

Abstract
Non-muscle myosin II (NMII) is a conserved force-producing cytoskeletal enzyme with important but poorly understood roles in cell migration. To investigate myosin heavy chain (MHC) phosphorylation roles in 3D migration, we expressed GFP-tagged NMIIA wild-type or mutant constructs in cells depleted of endogenous NMIIA protein. We find that individual mutation or double mutation of Ser-1916 or Ser-1943 to alanine potently blocks recruitment of GFP-NM-IIA filaments to leading edge protrusions in 2D, and this in turn blocks maturation of anterior focal adhesions. When placed in 3D collagen gels, cells expressing wild-type GFP MHC-IIA behave like parental cells, displaying robust and active formation and retraction of protrusions. However, cells depleted of NMIIA or cells expressing the mutant GFP MHC-IIA display severe defects in invasion and in stabilizing protrusions in 3D. These studies reveal an NMIIA-specific role in 3D invasion that requires competence for NMIIA phosphorylation at Ser-1916 and Ser-1943. In sum, these results demonstrate a critical and previously unrecognized role for NMIIA phosphorylation in 3D invasion.
Keywords: cell migration, cytoskeleton, extracellular matrix, invasion, myosin
Introduction
Mammalian cell migration in two dimensions (2D) is classically considered to be a multistep process that includes extension and attachment of a leading edge protrusion, coupled to formation of anterior sites of cell-substrate adhesions or focal adhesions. This is followed by cell body translocation and release of adhesions at the trailing edge of the cell and retraction of the cell posterior (1, 2). Some aspects of this traditional model are clearly valid in the more physiological three-dimensional (3D) setting, such as the fact that anterior protrusions do form focal adhesion-like structures when cells migrate in 3D collagen gels (3) and that microenvironmental stiffness and organization affect adhesion dynamics (4). However, it is also becoming clear that cancer cells in 3D environments and in tissue can display highly plastic migratory behavior, including switching between classic mesenchymal modes that are integrin-dependent and amoeboid modes that are integrin-independent (3, 5–8). The cytoskeletal motor protein non-muscle myosin II (NMII)2 is long recognized as participating in cell migration, but its exact roles and regulation in migration, particularly in 3D settings, remain poorly understood.
There are three distinct NMII heavy chain (MHC) genes (MYH9, MYH10, and MYH14) that encode MHC IIA, IIB, and IIC, respectively (9). MHC proteins homodimerize in cells and associate with two pairs of light chains (regulatory light chain (RLC) and essential light chain), to produce the intact NMII monomers referred to as NMIIA, NMIIB, and NMIIC. These monomers then assemble into bipolar filaments that associate with the actin cytoskeleton. The three isoforms have distinct expression patterns in tissues (10) and distinct biochemical properties (11, 12), suggesting unique roles in cellular processes. For example, during mesenchymal migration in 2D settings, NMIIA activity stabilizes anterior focal adhesions via unknown mechanisms, facilitating attachment of the anterior portion of the cell to the extracellular environment (13, 14). In both amoeboid and mesenchymal modes of migration, as well as a recently reported “piston” mode of migration, NMII activity is critical for anterior translocation of the nucleus through tight spaces (15–17).
NMII is classically activated by phosphorylation on RLC, which drives bipolar filament formation and activates motor activity (1). In addition to this RLC phospho-regulation, MHC IIA can be phosphorylated on several C-terminal residues, including a putative PKC target site at Ser-1916, and on a putative casein kinase II (CK-II) site at Ser-1943, within a 30-residue “non-helical tailpiece” at the very C terminus of the NMIIA tail (18–21). Studies in recent years have identified MHC phosphorylation at the tip of the NMII tail as having important roles in regulating NMII filament assembly dynamics and subcellular localization of NMII filaments (21–23). Additionally, Ser-1916 and Ser-1943 phosphorylations appear to be up-regulated during TGFβ-induced epithelial-mesenchymal transition (24), suggesting that these phosphorylation sites might be critical during invasion processes.
In this work, we have examined MHC IIA phosphorylation roles in 3D settings, uncovering previously unrecognized cooperative roles of Ser-1916 and Ser-1943 phosphorylation events as critical for upstream focal adhesion signaling, focal adhesion stabilization, and as critical for cells to extend protrusions and invade into 3D matrix. These studies support a model that adhesion stabilization and NMIIA-based force generation, although non-essential for 2D migration in many settings, are mechanistically linked with critical roles during invasive 3D migration.
Results
C-terminal Phosphorylation Sites Mediate NMIIA Recruitment to the Leading Edge and Focal Adhesion Maturation during Spreading on Matrix
In view of our earlier evidence for NMIIA C-terminal phosphorylation at Ser-1943 in regulating NMII filament localization (23), we asked whether expression of GFP MHC-IIA constructs mutated at either Ser-1943, Ser-1916, or at both sites in COS-7 cells would affect NMIIA recruitment to the leading edge during active spreading. We have previously demonstrated via mass spectroscopy that there is a transient elevation in phosphorylation on both Ser-1916 and Ser-1943 during spreading on matrix, suggesting that phosphorylation on these sites may affect NMIIA dynamics during spreading (29). COS-7 cells lack endogenous NMIIA (Fig. 1A), providing an excellent platform for analyzing the effects of expressing recombinant GFP fusion constructs. Plasmid constructs were generated carrying alanine mutations of each MHC phosphorylation site. In the case of the S1916A construct, the serine at Ser-1915 was also mutated to avoid possible spurious effects of any phosphorylation on this adjacent residue. Mutant constructs in this work are therefore referred to as GFP MHC-IIA S1915A/S1916A or GFP MHC-IIA-S1943A, or GFP MHC-IIA-3×A (indicating mutation of all three serines). COS-7 cell populations expressing wild-type GFP MHC-IIA and the mutant constructs were isolated via transient transfection and fluorescence-activated cell sorting (FACS). The FACS step allowed us to isolate cell populations with near-homogeneous expression levels, which were similar to NMIIA expression levels in other cell lines, such as HeLa and MDA-MB-231 (Fig. 1A). We fixed cells during active spreading, 60 min after plating on collagen I-coated glass, to create semi-synchronous populations of cells actively recruiting NMIIA filaments to protrusive margins. As in our earlier studies in HeLa cells (23), we found that the introduced wild-type GFP MHC-IIA was recruited robustly to the expanding lamellar margin when COS-7 cells spread on collagen I, colocalizing with lamellar F-actin (Fig. 1B, 2nd row). However, in all cells expressing the phosphorylation site mutants, the mutant protein displayed overassembly in the central regions of the cells during spreading and a near absence of recruitment to the spreading lamellae (Fig. 1B, lower panels). This defect was most dramatic in the GFP MHC-IIA 3×A mutant, indicating a cooperative role for the Ser-1916 and Ser-1943 phosphorylation sites in mediating NMIIA filament localization during protrusion and integrin/matrix engagement. This analysis supports the earlier studies from our group (23) and from the Bresnick and co-workers (22) that focused on roles of Ser-1943 phosphorylation, but it further demonstrates that Ser-1916 has critical roles in filament localization. Furthermore, these data demonstrate that in the absence of a pool of endogenous wild-type NMIIA, mutation of either site profoundly alters localization behavior of NMIIA in live cells (as opposed to modest localization defects observed in earlier expression studies performed in cells with a pool of endogenous wild-type NMIIA).
FIGURE 1.

C-terminal NMIIA phosphorylation sites are critical for recruitment of NMIIA to lamellar protrusions and for marginal paxillin phosphorylation during active cell spreading. A, whole-cell lysates of HeLa, MDA-MB-231, COS-7, and COS-7 cells expressing the indicated GFP MHC-IIA construct were subjected to Western blotting analysis with anti-MHC-IIA and anti-actin antibodies. B and C, parental COS-7 cells and COS-7 cells expressing the indicated GFP MHC-IIA construct were allowed to spread for 60 min on collagen I-coated glass, fixed, and stained with Alexa-568-phalloidin to visualize F-actin (violet) and DAPI to visualize nuclei (blue) and immunostained with anti-paxillin antibody (B) or anti-phospho-Tyr-118 paxillin antibody (C) (red). Scale bar, 20 μm. D, quantitation of paxillin versus phospho-paxillin staining in actively spreading cells. All images were obtained by confocal laser scanning microscopy and are from confocal slices taken within 2 μm of the substratum (n = 6 cells, data pooled from two different experiments performed on different dates). Data were plotted as mean ± S.D. *** indicated phospho-paxillin in GFP-MHC IIA differs from all other lines, p < 0.001.
