

Abstract
Most cases of Alzheimer's disease (AD) are sporadic, but a small percentage of AD cases, called familial AD (FAD), are associated with mutations in presenilin 1, presenilin 2, or the amyloid precursor protein. Amyloid precursor protein mutations falling within the amyloid-β (Aβ) sequence lead to a wide range of disease phenotypes. There is increasing evidence that distinct amyloid structures distinguished by amyloid conformation-dependent monoclonal antibodies have similarly distinct roles in pathology. It is possible that this phenotypic diversity of FAD associated with mutations within the Aβ sequence is due to differences in the conformations adopted by mutant Aβ peptides, but the effects of FAD mutations on aggregation kinetics and conformational and morphological changes of the Aβ peptide are poorly defined. To gain more insight into this possibility, we therefore investigated the effects of 11 FAD mutations on the aggregation kinetics of Aβ, as well as its ability to form distinct conformations recognized by a panel of amyloid conformation-specific monoclonal antibodies. We found that most FAD mutations increased the rate of aggregation of Aβ. The FAD mutations also led to the adoption of alternative amyloid conformations distinguished by monoclonal antibodies and resulted in the formation of distinct aggregate morphologies as determined by transmission electron microscopy. In addition, several of the mutant peptides displayed a large reduction in thioflavin T fluorescence, despite forming abundant fibrils indicating that thioflavin T is a probe of conformational polymorphisms rather than a reliable indicator of fibrillization. Taken together, these results indicate that FAD mutations falling within the Aβ sequence lead to dramatic changes in aggregation kinetics and influence the ability of Aβ to form immunologically and morphologically distinct amyloid structures.
Keywords: aggregation, Alzheimer disease, amyloid-β (Aβ), monoclonal antibody, peptide conformation
Introduction
The overwhelming majority of Alzheimer's disease (AD)2 cases are late-onset, occurring in individuals older than 65. Familial AD (FAD) is associated with mutations in presenilins 1 and 2 and the amyloid precursor protein (APP), which is sequentially cleaved by the enzymes β- and γ-secretase to yield the Aβ peptide (1). APP mutations leading to FAD either fall within the Aβ(1–43) region of the protein or are adjacent to the N or C terminus of the Aβ sequence. Typically, the mutations adjacent to the N terminus of the Aβ sequence lead to an increased production of all Aβ species, whereas the ones near the C terminus lead to an increase in the Aβ42/Aβ40 ratio (2, 3). There are also several FAD mutations that occur within the Aβ region of APP that have been reported in the literature. These amino acid substitutions have been reported to increase the amount of Aβ produced, increase the ratio of Aβ42 to Aβ40, increase the aggregation potential of the mutant Aβ variant, or promote the formation of particularly toxic conformations of aggregates, such as oligomers (1). For example, the recessive A2V mutation is reported to increase Aβ production (4), whereas the T43I mutation dramatically increases the Aβ42/Aβ40 ratio (5). The Osaka (E22Δ) mutation accelerates Aβ aggregation without affecting total Aβ levels (6). Other Aβ FAD mutations, such as the Italian (E22K) and Arctic (E22G) mutations, are believed to exert their pathogenic effects by inducing the formation of stable oligomers and protofibrils (7, 8). In addition, the Tottori (D7N), Flemish (A21G), and E22G mutations have been reported to lead to the adoption of distinct aggregate structures, as determined by ion mobility-based mass spectrometry (9).
One of the most interesting features of the FAD mutations within the Aβ sequence is that they lead to remarkable phenotypic diversity reminiscent of prion strain polymorphisms (1, 10). Some of the FAD mutations within the Aβ42 sequence lead to severe early-onset dementia and AD, whereas other FAD mutations that cluster at residues 21–23 lead to familial cerebral amyloid angiopathy (CAA), a disease distinct from AD characterized by microhemorrhages and often premature death (1, 11). Not only do several FAD Aβ mutations lead to CAA, which distinguishes these mutations from others causing only Alzheimer's dementia, but there is significant heterogeneity among these mutations in terms of pathological findings in patients (12–14). For instance, although most of these FAD mutations lead to hemorrhagic strokes, strokes are relatively rare in patients suffering from FAD due to the Arctic (E22G) and Iowa (D23N) mutations (13, 15). The A21G mutation is unique among the CAA-causing FAD mutations in that it increases total Aβ production in addition to causing biochemical and structural alterations in Aβ (16). Several CAA-associated mutations are associated with unique clinical findings. For instance, about half of the patients with CAA due to the Dutch (E22Q) mutation suffer from recurring seizures (17). Similarly, patients with CAA of the Piedmont (L34V) type suffer from paresthesia and impaired movements not seen in cerebral amyloid angiopathy due to other FAD mutations (18). Interestingly, the Dutch and Piedmont mutations differentially activate mitochondrial apoptotic pathways in endothelial and smooth muscle cells (19). Another example of a unique clinical feature associated with a particular FAD Aβ mutation is the distinctive presence of myoclonus and vocabulary and speech problems in patients suffering from CAA caused by the Iowa mutation (15). Although there is a large body of research on some of these FAD mutations, others have not been extensively studied, and there are conflicting reports regarding the effects of several FAD mutations (1).
