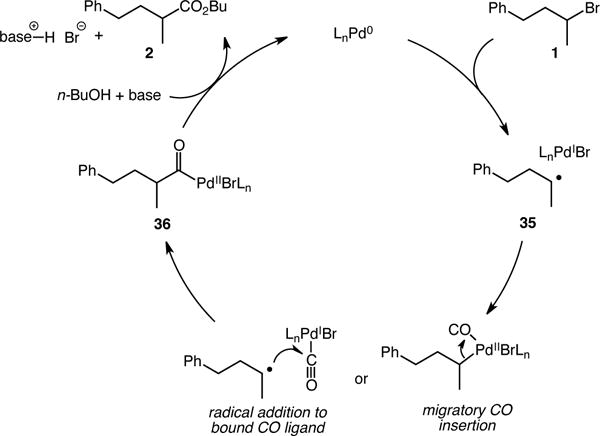
A mechanistic proposal consistent with our current studies is depicted in Scheme 2. The palladium catalyst irreversibly abstracts a bromine atom from the substrate (1), generating a carbon-centered radical (35) and a palladium(I) intermediate. This step is followed either by radical addition to a bound CO ligand, or CO migratory insertion of a putative alkylpalladium(II) intermediate formed by recombination of the carbon-centered radical and the catalyst. Both of these potential pathways have been proposed in carbonylation catalysis,17 and further studies are required to distinguish between the two possibilities. Either of these two mechanistic variants delivers an acylpalladium(II) species (36) which, upon nucleophilic displacement by butoxide, furnishes the product ester 2.
Scheme 2.
Plausible Catalytic Cycle for the Alkoxycarbonylation
In conclusion, we have developed a mild, low-pressure palladium-catalyzed alkoxycarbonylation applicable to diverse unactivated alkyl bromides. Employing a strongly donating NHC ligand enabled the development of a catalytic, fundamental C–C bond-forming transformation with simple alkyl halide building blocks which previously required harsh conditions, more reactive iodide substrates, and high pressures of carbon monoxide. Mechanistic investigations support a proposed hybrid or-ganometallic-radical pathway instead of more common two-electron transformations. Applications in complex organic synthesis and the development of enantioselective variants of the current reactions are underway.
Supplementary Material
Acknowledgments
This work was supported by Award No. R01 GM107204 from the National Institute of General Medical Sciences.
Footnotes
Supporting Information. Experimental procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Dekleva TW, Forster D. J Am Chem Soc. 1985;107:3565. [Google Scholar]; (b) Haynes A, Maitlis PM, Morris GE, Sunley GJ, Adams H, Badger PW, Bowers CM, Cook DB, Elliott PIP, Ghaffar T, Green H, Griffin TR, Payne M, Pearson JM, Taylor MJ, Vickers PW, Watt RJ. J Am Chem Soc. 2004;126:2847. doi: 10.1021/ja039464y. [DOI] [PubMed] [Google Scholar]
- 2.(a) Brennführer A, Neumann H, Beller M. Angew Chem Int Ed. 2009;48:4114. doi: 10.1002/anie.200900013. [DOI] [PubMed] [Google Scholar]; (b) Wu L, Fang X, Liu Q, Jackstell R, Beller M, Wu X-F. ACS Catal. 2014;4:2977. [Google Scholar]
- 3.(a) Frisch AC, Beller M. Angew Chem Int Ed. 2005;44:674. doi: 10.1002/anie.200461432. [DOI] [PubMed] [Google Scholar]; (b) Sumino S, Fusano A, Fukuyama T, Ryu I. Acc Chem Res. 2014;47:1563. doi: 10.1021/ar500035q. [DOI] [PubMed] [Google Scholar]
- 4.Non-metal-catalyzed carbonylations or carboxylations:; (a) Ryu I, Sonoda N. Angew Chem Int Ed. 1996;35:1050. [Google Scholar]; (b) Nagahara K, Ryu I, Komatsu M, Sonoda N. J Am Chem Soc. 1997;119:5465. [Google Scholar]; (c) Ryu I. Chem Soc Rev. 2001;30:16. [Google Scholar]; (d) Kobayashi K, Kondo Y. Org Lett. 2009;11:2035. doi: 10.1021/ol900528h. [DOI] [PubMed] [Google Scholar]; (e) Smith MB. March’s Advanced Organic Chemistry. 7th. John Wiley & Sons; Hoboken: 2013. p. 1132. [Google Scholar]
- 5.(a) Urata H, Maekawa H, Takahashi S, Fuchikami T. J Org Chem. 1991;56:4320. [Google Scholar]; (b) Fukuyama T, Nishitani S, Inouye T, Morimoto K, Ryu I. Org Lett. 2006;8:1383. doi: 10.1021/ol060123+. [DOI] [PubMed] [Google Scholar]; (c) Fusano A, Sumino S, Nishitani S, Inouye T, Morimoto K, Fukuyama T, Ryu I. Chem Eur J. 2012;18:9415. doi: 10.1002/chem.201200752. [DOI] [PubMed] [Google Scholar]
- 6.There are limited examples of catalytic carbonylative reactions of alkyl iodides at low pressure. For examples of reactions involving UV irradiation:; (a) Kondo T, Tsuji Y, Watanabe Y. Tetrahedron Lett. 1988;29:3833. [Google Scholar]; (b) Kondo T, Sone Y, Tsuji Y, Watanabe Y. J Organomet Chem. 1994;473:163. [Google Scholar]; Carbonylative Suzuki cross-couplings of alkyl iodides:; (c) Ishiyama T, Miyaura N, Suzuki A. Tetrahedron Lett. 1991;32:6923. [Google Scholar]
- 7.Liu Y, Cornella J, Martin R. J Am Chem Soc. 2014;136:11212. doi: 10.1021/ja5064586. [DOI] [PubMed] [Google Scholar]
- 8.Hartwig J. Organotransition Metal Chemistry: From Bonding to Catalysis. University Science Books; Sausalito, CA: 2010. pp. 301–320. [Google Scholar]
- 9.Bissember AC, Levina A, Fu GC. J Am Chem Soc. 2012;134:14232. doi: 10.1021/ja306323x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Secondary alkyl chlorides and tertiary alkyl bromides were unreactive under these conditions. Secondary alkyl iodides were viable substrates, albeit with decreased yields owing to their relative instability. See the Supporting Information for details.
- 11.See the Supporting Information for reaction details.
- 12.(a) Stille JK, Lau KSY. Acc Chem Res. 1977;10:434. [Google Scholar]; (b) Hills ID, Netherton MR, Fu GC. Angew Chem Int Ed. 2003;42:5749. doi: 10.1002/anie.200352858. [DOI] [PubMed] [Google Scholar]
- 13.Beckwith ALJ, Schiesser CH. Tetrahedron. 1985;41:3925. [Google Scholar]
- 14.A reaction with 1,1-diphenylethylene as a radical inhibitor delivered no carbonylation product; the mass of a radical addition product was observed via GC-MS. See the Supporting Information for details.
- 15.Loy RN, Sanford MS. Org Lett. 2011;13:2548. doi: 10.1021/ol200628n. [DOI] [PubMed] [Google Scholar]
- 16.(a) Roth H, Romero N, Nicewicz D. Synlett. 2016;27:714. [Google Scholar]; (b) Grimshaw J. Electrochemical Reactions and Mechanisms in Organic Chemistry. Elsevier; Amsterdam: 2000. [Google Scholar]
- 17.Hasanayn F, Nsouli NH, Al-Ayoubi A, Goldman AS. J Am Chem Soc. 2008;130:511. doi: 10.1021/ja072704f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.