Given the recognized role of NMII in stabilizing nascent focal adhesions at the anterior regions of migrating cells (6, 30–32), we asked whether expression of wild-type or mutant GFP MHC-IIA in COS-7 cells would affect focal adhesion localization and maturation during active spreading. Maturation was assessed by examination of paxillin localization and phosphorylation on Tyr-118, a marker of adhesion maturation (32, 33). In these actively spreading cells, total paxillin staining on the basal surface (measured via confocal slices 2 μm or less from the coverglass) was modestly increased in cells expressing GFP MHC-IIA constructs (Fig. 1, B, top row versus lower rows, and D). However, only cells expressing wild-type GFP MHC-IIA displayed prominent marginal paxillin adhesions during spreading (Fig. 1B, 2nd row). Examination of paxillin-Tyr-118 phosphorylation (named Y118-p) staining revealed strong marginal phosphorylation of paxillin in the wild-type GFP MHC-IIA cells (Fig. 1, C, 2nd row and D) that was absent in the NMIIA mutant lines (Fig. 1C, lower panels). This analysis reveals a striking requirement for NMIIA phosphorylation and for proper NMIIA localization in order for marginal paxillin Tyr-118 phosphorylation to occur.
To further define the mechanism by which NMIIA is required for adhesion stabilization, we asked whether NMIIA and its heavy chain phosphorylation sites are needed for upstream activation events in focal adhesion stabilization. During early stages of integrin clustering in response to matrix adhesion, focal adhesion kinase (FAK) is recruited via interaction with β-integrins, leading to autophosphorylation on Tyr-397. The tyrosine kinase SRC complexes with FAK, concomitant with trans-phosphorylation of FAK within the kinase domain activation loop (Tyr-576 and Tyr-577). Paxillin residue Tyr-118 is in turn a target of this kinase complex (34). We performed Western blotting analysis to test for roles of NMIIA and NMIIA heavy chain phosphorylation on these key upstream activation steps in focal adhesion assembly. During spreading, COS-7 cells rescued with GFP MHC-IIA displayed dramatically increased phosphorylation on paxillin Tyr-118, FAK Tyr-397, and Tyr-576/577 (Fig. 2A). We further tested the role of NMIIA on these phosphorylation events in the basal-like human breast cancer cell line MDA-MB-231, which normally express NMMIIA. We depleted NMIIA via shRNA and then reintroduced GFP MHC-IIA or the corresponding 3×A mutant. Depletion of endogenous NMIIA resulted in a substantial reduction in phosphorylation on paxillin Tyr-118, as well as on FAK Tyr-397 and Tyr-576–Tyr-577 during active spreading (Fig. 2B). In both the COS-7 setting and in MDA-MB-231 cells, the GFP MHC-IIA 3×A mutant did not efficiently rescue phosphorylation of any of these target sites. These experiments demonstrate that NMIIA and NMIIA phosphorylation on the C terminus of the tail have critical roles in activation of upstream signaling that leads to focal adhesion stabilization.
FIGURE 2.
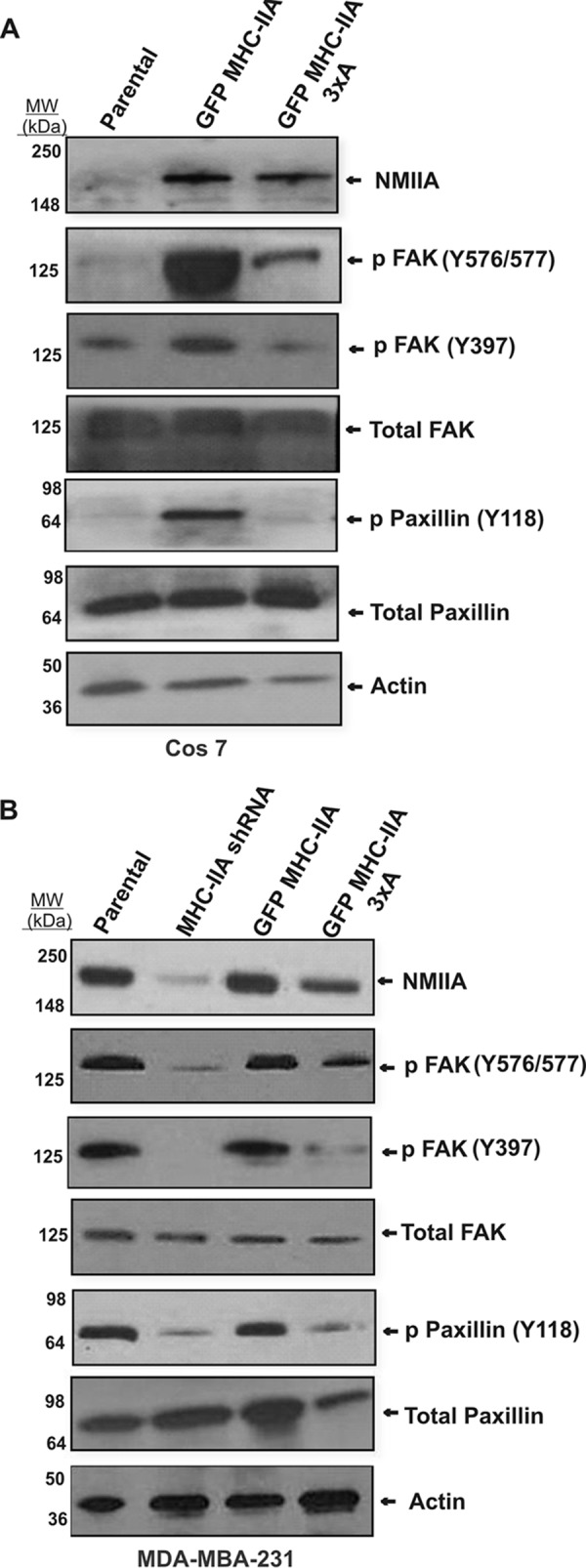
NMIIA is required for upstream signaling in focal adhesion maturation. A, COS-7 cells carrying indicated plasmid constructs were allowed to spread on fibronectin-coated cover glass for 60 min and then harvested for Western blotting analysis with indicated antibodies. B, MDA-MB-231 cells were subjected to lentivirus-based shRNA depletion of NMIIA. The shRNA cells were then transfected with indicated NMIIA constructs (right two lanes) and subjected to spreading analysis as in A.
We also evaluated paxillin phosphorylation in cells plated for 24 h to assess NMIIA roles in cells in steady-state adhesion. In contrast to the spreading condition, when COS-7 cells were plated on collagen I-coated glass for 24 h, NMIIA distribution was more variable as compared with actively spreading cells, but the GFP MHC-IIA 3×A construct still tended to be absent from the cell margin. Parental cells lacking NMIIA and cells expressing GFP MHC-IIA displayed some marginal enrichment for paxillin but also had significant basal paxillin puncta in internal regions (Fig. 3A). However, GFP MHC-IIA 3×A cells persistently accumulated paxillin in the central regions of the cells, and marginal paxillin-positive adhesions were less abundant (Fig. 3A, lower panels). Staining for paxillin-Y118-p revealed a distinct increase in the presence of this marker of focal adhesion maturation in cells transfected with either GFP MHC-IIA construct, but in contrast to cells undergoing active spreading, this staining was not restricted to the cell margin (Fig. 3, B and C). Earlier studies showed that the global inhibition of NMII activity with blebbistatin prevented paxillin phosphorylation and adhesion maturation in fibroblasts (33). The dramatic stimulation of paxillin phosphorylation in COS-7 cells upon introduction of GFP-NMIIA constructs shown here demonstrates that this effect is mediated by NMIIA, as COS-7 cells already express NMIIB.