There is an increasing understanding of the conformational diversity of amyloid Aβ oligomer and fibril structures both in vitro and in vivo that may underlie their pathological phenotypic diversity (20, 21). This is of particular interest here because of the possibility that conformational variation may explain the phenotypic variation associated with FAD mutations within Aβ. Conformation-dependent aggregation-specific monoclonal antibodies can distinguish multiple conformations of amyloid that accumulate at specific times and in unique locations in human AD and transgenic mouse brains and in vitro (22–25). For instance, mOC78 recognizes intraneuronal amyloid at early times and the core of neuritic plaques at later stages, indicating that neuritic plaques may arise from the death of neurons that contain aggregated intraneuronal amyloid (23, 25), whereas mOC31 uniquely recognizes a subset of vascular amyloid (25, 27).
We have compared the effects of a comprehensive number of FAD amino acid substitutions in Aβ40 on the aggregation state and conformation of Aβ using a panel of amyloid conformation-dependent monoclonal antibodies we have described previously (22, 25). Here, we report that most of the FAD mutations lead to a dramatic increase in the rate of aggregation, differences in maximum thioflavin T (ThT) fluorescence intensity, and sizes of aggregates. These mutations also alter the conformational trajectory of Aβ to aggregate into specific conformations recognized by mOC antibodies. The different FAD mutations also result in differences in the ability of Aβ to form fibrils and in the morphology of the fibrils that are formed, as seen by transmission electron microscopy (TEM). To our knowledge, this is the first study to examine such a large number of Aβ peptides containing FAD mutations as an ensemble and under carefully controlled aggregation conditions. As such, it allows for comparisons among the different peptides without the confounding differences in methodology or experimental technique that may arise from comparing results from smaller independent studies. Taken together, our results demonstrate that single amino acid Aβ mutations associated with FAD lead to alterations in the structure of Aβ, which are associated with differential aggregation kinetics, morphologies, and immunological reactivity patterns. These conformational changes may underlie the pathological polymorphism of FAD. However, some mutations have no discernable effect on aggregation or conformation of the peptide and may exert their effects by causing the misfolding of APP and necessitating its degradation to prevent aggregation.
Results
Most FAD Mutations Lead to an Increase in the Aggregation Kinetics of Aβ and Its Interaction with Thioflavin T
The 11 mutations studied and their reported pathologic effects and disease phenotypes are summarized in Table 1. To study the aggregation kinetics of the different Aβ variants and to compare them with WT Aβ40, we began an aggregation reaction using condition A (resuspension in NaOH, followed by dilution in phosphate buffer, pH 7.4), immediately transferred the reaction mixtures to a 96-well plate, and recorded ThT fluorescence measurements from the plate over a 40-h time course. Readings were obtained every 10 min, and the results are presented as the mean of triplicate absorbance values for each time point (Fig. 1). Most of the FAD mutations cause a decrease in the lag time and an increase in the rate of aggregation, including E22G, L34V, D7N, E22K, A2V, and H6R. This effect is not restricted to mutations falling within a specific region of the Aβ sequence, as the mutations leading to increases in the rate of aggregation may be N- or C-terminal, or fall within the mid-region of Aβ. The A21G and E22Q mutations exhibit longer lag times, whereas E22Δ rapidly aggregates into species with very low ThT binding (Fig. 1b). We can also divide the Aβ variants into two broad groups based on their maximum ThT fluorescence levels. Whereas the D7N, E22G, E22K, D23N, and L34V variants had higher maximum fluorescence values than those obtained with the WT peptide, the remaining FAD mutant Aβs (A2V, H6R, A21G, E22Δ, and E22Q) had maximum fluorescence values approximately the same as or lower than that obtained with WT Aβ (Fig. 1). Among the higher fluorescing group, the E22G variant has the shortest lag phase, followed by the L34V, E22K, D23N, and D7N variants (Fig. 1a). There was also some variation in the maximum ThT fluorescence within this highly specific fluorescence group, with the E22G and L34V mutants exhibiting higher ThT fluorescence values than the E22K, D23N, and D7N peptides. We observed biphasic aggregation courses for the WT, H6R, and D7N peptides, which may be associated with the initial formation of structures with weak ThT binding followed by a conformational conversion to structures with higher ThT binding. Alternatively, the biphasic aggregation courses may indicate the presence of kinetically stable oligomeric intermediates, followed by a secondary nucleation event. Although such biphasic ThT fluorescence curves are not uncommon, the fact that only three of the 11 Aβ peptides display this aggregation pattern indicates that FAD mutations lead to changes in the aggregation dynamics of Aβ. Our data are consistent with reports of ThT binding to distinct β-sheet-rich structures (28) and suggest the formation of aggregates with differential conformations by the FAD mutant Aβ variants. The ThT fluorescence curve for the E22K variant was particularly interesting due to the fact that the fluorescence values for this peptide gradually decreased over the 40-h time course after reaching a maximum at 3 h. This particular trend may indicate the slow conformational conversion of the aggregates of this peptide into an alternative structure that displays lower ThT binding and fluorescence. The results obtained with the E22Δ mutant Aβ are particularly striking, as this peptide demonstrates very low ThT fluorescence in this assay and yet forms abundant fibrils, as observed by electron microscopy (Fig. 2). Our results are consistent with previous reports describing the rapid aggregation of this peptide into a unique fibril structure with very low specific ThT fluorescence (29, 30). The atypical nature of E22Δ Aβ aggregates is further demonstrated by the fact that brains of patients suffering from FAD associated with this mutation reveal little Pittsburgh compound B binding in positron emission tomography scans despite the tendency of the peptide to aggregate aggressively (31, 32).
TABLE 1.
Summary of FAD mutations studied and their reported pathological effects and disease phenotypes
| Name | Mutation | Summary of reported effects | Reported effect on Aβ | Ref. |
|---|---|---|---|---|
| A2V | A2V | Pathological only in homozygous individuals, anti-amyloidogenic in presence of WT Aβ, large perivascular aggregates, severe cerebral and cerebellar pathology, neostriatum is spared | Enhanced production and accelerated aggregation | 4, 47, 48 |
| English | H6R | Enhanced cellular toxicity of oligomers, more efficient seeding ability | Unchanged total Aβ levels, accelerated fibril formation with no increase in protofibril levels, enhanced elongation phase with no effects on nucleation | (49, 50) |
| Tottori | D7N | Enhanced cellular toxicity of oligomers, more efficient seeding ability | Unchanged total Aβ levels, accelerated fibril formation with no increase in protofibril levels, enhanced elongation phase with no effects on nucleation | 49, 50 |
| K16N | K16N | Located at the Aβ α-cleavage site, only toxic in the presence of WT Aβ, forms proteolysis-resistant aggregates, K16N Aβ42 inhibits fibrillation of WT Aβ42 | Increased production | 51 |
| Flemish | A21G | Accumulates in smooth muscle cells of brain vasculature, causes severe CAA, not toxic to vascular smooth muscle cells in vitro, proteolysis-resistant aggregates | Increased secreted Aβ, slow fibrillation | 16, 52, 53–55 |
| Osaka | E22Δ | Forms fibrillar bundles, enhanced ThT fluorescence of fibrils, reversal of toxicity profiles of Aβ40 and Aβ42 | Reduced secreted Aβ in favor of intracellular aggregate formation, accelerated aggregation | 6, 56–58 |
| Dutch | E22Q | Forms vascular amyloid and displays reduced aggregation in brain parenchyma, proteolysis-resistant aggregates, causes severe CAA with seizures in half of cases, oligomers toxic to endothelial and vascular smooth muscle cells, enhances MMP-2 production by endothelial cells, enhanced membrane binding | Increases aggregation without altering APP processing | 16, 17, 49, 53, 54, 58–61 |
| Italian | E22K | CAA with no neurofibrillary involvement, proteolysis-resistant aggregates | Enhanced aggregation of stable oligomers | 7, 54, 58, 62 |
| Arctic | E22G | CAA with rare stroke incidence, proteolysis-resistant aggregates, smaller oligomers than WT Aβ, more toxic to cells than WT Aβ, Congo red-negative parenchymal deposits composed of N-terminally truncated Aβ | Enhanced protofibril formation, lower plasma levels of Aβ | 8, 13, 26, 63, 64 |
| Iowa | D23N | CAA with reduced incidence of stroke complicated by myoclonus and language and speech problems, toxic to smooth muscle cells in vitro | Increased rate of fibril formation, APP processing is unaffected | 13, 15, 16, 46, 61 |
| Piedmont | L34V | CAA characterized by paresthesia, movement disorders, and an absence of neurofibrillary pathology; vascular aggregates; absence of plaques; oligomers toxic to endothelial cells; increased MMP-2 production in endothelial cells | Hypothesized to enhance aggregation into β-sheets | 10, 18, 19, 59, 62 |
FIGURE 1.