FIGURE 3.
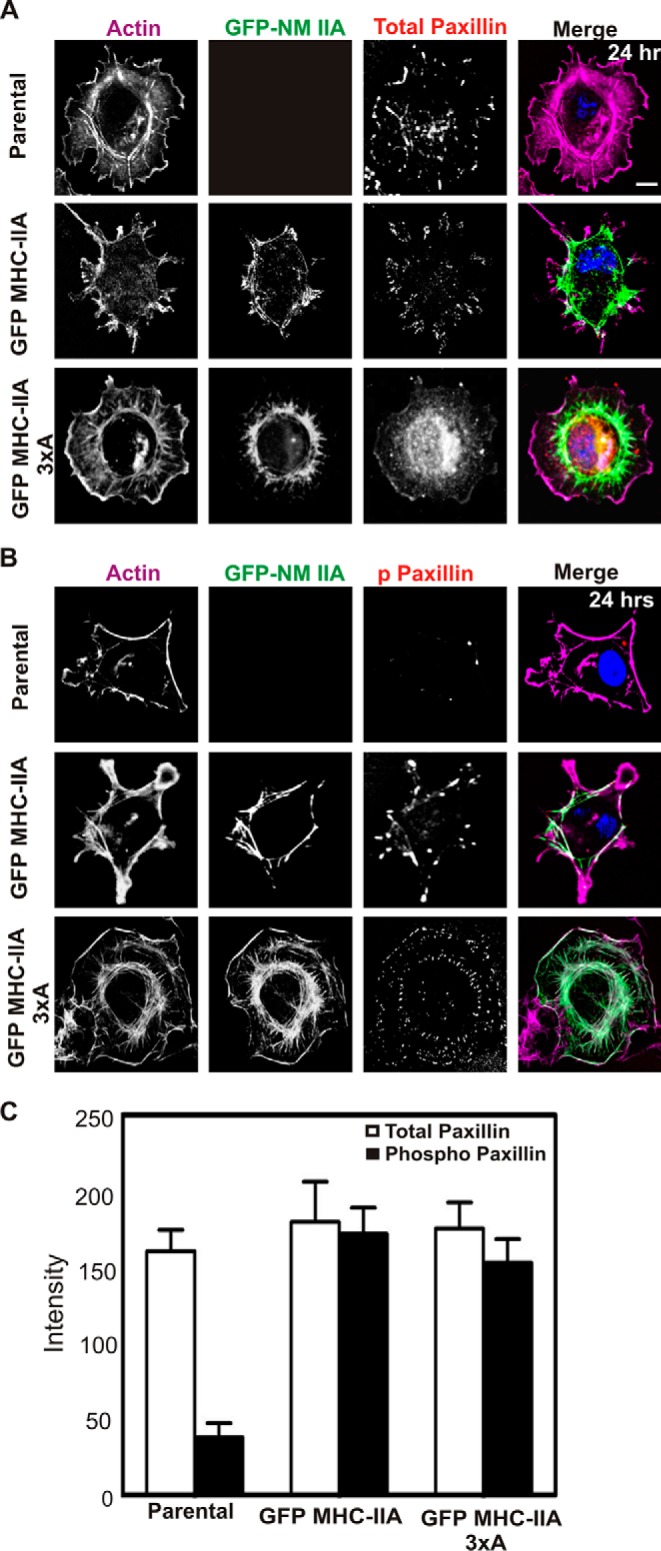
Localization of GFP-NMIIA phosphorylation site mutants and paxillin phosphorylation at 24-h plating in COS-7 cells. Parental COS-7 cells and COS-7 cells expressing GFP MHC-IIA or GFP MHC-IIA 3×A were plated for 24 h on collagen I-coated glass, fixed, and stained with Alexa-568-phalloidin to visualize F-actin (violet) and DAPI to visualize nuclei (blue) and immunostained with anti-paxillin antibody (A) or anti-phospho-Tyr-118 paxillin antibody (B) (red). Scale bar, 20 μm. C, for A and B, each image represents five 0.2-μm confocal Z-sections, collapsed, from the attached base of the cell. Signal intensity in this section of the cell was quantitated and reported as arbitrary units. Data are plotted as mean ± S.D. n = 6 cells for each line, and data were pooled from experiments performed on two different dates. At this 24-h plating time, phospho-paxillin signal for GFP MHC IIA and GFP MHC-IIA 3×A displayed no statistically significant difference.
In sum, spreading analysis demonstrates the following: (i) that introduction of GFP MHC-IIA into cells that normally lack this protein results in accurate recruitment of the GFP MHC-IIA to leading edge protrusions, behavior typically seen for endogenous NMIIA in other cell types; (ii) that introduction of wild-type GFP MHC-IIA into COS-7 cells dramatically stimulates leading edge focal adhesion maturation that is not normally present in these cells; and (iii) that NMIIA heavy chain phosphorylation on both Ser-1916 and Ser-1943 is critical both for lamellar localization of the GFP MHC-IIA and for NMIIA-driven maturation of leading edge focal adhesions.
NMIIA Phosphorylation Sites Are Critical for 3D Invasion but Not for 2D Migration
Although the cells expressing GFP MHC-IIA mutants displayed spreading rates similar to parental cells or wild-type GFP MHC-IIA cells in the 2D setting, we speculated that NMIIA phosphorylation might have a more critical role on lamellar protrusion in a setting where the external microenvironment offers resistance to protrusion extension. To test this idea, we switched to the mouse basal-like mammary gland cancer line 4T1 that displays robust 3D invasive behavior (16). Lentivirus-based shRNA, directed against the 3′-untranslated region of the MYH9 transcript, was used to deplete endogenous NMIIA. Cells were then transfected with wild-type GFP MHC-IIA or with phosphorylation site mutants. Transiently transfected populations were obtained via FACS that displayed levels of GFP MHC-IIA similar to the NMIIA expression level of the parental line (Fig. 4A). Modified scratch wound migration assays on collagen I-coated surfaces were performed with these cell populations (Fig. 4, B and C). Although the reduced migration rate of the GFP MHC-IIA 3×A was statistically significant (p = 0.01) relative to parental or MHC-IIA shRNA cells, the difference in migration rate among all the cell lines was modest in this 2D setting.
FIGURE 4.