Aggregation kinetics of WT and FAD mutant Aβ peptides. a, ThT fluorescence profiles for WT Aβ and 10 Aβ variants containing FAD mutations. b, expanded view of the ThT fluorescence data for selected variants with maximum fluorescence values similar to that of WT Aβ. The peptides were aggregated under condition A (NaOH/phosphate buffer method). ThT fluorescence intensities for each time point were obtained in triplicate and are presented as the mean of the three values. This experiment was performed in duplicate with similar results.
FIGURE 2.

Morphologies of WT and FAD mutant Aβ peptides. TEM images of the aggregates formed by WT Aβ40 and 11 Aβ40 FAD variants under condition A (resuspension in NaOH followed by dilution in phosphate buffer) after 10 days are shown. This experiment was performed twice, with similar results, and representative images from three technical replicates per peptide are displayed. Scale bar, 10 nm and is the same for all panels.
FAD Mutant Aβ Fibrils Have Different Morphologies
We used TEM to study the morphologies of the aggregates formed by WT Aβ40 and the 11 FAD variants under condition A after 10 days (Fig. 2). Whereas most of the samples contained amyloid fibrils as observed by TEM, we were unable to observe fibrillar aggregates with the K16N, E22K, and A21G peptides. Instead, we observed amorphous aggregates of different morphologies for these peptides. The D23N, E22Δ, D7N, A2V, E22Q, and WT peptides formed both fibrillar aggregates and amorphous structures with different morphologies, whereas the E22G, L34V, and H6R peptide samples contained mainly amyloid fibrils (Fig. 2). The fibrillar species formed by the different variants had different morphological characteristics. The D23N, D7N, E22Δ, and H6R variants formed dense networks of bundled fibrillar structures. The H6R peptide also formed long bundles of amyloid fibrils, similar to those formed by the E22Q mutant and the WT peptide (Fig. 2). In addition to forming dense fibrillar networks, the E22Δ variant formed individual amyloid fibrils, similar to those formed by the E22G, L34V, and A2V peptides (Fig. 2). It is important to note that whereas Fig. 2 displays representative images of the aggregates formed by the different peptides, the abundances of these aggregates were variable between the different peptides. For instance, the A21G peptide formed fewer fibrils of typical morphology, and the E22K peptide led to the formation of fewer bundled fibrils than observed with the other peptides. The relative abundances of the fibrils formed by the different peptides are indicated in Table 2.
TABLE 2.
Summary of the characteristics of aggregates formed by WT and FAD mutant Aβ40 peptides
| Aβ variant | ThT at 48 h | Morphology by TEM at 10 days |
|---|---|---|
| WT | 344.055 | Long bundled fibrils, amorphous aggregates |
| A2V | 1156.573 | Abundant fibrils of at least two different morphologies, globular aggregates |
| H6R | 521.1033 | Long and short bundled fibrils |
| D7N | 628.1713 | Short straight fibrils, amorphous aggregates |
| K16N | −22.117 | No fibrils, amorphous aggregates |
| A21G | 76.77067 | Few typical fibrils, amorphous aggregates |
| E22Δ | 138.4323 | Abundant fibrils of two different morphologies, fibril bundles, amorphous aggregates |
| E22Q | 56.40767 | Abundant long bundled fibrils, abundant amorphous aggregates |
| E22K | 518.2293 | Few fibrils in bundles, non-fibrillar aggregates |
| E22G | 781.5587 | Abundant fibrils |
| D23N | 222.0607 | Fibril bundles, amorphous aggregates |
| L34V | 590.133 | Short straight fibrils |
ThT Fluorescence Is Not an Accurate Reflection of Amyloid Fibril Content
Because the E22Δ fibrils display dramatically reduced ThT fluorescence, we performed ThT fluorescence measurements on all of the samples used for the TEM studies. As expected from the kinetic ThT fluorescence experiments, our results indicate that the different variants form aggregates with differential ThT fluorescence values at the 48-h time point (Table 2). However, and more importantly, we observe that high ThT fluorescence values are not always indicative of the presence of amyloid fibrils. Whereas the A21G and K16N samples had low ThT fluorescence values and were largely devoid of fibrils (Fig. 2), the E22Δ and E22Q variants formed abundant fibrillar aggregates despite having low ThT fluorescence values. Conversely, the E22K peptide displayed relatively high ThT fluorescence values but formed mostly amorphous aggregates and very few amyloid fibrils (Fig. 2). The results of the TEM experiment and the ThT fluorescence values for each peptide are summarized in Table 2.