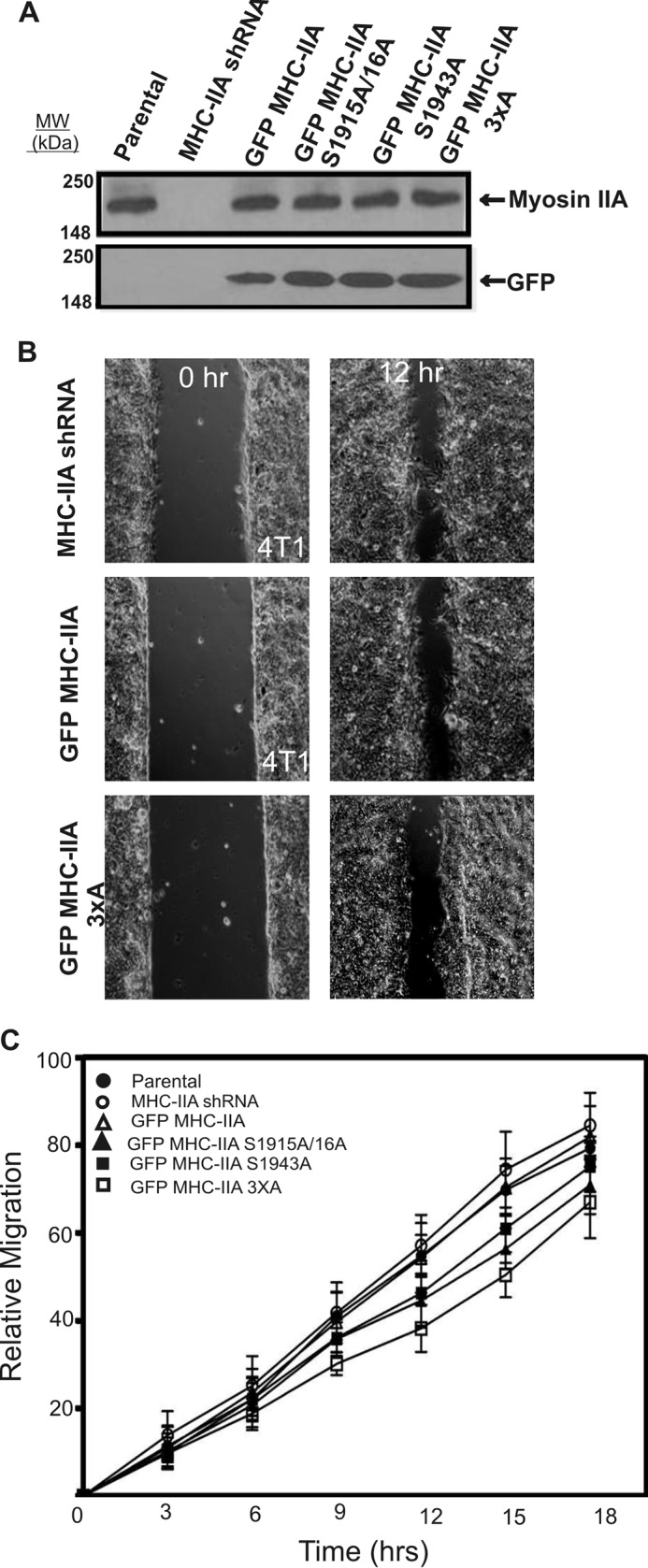
NMIIA phosphorylation is not critical for 2D migration in 4T1 mammary gland carcinoma cells. A, whole-cell lysates of parental 4T1 cells, MHC-IIA shRNA cells, and MHC-IIA shRNA cells expressing the indicated GFP MHC-IIA construct were subjected to Western blotting analysis with anti-MHC-IIA and anti-GFP antibodies. B, 4T1 cell series was analyzed for 2D migration using a modified scratch wound assay on collagen I-coated glass surfaces and time-lapse imaging. Sample images from parental GFP MHC-IIA-expressing and GFP MHC-IIA 3×A-expressing 4T1 cells are shown at t = 0 (left column) and t = 12 h (right column). C, quantification of 2D migration derived from time-lapse imaging reveals modest change upon MHC-IIA shRNA or upon re-expression of NMIIA phosphorylation site mutants. Data were plotted as mean ± S.D., n = 9 fields of view, with three fields of view quantified from each of three separate experiments performed on different dates. Migration time points were analyzed by one-way analysis of variance (Tukey-Kramer test).
To examine roles of NMIIA and of NMIIA phosphorylation for 4T1 migration in 3D, we performed collagen I invasion assays. Depletion of NMIIA resulted in a severe defect in 3D invasion that could be fully rescued by introduction of wild-type GFP MHC-IIA (Fig. 5, A, top three panels, and B). In contrast, none of the three mutant constructs, GFP MHC-IIA S1915A/S1916A, GFP MHC-IIA S1943A, or GFP MHC-IIA 3×A, could fully rescue this invasion defect (Fig. 5, A, lower panels, and B). These results demonstrate that whereas NMIIA and NMIIA phosphorylation are largely dispensable for migration in the 2D scratch wound setting, NMIIA, and the ability to phosphorylate NMIIA at Ser-1916 and Ser-1943, has a profound role in mediating sustained 3D invasion.
FIGURE 5.

NMIIA phosphorylation is critical for 3D invasion and contraction. A, invasion into 3D collagen gels. The indicated cell lines were plated on a glass bottom dish, overlaid with 3 mg/ml collagen I, and then incubated with complete medium for 18 h, at which point samples were fixed and stained with DAPI. Confocal Z-stacks were collected to quantify migration vertically into the gel. Shown are XZ images for sample Z-stacks from each cell line. B, to quantify invasion, the percent of DAPI signal greater than 40 μm away from the coverslip was measured for 24 fields of view from three independent experiments performed on different dates. Data are plotted as mean ± S.D. C, invasion into collagen gels was quantified in 10-μm increments for each cell line as fine resolution metric for invasion. Fluorescence intensity (DAPI) in each subsequent 10-μm Z slice is reported on the y axis. Data were derived from the same set of experiments described in B. D and E, collagen gel contraction assays. Scattered graph of MDA-MB-231 (D) and 4T1 (E) cells showing collagen gel size for parental cells, MHC-IIA shRNA cells, and MHC-IIA shRNA cells expressing the indicated GFP MHC-IIA construct, after 24 h of embedding cells in 2 mg/ml collagen. MDA-MB-231 cells and 4T1 cells lines, n = 9 and n = 6, respectively, two independent experiments were performed on different days. Data are plotted as mean ± S.D. Statistical analyses were performed with one-way analysis of variance (Dunnett's multiple comparison test).
We hypothesized that phosphorylation at Ser-1916 and at Ser-1943 might function in an additive manner to facilitate invasion. Therefore, we asked whether the double phosphorylation site mutant conferred a more severe invasion defect than the single S1916A or S1943A mutant by calculating collagen invasion distance for each cell line in 10-μm increments (Fig. 5C). Surprisingly, the single mutants displayed just as severe an invasion defect as the double mutant, with very little invasion occurring past 20 μm for any of these three cell lines. This result suggests that each phosphorylation site may play a specific and distinct role in NMIIA functions needed for invasion.
We hypothesized that the defects observed in focal adhesion maturation during initial engagement with matrix in cells lacking fully functional NMIIA (Fig. 1) might result in defects in matrix engagement that could underlie defective 3D invasion. As a test of this idea, we performed collagen gel contraction assays with both the MDA MB 231 and 4T1 cell lines. Cells were suspended in collagen (2 mg/ml) in growth medium and incubated 24 h, followed by quantitation of gel contraction. Depletion of NMIIA resulted in defect in contraction in both the MDA MB 231 (Fig. 5D) and in 4T1 (Fig. 5E) models. This defective contraction was rescued upon reintroduction of wild-type GFP MHC-IIA in both cell lines. However, none of the three MHC phosphorylation site mutants was able to rescue contraction as fully as the wild-type construct. In sum, this assay reveals a defect in the phosphorylation site mutants with respect to generation of contractile force relative to the external matrix.
Protrusion Formation in 3D Requires NMIIA and the Ability to Phosphorylate Ser-1916 and Ser-1943
To further dissect the roles and isoform specificity of NMII in 3D invasion, we generated additional 4T1 cell populations depleted of NMIIB via shRNA (MHC-IIB shRNA cells) (Fig. 6A). As we recently reported (16), NMIIB is expressed in 4T1 cells and has critical roles in long term traction force maintenance and in nuclear translocation through tight spaces during invasive migration. We performed time-lapse phase-contrast imaging of the engineered NMIIA and NMIIB 4T1 cell lines and compared their protrusion dynamics on 2D surfaces versus when embedded in 3D collagen gels. When cells were imaged on collagen I-coated 2D surfaces, morphology and dynamics of protrusion appeared similar between parental cells, MHC-IIB shRNA cells, MHC-IIA shRNA cells, and MHC-IIA shRNA cells rescued with wild-type GFP MHC-IIA or cells rescued with GFP MHC-IIA 3×A (Fig. 6, B, top row and C, and supplemental Movies S1–S5).
FIGURE 6.

NMIIA has a critical role in allowing cells to make protrusions in 3D environments. A, whole-cell lysates of parental 4T1 cells and 4T1 cells depleted of MHC-IIA or MHC-IIB were subjected to Western blotting analysis. B, representative frames from time-lapse imaging of parental, MHC-IIA-shRNA cells, and MHC-IIA-shRNA cells expressing GFP MHC-IIA, GFP MHC-IIA 3×A, or MHC-IIB shRNA cells performed on 2D collagen-coated glass surfaces or in 3D in 3 mg/ml collagen gels. Representative images of the cells 12 h after plating are shown in 2D (B, top row, scale bar, 100 μm) and 3D (B, bottom row, scale bar, 50 μm). The number of new protrusions formed in 2D (C, n = 12) and 3D (D, n = 12) for each cell population was quantified over a 3-h window, starting 12 h after plating. MHC IIB shRNA cell protrusion events were not statistically different from either parental or GFP MHC-IIA. E, maximum cell length in 3D collagen was quantified 12 h after plating (E, n = 30). For both conditions, three independent experiments were performed on different dates. C–E, data are plotted as mean ± S.D.