Aβ40 Variants Associated with FAD Vary in Their Abilities to Fold into Immunologically Distinct Structures When Aggregated under Different Conditions
We investigated whether FAD point mutations located within the Aβ sequence lead to the adoption of unique Aβ conformations not observed with WT Aβ. To answer this question, we aggregated WT Aβ40 and the 11 FAD-associated Aβ40 variants under three different conditions over a 10-day time course. The three aggregation conditions were selected based on previous data indicating that they lead to the adoption of conformations with different immunological reactivity patterns. We studied the time course of these immunoreactivity patterns using conformation-dependent polyclonal antiserum OC (33) and mOC antibodies (25). These antibodies have been shown to have differential reactivity patterns against different amyloid conformations in vitro and against AD human and mouse model brain tissue. The in vivo reactivity patterns of the representative mOC antibodies presented in this study are summarized in Table 3. We observed that the three aggregation conditions dramatically alter the immunoreactivity profiles of the different Aβ peptides over the 10-day time course (Figs. 3 and 4).
TABLE 3.
Characteristics of representative mOC antibodies presented in this study
| Antibody name | Human AD brain reactivity | 14-month-old 3×Tg mouse brain reactivity |
|---|---|---|
| mOC1 | Frontal cortex plaques | Layer V cortical neurons, subiculum plaques |
| mOC29 | None | None |
| mOC31 | Vascular amyloid | None |
| mOC78 | Intracellular/nuclear, frontal cortex plaques | Layer V cortical neurons |
| mOC86 | Frontal cortex plaques | Cortical plaques |
| mOC87 | Frontal cortex plaques | Layer V cortical neurons |
| mOC98 | Frontal cortex plaques | Layer V cortical neurons |
| mOC108 | None | None |
| mOC116 | Frontal cortex plaques | Layer V cortical neurons, hippocampal plaques |
FIGURE 3.

Immunological analysis of aggregation by dot blotting at representative time points. Dot blotting results from WT Aβ and 11 Aβ variants containing FAD mutations aggregated using condition A (NaOH/phosphate buffer method) (a), condition B (HFIP/water method) (b), and condition c (NaOH/HEPES/NaCl method) (c). The peptides were aggregated over a 10-day time course, and the selected time points were probed for their immunological reactivity with the indicated antibodies. This figure displays only the time 0 and the 3- and 10-day time points of the aggregation reactions. The full data are presented in Fig. 4. These experiments were performed in duplicate with similar results.
FIGURE 4.

Immunological analysis of aggregation by dot blotting. Full set of dot blotting data from WT Aβ and 11 Aβ variants containing FAD mutations aggregated using condition A (NaOH/phosphate buffer method) (a), condition B (HFIP/water method) (b), and condition c (NaOH/HEPES/NaCl method) (c). The peptides were aggregated over a 10-day time course, and the selected time points were probed for their immunological reactivity with the indicated antibodies. Fig. 3 is an abbreviated version of this figure included to enable easier interpretation of the data. These experiments were performed in duplicate with similar results.
When probed with the majority of the antibodies, WT Aβ aggregated under condition A formed immunopositive aggregates early in the time course, with weakening antibody reactivity at later time points, although it did not react with some antibodies under these conditions (Figs. 3a and 4a). Under the same aggregation conditions, the K16N variant failed to form aggregates that significantly reacted with any of the mOC antibodies even though the site of the mutation is well outside the epitopes (residues 2–12) of the vast majority of the antibodies (25). Only weak reactivity with mOC87 at the 6-h and 1-ay time points was observed. In contrast, the D23N, E22K, and E22Δ peptides adopted stable conformations that reacted with a majority of the mOC antibodies. An interesting exception to this pattern was observed with mOC116, which recognizes the E22Δ peptide only at early and late time points, with very weak or no reactivity at intermediate time points. Similar to the three variants discussed above, the E22Q, E22G, A21G, and D7N peptides form aggregates that in most cases maintain their immunoreactivity throughout the 10-day time course, with few instances of time-dependent changes in reactivity strength. In contrast to aggregates of the D23N, E22K, and E22Δ mutants, aggregates formed by E22Q, E22G, A21G, and D7N peptides are only recognized restricted groups of mOC antibodies. The A2V, H6R, and L34V peptides generally display similar aggregation kinetics to the WT peptide, with antibody reactivity present early on and diminishing through the remainder of the reaction time course. However, compared with WT Aβ, aggregates of these three variants are recognized by a more limited group of antibodies. In addition, they have weaker immunological reactivity than WT Aβ in general. Interestingly, we observe weak mOC78 reactivity with aggregates of H6R Aβ at later time points, which are unreactive with the other mOC antibodies.
WT Aβ40 aggregated under condition B (resuspension in hexafluoroisopropanol (HFIP) and dilution in water) was immunoreactive at very early time points, showed no or very weak reactivity from the 2-h to 3-day intermediate time points, and adopted structures recognized by mOC antibodies again from the 5-day time point onward (Figs. 3b and 4b). When probed with most of the mOC antibodies, the D23N, E22K, E22Δ, and E22G Aβ variants displayed similar immunological profiles to that of WT Aβ, with an absence of immunoreactivity at intermediate time points. Although the K16N peptide also had similar aggregation kinetics to WT Aβ40, aggregates formed by this variant were only recognized by about half of the mOC antibodies. The A2V, H6R, D7N, A21G, E22Q, and L34V mutant peptides show similar aggregation kinetics to the WT peptide with the distinction that their early time points are only reactive with a subset of the mOC antibodies, whereas the later time point aggregates from the same samples react with a much wider range of antibodies. This observation implies structural differences between the early versus late time point aggregates of the FAD mutant Aβ peptides.
Aggregating Aβ40 under condition C (resuspension in NaOH and dilution in HEPES/NaCl buffer, pH 6) largely abolishes the time-dependent reactivity patterns observed in conditions A and B, with only a few observable instances of kinetic changes. As a result, this condition is useful as a control in studying the intrinsic abilities of the different mOC antibodies to recognize the Aβ40 variants without the additional variable of aggregation kinetics. For instance, the K16N peptide folds into an aggregate structure that is only recognized by four of the nine representative mOC antibodies in Figs. 3c and 4c (mOC78, -86, -87, and -98). Similarly, the A2V, H6R, L34V, D7N, and A21G peptides only react with a subset of the mOC antibodies. Conversely, the D23N, E22Δ, E22Q, and E22G peptides reliably react with the vast majority of the mOC antibodies throughout the 10-day time course when aggregated under this condition (Figs. 3c and 4c). Interestingly, whereas the widely used Aβ antibody 4G8 is able to react with WT Aβ and 10 of the FAD mutant Aβs in this aggregation condition, it is unable to recognize aggregates formed by the E22K peptide even though it recognizes all of the other mutations at this position, suggesting the presence of a unique conformation in the structures adopted by this Aβ variant. Likewise, mOC86 recognizes all of the Aβ peptides studied here except for the L34V variant, again implying the adoption of a unique amyloid fold by this peptide.
FAD Mutations Alter the Ability of Aβ40 to Aggregate into Distinct SDS-resistant Structures Characterized by Unique Molecular Weights and Exposed Epitopes
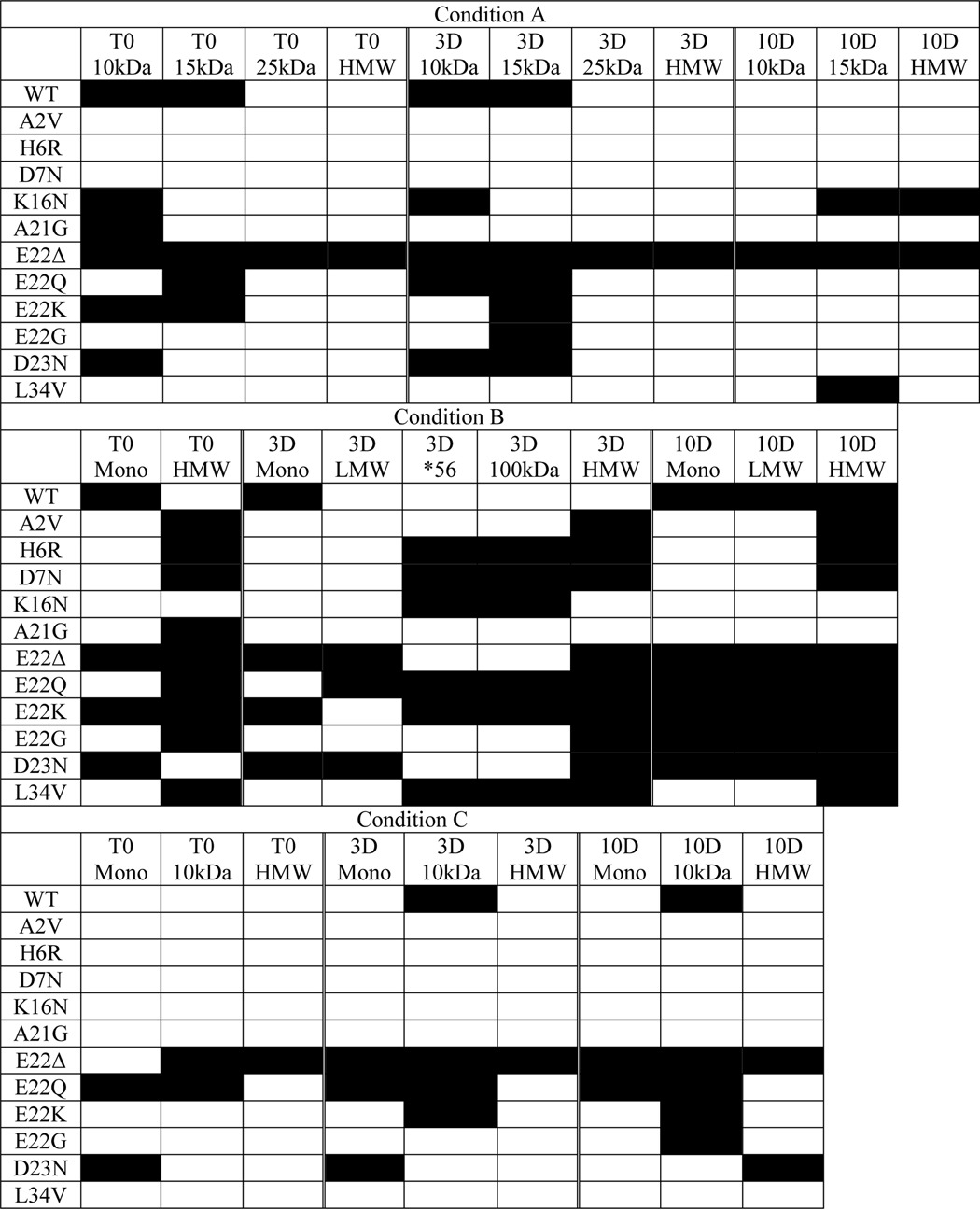