In 3D collagen gels parental cells and cells expressing wild-type GFP MHC-IIA extended and retracted protrusions robustly. However, severe motility defects were observed in 3D in both NMIIA-shRNA cells and in cells expressing GFP MHC-IIA 3×A. NMIIA-shRNA cells with or without expression of GFP MHC-IIA 3×A remained mostly spherical, with dramatically less protrusion formation. In time-lapse movies, thin protrusions are visible that extend a short distance beyond the circular perimeter of the cell and then retract (Fig. 6, B, lower row, and D, and supplemental Movies S6–S10).
Notably, MHC-IIB shRNA cells were still dynamic both in 2D and in 3D collagen I gels, producing protrusions at frequencies similar to cells expressing wild-type GFP MHC-IIA (Fig. 6B and supplemental Movies S5 and S10). Quantitation of protrusion frequency in 3D (Fig. 6D) and maximal cell length in 3D (a static proxy for ability to generate sustained protrusions; Fig. 6E) both demonstrate a requirement for NMIIA and NMIIA phosphorylation in 3D protrusion formation and dynamics. Furthermore, these data demonstrate that NMIIB is not required for protrusion formation nor for protrusion dynamics in collagen gels.
NMIIA and NMIIA Phosphorylation Are Needed for 3D Protrusion Only in Stiff Collagen Gels
We hypothesized that NMIIA and NMIIA phosphorylation were potentially necessary for persistent matrix engagement and persistent stabilization of anterior protrusions when cells are confronted with mechanical resistance. Indeed, close examination of time-lapse images reveals transient initiation of thin protrusions in NMIIA-shRNA cells and GFP MHC-IIA 3×A cells, which extend a short distance beyond the circular perimeter of the cell and then retract (e.g. supplemental Movies S7 and S9). To determine whether stiffness, as opposed to some other feature of the 3D collagen environment, created the need for NMIIA in 3D protrusion formation, we examined protrusion dynamics of 4T1 cells embedded in collagen I gels of varying densities. Parental 4T1 cells, NMIIA shRNA cells, NMIIA shRNA cells rescued with GFP MHC-IIA or GFP MHC-IIA 3×A, and NMIIB shRNA cells were embedded in soft 1.0 mg/ml collagen gels. In this setting, all lines behaved similar to each other and displayed dynamics similar to when they were plated on 2D collagen-coated surfaces, with extended morphology (Fig. 7, A, and B, left set of bars) and robust protrusion formation (Fig. 7C, left set of bars). In contrast, when these cells were imaged in stiffer 2.5 mg/ml collagen gels, a critical role for NMIIA and for NMIIA phosphorylation at Ser-1916 and Ser-1943 was apparent (Fig. 7, B and C, center set of bars). In 2.5 mg/ml collagen, NMIIA shRNA cells, and GFP MHC-IIA 3×A cells could not efficiently form protrusions, whereas cells expressing wild-type GFP MHC-IIA formed frequent and long protrusions similar to the parental. Cells depleted of NMIIB formed protrusions efficiently in both 1.0 and 2.5 mg/ml collagen. When cells were imaged in very stiff 5.0 mg/ml collagen gels, protrusive activity was largely eliminated in all cell populations (Fig. 7, A, bottom panel, and B and C, right set of bars).
FIGURE 7.

Role of NMIIA and NMIIA phosphorylation in 3D protrusion formation depends upon the stiffness of the extracellular environment. A, time-lapse imaging of the indicated 4T1 cell lines embedded in collagen-I gels of either 1.0, 2.5, or 5 mg/ml was performed. Representative images 12 h post-plating are shown. Scale bar, 50 μm. B and C, maximum cell length (B) and number of protrusions/3 h (C) for all cell populations at each collagen concentration was quantitated. Data are plotted as mean ± S.D. *** indicates p < 0.001 representing significant increase or decrease, n = 30 and n = 12 cells for each cell type in (B and C), respectively. Individual measures were made on data sets from three independent dates.
To determine how NMIIA influenced protrusion behavior, we quantified the rate of extension of individual protrusions into the collagen gels at each collagen density by measuring protrusion lengths versus time from time lapse images (Fig. 8A). In 1.0 mg/ml collagen, cells expressing wild-type GFP MHC-IIA displayed slightly faster protrusion rates than the other three lines (Fig. 8B). In 2.5 mg/ml collagen, a severe defect in persistent extension was clearly revealed in the NMIIA shRNA cells and in cells expressing the GFP MHC-IIA 3×A mutant, as compared with cells expressing wild-type GFP MHC-IIA or NMIIB shRNA cells (Fig. 8C). In 5 mg/ml collagen gels, protrusions longer than the cell body diameter were extremely rare in all four cell populations (Fig. 8D).
FIGURE 8.

Rate of protrusion elongation depends upon the stiffness of the extracellular environment. A, time lapse images of protrusion extension by indicated cell lines in 2.5 mg/ml collagen. Protrusion lengths were measured from consecutive images to quantify extension rates. White arrows indicate position of initial protrusion event, and white arrowheads indicate tip of the protrusion at sequential time points. Protrusions are much more visible if one views the supplemental time-lapse movies. B–D, protrusion rates were quantitated by measuring length of each protrusion every 3 min for 60 min from cells embedded in collagen gels of 1.0 mg/ml (B), 2.5 mg/ml (C), or 5 mg/ml (D). Data are plotted as mean ± S.D. n = 12 cells for each condition; data were pooled from experiments performed on three different dates.
In sum, these experiments demonstrate a stiffness-dependent and isoform-specific requirement for NMIIA and for NMIIA phosphorylation sites for formation of protrusions in 3D and demonstrate that the ability of NMIIA to drive formation of protrusions in 3D is lost when the 3D environment becomes excessively stiff.
Discussion
Although heavy chain phosphorylation on mammalian NMII was first observed over 2 decades ago (18, 19, 35), the pathways driving this phosphorylation, and the biochemical and cellular roles of heavy chain phosphorylation remain poorly understood. The presence of multiple NMII isoforms in mammalian cells as well as the presence of multiple phosphorylation sites on both MHC IIA and MHC IIB (21, 36, 37) suggest complex regulatory pathways and complex roles of MHC II phosphorylation in mammalian systems. It is clear that C-terminal phosphorylation functions to accelerate turnover of previously assembled filaments, thus allowing recruitment and assembly of NMIIA filaments to more anterior locations in cells migrating in 2D settings (21–23). Earlier studies have demonstrated that Ser-1943 phosphorylation, possibly via casein kinase II, lies downstream from EGF receptor signaling during chemotaxis (22) and that Ser-1916 phosphorylation, via Rac1 and PKC signaling, has critical roles in the capture of individual NMIIA minifilaments into nascent leading edge adhesions (38). In sum, previous work demonstrates complex roles for NMIIA phosphorylation in filament dynamics and adhesion maturation, with separate signaling pathways linked to Ser-1916 versus Ser-1943 phosphorylation. The spreading analysis presented here demonstrates that C-terminal NMIIA phosphorylation on both sites functions cooperatively to control recruitment of NMIIA to leading edge protrusions and that this recruitment in turn is essential for normal focal adhesion maturation.
The requirement for mammalian NMIIA for cell migration in 2D appears variable, with impact of shRNA treatments on cell velocity varying widely in different studies (39–42). In contrast to this variable importance of NMIIA for 2D migration, evidence is consistent (i) in that the NMIIA isoform shows strong recruitment to anterior protrusions during 2D migration and (ii) that NMIIA has a strong and isoform-specific role in stabilizing nascent focal adhesions at the anterior of cells migrating in 2D (14, 30, 43–45). We hypothesized that the limited and variable impact of NMIIA depletion on migration rate in 2D studies may relate to the lack of resistance a cell encounters during 2D migration on rigid surfaces. On rigid 2D surfaces, the largest source of resistance to migration is likely to be the large and stable focal adhesions that develop in this setting and that are formed in an NMIIA-dependent manner. Recent studies examining NMII roles for 3D migration suggest a more consistent requirement for NMII functions for invasion (24, 28, 46, 47).