To further study the differences in the size distributions of SDS-resistant amyloid structures formed by the FAD mutant Aβs, we carried out Western blotting experiments on samples from the time 0 and the 3- and 10-day time points of the aggregation reactions described above. Although we have previously reported that SDS treatment can reveal, hide, or have no effect on the epitopes recognized by the different mOC antibodies (25), Western blotting allowed us to study the size distributions of the structures formed by the different FAD peptides under a different set of conditions than those used in the dot blotting experiments. Representative results from one of two replicates are shown (Fig. 5). Although the conditions used for Western blotting may not reflect physiological circumstances, Western blotting is widely used to characterize oligomeric species, and it is a reliable method for comparing relative size distributions. Here, we present data obtained using antibody mOC87. This antibody reacts with a broad size range of structures in vitro, has robust reactivity with human AD and transgenic AD mouse model brain tissue (25), and is unaffected by the FAD amino acid substitutions used (Fig. 5). Aggregating WT Aβ40 under condition A leads to the appearance of oligomers of 10–15-kDa molecular mass at the time 0 and 3-day time points. As expected from the dot blotting results, the 10-day time point of this preparation shows no reactivity with mOC87 (Fig. 5a). Also consistent with the dot blotting results, the A2V and H6R peptides are unable to form aggregates with the specific conformation recognized by mOC87 under condition A. Aggregates formed by A21G and D7N Aβs are also not recognized by mOC87 in our Western blotting experiments, even though they show reactivity with this antibody in the dot blotting assay, indicating that the epitope is not stable in these variants under the conditions used for Western blotting. This observation supports the idea that subjecting aggregated Aβ to SDS-PAGE may alter its conformation and is consistent with previously published data indicating that mOC antibody reactivity patterns are altered by SDS-PAGE treatment (25). Although the K16N variant is unreactive with mOC87 in the dot blotting assay, it displays increasing aggregation over time when probed by Western blotting. Specifically, this peptide forms immunopositive aggregates of ∼10 kDa at time 0 and the 3-day time point, which then disappear and are replaced with a distinct band at ∼15 kDa at the 10-day time point. There are also a wide range of high molecular mass aggregates at the 10-day time point. The E22K, E22G, and D23N mutant Aβs behave very similarly to WT Aβ in terms of aggregation kinetics and the size range of the mOC87-positive aggregates formed. The E22Q peptide shows similar kinetics to these variants but forms immunopositive aggregates with a wider range of molecular weights. The E22Δ peptide forms highly reactive aggregates appearing as distinct bands of 10–15-kDa, as well as higher molecular mass aggregates, which are present at all three time points tested. Under condition A, the L34V peptide shows an aggregation kinetic pattern that is unique among the 12 peptides tested, with mOC87 reactivity of ∼10 kDa appearing only at the 10-day time point (Fig. 5a).