In this study, we demonstrate that NMIIA is essential for invasive migration in dense 3D collagen gels but not soft 3D gels, suggesting a force-generating role for NMIIA in this setting. We further establish that NMIIA is specifically necessary for allowing cells to achieve sustained extension of nascent protrusions into the dense extracellular matrix. Strikingly, NMIIA is dispensable for this extension function both in 2D and in extremely soft 3D collagen gels (e.g. 1.0 mg/ml). Our studies furthermore show that NMIIA phosphorylation on both Ser-1916 and Ser-1943 is critical for this density-dependent role of NMIIA in 3D protrusiveness. We suggest, based on the behavior of the S1916A and S1943A mutant constructs in COS-7 cells, that in the 3D setting cells must phosphorylate these sites to relocalize NMIIA toward the leading edge as a protrusion is initiated and that this localization is critical for focal adhesion maturation and stabilization, which in turn may be critical for stabilization of nascent protrusions and for persistent extension of those protrusions into the surrounding matrix. This proposed mechanism is critical when protrusions encounter resistance from the extracellular matrix but is dispensable in soft matrix or in 2D settings where protrusions are extending into buffer. Further studies are needed to test this model.
Experimental Procedures
Cell Culture and Transfection
COS-7 cells (African green monkey kidney fibroblast-like (25)) and 4T1 cells (mouse basal-like breast cancer cells (26)) were maintained in Dulbecco's modified Eagle's medium and RPMI 1640 medium, respectively, with 10% fetal bovine serum and 1% penicillin/streptomycin cells. MDA-MB 231 cells were obtained from the ATCC and used within 4–5 passages. Cells were maintained at 37 °C, 5% CO2. Polyclonal COS-7 stable cell populations were created by transfecting 2 μg of plasmid DNA in 5 × 105 COS-7 cells using Lonza Group Amaxa Nucleofector system (Solution Kit R, program W-001) and selecting with 800 μg/ml geneticin. Typically 100–200 resistant colonies formed upon selection, which were collectively expanded and subjected to fluorescence-activated cell sorting (FACS) to obtain populations expressing uniform GFP MHC-IIA fluorescence. Populations were used for experiments within 3–4 passages. Transfection in a 70% confluent plate with 4T1 cells were carried out by using Lipofectamine (Life Technologies, Inc.) according to the manufacturer's instructions in 4T1 NMIIA-depleted cells. Colonies were isolated resistant to both 800 μg/ml geneticin and 5 μg/ml puromycin (selection for MHC IIA lentiviral construct insertion). As with COS-7 lines, ∼100–200 colonies obtained from transfection were collectively expanded to avoid clonal effects and subjected to FACS to obtain uniform expression level of GFP fusion proteins.
Lentivirus-based shRNA
NMIIA and -IIB expression was depleted using Mission shRNA pLK0.1 lentiviral plasmids (Sigma; TRCN0000304377 and TRCN0000110555 targeting NMIIA and NM IIB, respectively). The pLK0.1 plasmid was co-transfected with pCMV-R8.2 and pCMV-VSVG helper plasmids into HEK-293T cells via the Lipofectamine 2000 transfection agent (Life Technologies, Inc., catalogue no. 11668027). Transfected cells were then grown in DMEM containing 10% FBS and 1% penicillin/streptomycin for 24 h at 37 °C, 5% CO2. Media containing virus was then harvested and centrifuged for 10 min on setting 4 of a clinical specimen centrifuge. 4T1 cells were then infected by incubating with virus-containing media for 24 h. Cells were selected for positive shRNA infection through puromycin selection at 10 μg/ml for 5 days and selection conditions were then maintained at 5 μg/ml. Infection rates were typically >90%, thus generating shRNA knockdown cell populations derived from thousands of independent infection events.
Chemicals and Antibodies
Transfecting reagents were purchased from Amaxa Biosystems USA and Life Technologies, Inc. Primary antibodies used were all commercial products generated in rabbits as follows: GFP (Invitrogen, A-6455);NMIIA (Biomedical Technologies, catalogue no. BT-567); NM IIB (Covance, catalogue no. PRB-445P); phosphospecific paxillin Y118-p (Cell Signaling catalogue no. 2541S); total paxillin (Cell Signaling catalogue no. 2542); phospho-FAK (Cell Signaling catalogue no. 8556, 3281); total FAK (Cell Signaling catalogue no. 13009); and actin (Sigma, catalogue no. A2066); GAPDH (Santa Cruz Biotechnology, catalogue no. sc-25778). We note that the absence of NMIIA protein in COS-7 cells was also confirmed via Western blotting with a Cell Signaling antibody (catalogue no. 3403) that reacts with an NMIIA C-terminal epitope that is 100% conserved in the African green monkey MYH9 coding region. Peroxidase-conjugated secondary antibodies were from Thermo Fisher Scientific. Fibronectin was from Sigma. Samples were mounted in Vectashield (Vector Laboratories, H-1200). Alexa Fluor® 568 phalloidin, Alexa Fluor® 647 phalloidin, and 4′,6-diamidino-2-phenylindole nucleic acid stain were obtained from Molecular Probes, Inc. SuperFemto Western blotting reagents were obtained from (Pierce”). Protease inhibitor (catalogue no. P2714) and phosphatase inhibitor mixtures 2 and 3 were obtained from Sigma (catalogue nos. P0044 and P5726).
Creation of NMIIA Phosphorylation Site Mutants
Plasmid pEGFP-NMHC-IIA-C3 expressing the full-length human myosin heavy chain under the control of a constitutive cytomegalovirus (CMV) promoter, with EGFP fused at the N terminus of the NM-IIA coding region and G418/kanamycin, was used to express wild-type GFP-NM-IIA and other mutants. The plasmid pEGFP-MHC-IIA-C3 and the pEGFP-MHC-IIA S1943A phosphorylation site mutant plasmids were gifted from Dr. Anne Bresnick, and have been described previously (22, 23). To make the plasmids pEGFP-NMHC-IIA S1916A and pEGFP-MHC-IIA 3×A, synthetic DNA segments were purchased from DNA 2.0 with the desired point mutations, with 5′ XhoI and 3′ SalI sites. These were then digested and ligated into pEGFP MHC-IIA-C3 using XhoI-SalI. All DNA segments subjected to PCR were sequenced to confirm the absence of PCR-generated errors. For both plasmids carrying the S1916A mutation, the adjacent serine at Ser-1915 was also mutated to alanine to prevent any in vivo phosphorylation occurring at this adjacent residue.
Western Blottings
Cells were lysed directly by the addition of 2× Laemmli sample buffer containing protease and phosphatase inhibitors and incubated on ice for 3–5 min. The cell lysates were then subjected to brief cup sonication to reduce DNA viscosity, heated at 95 °C for 2 min, and stored frozen until use. Protein samples were subjected to SDS-PAGE, transferred to polyvinylidene difluoride membrane (Immobilon catalogue no. IPVH00010), blocked in 3% BSA, and incubated with anti-rabbit polyclonal antibodies at 4 °C overnight. Blots were washed and incubated with horseradish peroxidase-conjugated secondary antibodies at room temperature for 2 h, and bands were visualized by SuperSignal West Pico Luminol/Enhancer solution.
Cell Spreading Assays
COS-7 cells growing on tissue culture plates were collected and plated on fibronectin-coated (20 μg/ml) or collagen-I-coated (20 μg/ml) glass coverslips for NMII localization or focal adhesion imaging, respectively. Cells were allowed to spread in growth medium for 60 min at 37 °C. Cells were fixed in PBS supplemented with 2 mm MgCl2, 2 mm EGTA, and 4% formaldehyde for 30 min at room temperature, permeabilized for 5 min at room temperature, blocked, and then incubated with primary antibodies for 2 h at room temperature followed by incubating with Alexa-conjugated secondary antibodies (Molecular Probes, Inc.) for 1 h at room temperature. Samples were washed and incubated with 4′,6-diamidino-2-phenylindole (20 μg/ml) solution for 15 min at room temperature. Images were acquired on a Leica SP2 confocal microscope equipped with a ×63/1.4 NA oil objective.
2D Cell Migration Assays
Migration was performed using a modified scratch wound assay as described previously (23). Briefly, 4T1 cells transfected with GFP MHC-IIA constructs were plated at high density on glass coverslips in collagen-I-coated six-well tissue culture plates that had ∼2-mm-wide strip of polydimethylsiloxane strip placed across the middle on top of the collagen coating. After incubation cells at 37 °C for 16 h, the polydimethylsiloxane strip was removed, creating a well defined wound margin. Phase-contrast images were recorded every 5 min for 18 h with a ×10 objective, and the cells were maintained at 37 °C for the duration of the experiment. The distance migrated by the leading edge of the cell sheet was measured at the indicated time points after initiating migration. The supplemental Movies were assembled using ImageJ (National Institutes of Health).
3D Collagen Invasion Assay
Invasion assays were performed using a modified version of an earlier method (27), except that we observed 4T1 cells invaded collagen gels robustly regardless of whether or not macrophages were present, so we omitted macrophages from the current invasion assays. Briefly, 4T1 MHC-IIA shRNA cells and cells lines transfected with a GFP-myosin construct labeled with CellTracker Green were plated overnight on 35-mm glass bottomed plates (LabTek) in complete RPMI 1640 medium. Cells were overlaid with an ∼1-mm-thick layer of 3 mg/ml collagen-I (BD Biosciences) and then overlaid with RPMI 1640 complete medium (10% serum). After 24 h, cells were fixed with 4% formaldehyde, and optical Z-sections were collected using laser scanning confocal microscopy. Z-sections were collected every 5 μm to a distance of 100 μm into the gel. Quantification of invasion done by calculating total florescence intensities above 40 μm from the bottom divided by total fluorescence in all Z-sections (100 um).
2D and 3D Protrusion Assays
For 2D protrusion assays, 10-μm-slide chemotaxis 3D chambers (ibidi, catalogue number 80326) were coated with 20 μg/ml collagen-I (BD Biosciences) for 1 h. Plates were then washed gently with 1× PBS and seeded with a low density of cells. For 3D protrusion assays, collagen gels with different stiffness were prepared on ice by mixing collagen-I in RPMI 1640 medium. The pH was neutralized with NaOH and made isotonic with 10× PBS before mixing with cells. Cells were suspended in required concentration of collagen (on ice) and then plated in the ibidi chambers. Gels were polymerized at 37 °C in the tissue culture incubator for 30 min, then transferred to the microscope, and imaged in a heated CO2 chamber. These polymerization conditions favor formation of web-like reticular networks of collagen, as opposed to bundled fibers (4). Cells on 2D surfaces and in 3D collagen were imaged at ×10 magnification for 18 h, with images collected every 10 min. Maximum length of cell (longest chord between two point along the perimeter) was measured in the 3D collagen gel after 12 h of plating using ImageJ. Number of protrusions was quantified from a 3-h window (12–15 h after plating for all samples). Sequential video frames were examined manually, and any cell body extension greater than the diameter of the cell body itself was scored as a protrusion.
Collagen Gel Contraction Assay
Cells were trypsinized and resuspended in collagen solution (2 mg/ml) in growth medium yielding the final concentration of 500,000 cells/ml. Cell suspension embedded in collagen were poured 1 ml into each well of a 12-well tissue culture plate and incubated at 37 °C for 1 h to facilitate gelling. After gelation, the gels were released from the surface of the culture well by rimming the edges with a sterile pipette tip. Gel diameters were digitally photo-documented at 24 h. Quantification of relative areas after contraction was quantified using image software (rsb.info.nih.gov).
Statistical Analysis
Statistical analyses were performed in Prism 5 (GraphPad). Data were analyzed with one-way analysis of variance (Tukey-Kramer test) unless otherwise stated. A p value < 0.05 was considered statistically significant. *** signifies significance at p < 0.001; ** signifies significance at p value of 0.001 to 0.01, and * signifies significance at p value of 0.01 to 0.05. Data are presented as the mean ± S.D.
Author Contributions
V. R. conducted the majority of all experiments described. V. R. also participated in planning and experimental design and wrote the manuscript. D. G. T. participated in experimental design and performance of some of the invasion experiments in this project and helped establish technology for shRNA treatments. J. R. B. established initial discovery of impaired invasion upon myosin gene depletion, helped establish lentiviral methods for depleting myosin expression in cells lines needed for these studies, and contributed to experimental design planning throughout the project. T. T. E. directed and consulted in planning, implementing, and troubleshooting all studies reported here. T. T. E. planned the experimental focus within which laboratory members pursued the studies reported here. T. T. E. helped write and edit all drafts of the manuscript.
Supplementary Material
This work was supported by National Institutes of Health Grant GM50009. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Movies S1–S10.
- NMII
- non-muscle myosin II
- EGFP
- enhanced GFP
- RLC
- regulatory light chain
- FAK
- focal adhesion kinase.
References
- 1. Vicente-Manzanares M., Ma X., Adelstein R. S., and Horwitz A. R. (2009) Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 10, 778–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Friedl P., and Wolf K. (2009) Proteolytic interstitial cell migration: a five-step process. Cancer Metastasis Rev. 28, 129–135 [DOI] [PubMed] [Google Scholar]
- 3. Kubow K. E., Conrad S. K., and Horwitz A. R. (2013) Matrix microarchitecture and myosin II determine adhesion in 3D matrices. Curr. Biol. 23, 1607–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Doyle A. D., Carvajal N., Jin A., Matsumoto K., and Yamada K. M. (2015) Local 3D matrix microenvironment regulates cell migration through spatiotemporal dynamics of contractility-dependent adhesions. Nat. Commun. 6, 8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Friedl P., and Alexander S. (2011) Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147, 992–1009 [DOI] [PubMed] [Google Scholar]
- 6. Gardel M. L., Schneider I. C., Aratyn-Schaus Y., and Waterman C. M. (2010) Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 26, 315–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fraley S. I., Feng Y., Krishnamurthy R., Kim D. H., Celedon A., Longmore G. D., and Wirtz D. (2010) A distinctive role for focal adhesion proteins in three-dimensional cell motility. Nat. Cell Biol. 12, 598–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Friedl P., Sahai E., Weiss S., and Yamada K. M. (2012) New dimensions in cell migration. Nat. Rev. Mol. Cell Biol. 13, 743–747 [DOI] [PubMed] [Google Scholar]
- 9. Conti M. A., and Adelstein R. S. (2008) Nonmuscle myosin II moves in new directions. J. Cell Sci. 121, 11–18 [DOI] [PubMed] [Google Scholar]
- 10. Golomb E., Ma X., Jana S. S., Preston Y. A., Kawamoto S., Shoham N. G., Goldin E., Conti M. A., Sellers J. R., and Adelstein R. S. (2004) Identification and characterization of non-muscle myosin II-C, a new member of the myosin II family. J. Biol. Chem. 279, 2800–2808 [DOI] [PubMed] [Google Scholar]
- 11. Rosenfeld S. S., Xing J., Chen L. Q., and Sweeney H. L. (2003) Myosin IIb is unconventionally conventional. J. Biol. Chem. 278, 27449–27455 [DOI] [PubMed] [Google Scholar]
- 12. Wang F., Kovacs M., Hu A., Limouze J., Harvey E. V., and Sellers J. R. (2003) Kinetic mechanism of non-muscle myosin IIB: functional adaptations for tension generation and maintenance. J. Biol. Chem. 278, 27439–27448 [DOI] [PubMed] [Google Scholar]
- 13. Vicente-Manzanares M., Zareno J., Whitmore L., Choi C. K., and Horwitz A. F. (2007) Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J. Cell Biol. 176, 573–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Doyle A. D., Kutys M. L., Conti M. A., Matsumoto K., Adelstein R. S., and Yamada K. M. (2012) Micro-environmental control of cell migration–myosin IIA is required for efficient migration in fibrillar environments through control of cell adhesion dynamics. J. Cell Sci. 125, 2244–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lämmermann T., Bader B. L., Monkley S. J., Worbs T., Wedlich-Söldner R., Hirsch K., Keller M., Förster R., Critchley D. R., Fässler R., and Sixt M. (2008) Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature 453, 51–55 [DOI] [PubMed] [Google Scholar]
- 16. Thomas D. G., Yenepalli A., Denais C. M., Rape A., Beach J. R., Wang Y. L., Schiemann W. P., Baskaran H., Lammerding J., and Egelhoff T. T. (2015) Non-muscle myosin IIB is critical for nuclear translocation during 3D invasion. J. Cell Biol. 210, 583–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Petrie R. J., Koo H., and Yamada K. M. (2014) Generation of compartmentalized pressure by a nuclear piston governs cell motility in a 3D matrix. Science 345, 1062–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kawamoto S., Bengur A. R., Sellers J. R., Adelstein R. S. (1989) In situ phosphorylation of human platelet myosin heavy and light chains by protein kinase C. J. Biol. Chem. 264, 2258–2265 [PubMed] [Google Scholar]
- 19. Ludowyke R. I., Peleg I., Beaven M. A., and Adelstein R. S. (1989) Antigen-induced secretion of histamine and the phosphorylation of myosin by protein kinase C in rat basophilic leukemia cells. J. Biol. Chem. 264, 12492–12501 [PubMed] [Google Scholar]
- 20. Moussavi R. S., Kelley C. A., and Adelstein R. S. (1993) Phosphorylation of vertebrate non-muscle and smooth muscle myosin heavy chains and light chains. Mol. Cell. Biochem. 127, 219–227 [DOI] [PubMed] [Google Scholar]
- 21. Dulyaninova N. G., and Bresnick A. R. (2013) The heavy chain has its day: regulation of myosin-II assembly. Bioarchitecture 3, 77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dulyaninova N. G., House R. P., Betapudi V., and Bresnick A. R. (2007) Myosin-IIA heavy-chain phosphorylation regulates the motility of MDA-MB-231 carcinoma cells. Mol. Biol. Cell 18, 3144–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Breckenridge M. T., Dulyaninova N. G., and Egelhoff T. T. (2009) Multiple regulatory steps control mammalian non-muscle myosin II assembly in live cells. Mol. Biol. Cell 20, 338–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beach J. R., Hussey G. S., Miller T. E., Chaudhury A., Patel P., Monslow J., Zheng Q., Keri R. A., Reizes O., Bresnick A. R., Howe P. H., and Egelhoff T. T. (2011) Myosin II isoform switching mediates invasiveness after TGF-β-induced epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. U.S.A. 108, 17991–17996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gluzman Y. (1981) SV40-transformed simian cells support the replication of early SV40 mutants. Cell 23, 175–182 [DOI] [PubMed] [Google Scholar]
- 26. Aslakson C. J., and Miller F. R. (1992) Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 52, 1399–1405 [PubMed] [Google Scholar]
- 27. Goswami S., Sahai E., Wyckoff J. B., Cammer M., Cox D., Pixley F. J., Stanley E. R., Segall J. E., and Condeelis J. S. (2005) Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 65, 5278–5283 [DOI] [PubMed] [Google Scholar]
- 28. Beadle C., Assanah M. C., Monzo P., Vallee R., Rosenfeld S. S., and Canoll P. (2008) The role of myosin II in glioma invasion of the brain. Mol. Biol. Cell 19, 3357–3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Betapudi V., Gokulrangan G., Chance M. R., and Egelhoff T. T. (2011) A proteomic study of myosin II motor proteins during tumor cell migration. J. Mol. Biol. 407, 673–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Choi C. K., Vicente-Manzanares M., Zareno J., Whitmore L. A., Mogilner A., and Horwitz A. R. (2008) Actin and α-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat. Cell Biol. 10, 1039–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parsons J. T., Horwitz A. R., and Schwartz M. A. (2010) Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 11, 633–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Webb D. J., Donais K., Whitmore L. A., Thomas S. M., Turner C. E., Parsons J. T., and Horwitz A. F. (2004) FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 6, 154–161 [DOI] [PubMed] [Google Scholar]
- 33. Pasapera A. M., Schneider I. C., Rericha E., Schlaepfer D. D., and Waterman C. M. (2010) Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J. Cell Biol. 188, 877–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mitra S. K., and Schlaepfer D. D. (2006) Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 18, 516–523 [DOI] [PubMed] [Google Scholar]
- 35. Adelstein R. S., Beaven M. A., Bengur A. R., Kawamoto S., Ludowyke R. I., Peleg I., and Sellers J. R. (1989) In situ phosphorylation of human platelet and rat basophilic leukemia cell (RBL-2H3) myosin heavy chain and light chain. Adv. Exp. Med. Biol. 255, 289–297 [DOI] [PubMed] [Google Scholar]
- 36. Rosenberg M., and Ravid S. (2006) Protein kinase Cγ regulates myosin IIB phosphorylation, cellular localization, and filament assembly. Mol. Biol. Cell 17, 1364–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Even-Faitelson L., and Ravid S. (2006) PAK1 and aPKCζ regulate myosin II-B phosphorylation: a novel signaling pathway regulating filament assembly. Mol. Biol. Cell 17, 2869–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pasapera A. M., Plotnikov S. V., Fischer R. S., Case L. B., Egelhoff T. T., and Waterman C. M. (2015) Rac1-dependent phosphorylation and focal adhesion recruitment of myosin IIA regulates migration and mechanosensing. Curr. Biol. 25, 175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Babbin B. A., Koch S., Bachar M., Conti M. A., Parkos C. A., Adelstein R. S., Nusrat A., and Ivanov A. I. (2009) Non-muscle myosin IIA differentially regulates intestinal epithelial cell restitution and matrix invasion. Am. J. Pathol. 174, 436–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Even-Ram S., Doyle A. D., Conti M. A., Matsumoto K., Adelstein R. S., and Yamada K. M. (2007) Myosin IIA regulates cell motility and actomyosin-microtubule crosstalk. Nat. Cell Biol. 9, 299–309 [DOI] [PubMed] [Google Scholar]
- 41. Betapudi V., Licate L. S., and Egelhoff T. T. (2006) Distinct roles of non-muscle myosin II isoforms in the regulation of MDA-MB-231 breast cancer cell spreading and migration. Cancer Res. 66, 4725–4733 [DOI] [PubMed] [Google Scholar]
- 42. Jorrisch M. H., Shih W., and Yamada S. (2013) Myosin IIA deficient cells migrate efficiently despite reduced traction forces at cell periphery. Biol. Open 2, 368–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kolega J. (1998) Cytoplasmic dynamics of myosin IIA and IIB: spatial ‘sorting’ of isoforms in locomoting cells. J. Cell Sci. 111, 2085–2095 [DOI] [PubMed] [Google Scholar]
- 44. Vicente-Manzanares M., Newell-Litwa K., Bachir A. I., Whitmore L. A., and Horwitz A. R. (2011) Myosin IIA/IIB restrict adhesive and protrusive signaling to generate front-back polarity in migrating cells. J. Cell Biol. 193, 381–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kuo J. C., Han X., Hsiao C. T., Yates J. R. 3rd., and Waterman C. M. (2011) Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for β-Pix in negative regulation of focal adhesion maturation. Nat. Cell Biol. 13, 383–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shih W., and Yamada S. (2010) Myosin IIA dependent retrograde flow drives 3D cell migration. Biophys. J. 98, L29–L31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Raab M., Swift J., Dingal P. C., Shah P., Shin J. W., and Discher D. E. (2012) Crawling from soft to stiff matrix polarizes the cytoskeleton and phosphoregulates myosin-II heavy chain. J. Cell Biol. 199, 669–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.