FIGURE 5.

Western blotting analysis of aggregation. Western blotting results from WT Aβ and 11 Aβ variants containing FAD mutations aggregated under conditions A, B, and C (a–c, respectively). Samples from time 0 and the 3- and 10-day time points were subjected to SDS-PAGE and probed with the widely reacting representative antibody mOC87. These experiments were performed in duplicate with similar results.
Under aggregation condition B, WT Aβ40 shows weak monomer reactivity with mOC87 at time 0 and the 3-day time point, with high molecular mass reactivity appearing at the 10-day time point (Fig. 5b). Interestingly, the WT peptide is unique in producing this pattern under this condition. The A2V, H6R, E22G, and L34V peptides display high molecular mass reactivity that increases in intensity by the 10-day time point, although A2V reactivity is very weak, and E22G and L34V react in a more robust manner than the other two peptides. The E22G peptide also shows low molecular mass reactivity at the 10-day time point. D7N Aβ displays high molecular mass reactivity with mOC87 at all three time points, with the 3-day time point having weaker reactivity than the time 0 and 10-day time points. The A21G peptide displays weak high molecular mass reactivity only at time 0 and is unreactive at later time points. The E22K and E22Δ peptides show very similar immunoreactivity and kinetic patterns characterized by monomer reactivity at all three time points, with weakening reactivity at the intermediate 3-day time point. These peptides also show abundant high molecular mass reactivity that increases over time. The E22Q and D23N mutants' reactivity patterns are similar and are characterized by increasing reactivity in both low molecular mass and high molecular mass size ranges with incubation time. Perhaps the most interesting result under this aggregation condition was obtained using the K16N variant. This peptide reacts with mOC87 only as a distinct oligomeric band of ∼100 kDa, which is only present at the intermediate 3-day time point. Curiously, this band is also apparent in the H6R, D7N, E22K, E22Q, and L34V samples and is only present at the 3-day time point. The ∼100-kDa band seems to be accompanied by a weakly reactive band at ∼60 kDa, which may be indicative of the presence of Aβ *56 (Fig. 5b), which is a dodecameric species of Aβ associated with memory loss in Tg2576 mice (34) and with cognitive impairment and aging in humans (35).
Aggregation of WT Aβ40 using condition C results only in weak mOC87 immunoreactivity of an ∼10-kDa molecular mass band at the 3- and 10-day time points (Fig. 5c). The A2V, H6R, D7N, K16N, A21G, and L34V peptides fail to react with mOC87 at any of the time points studied under this aggregation condition. The E22G and E22K peptides show reactivity patterns similar to that of WT Aβ40. As in the other aggregation reactions, the E22Δ mutant shows the strongest reactivity here, characterized by consistent and robust reactivity in the high molecular mass range throughout the entire reaction time course. This peptide also displays monomer and ∼10-kDa reactivity slightly increasing in intensity over time. The E22Q variant was also unique in its reactivity pattern, which was in the monomer to ∼10-kDa range persisting at similar levels throughout the reaction time course, but with the highest reactivity at time 0. The D23N peptide showed yet another unique pattern with weak monomer reactivity at the earlier time points and high molecular mass reactivity appearing at the 10-day time point (Fig. 5c). The results from the Western blotting experiments are summarized in Table 4.
TABLE 4.
Summary of mOC87 Western blotting reactivity patterns of WT and FAD mutant Aβ40 peptides aggregated under conditions A–C
Images of the Western blottings are presented in Fig. 4. Filled box indicates reactivity. *56 = Aβ*56; HMW = high molecular weight aggregates; LMW = low molecular weight oligomers; Mono = monomer.
Point Mutations within an Antibody's Apparent Linear Epitope Have Little Effect on Antibody Reactivity
We observed that the existence of a point mutation within an antibody's apparent linear epitope (25) does not necessarily abolish that antibody's reactivity with the Aβ variant. For instance, mOC98, which displays a discontinuous epitope localized to residues 2–7 and 22–28 of Aβ (25), recognizes the H6R, D7N, E22K, E22Δ, E22Q, E22G, and D23N variants under all aggregation conditions by dot blot, even though all of these peptides contain mutations that fall within the apparent linear epitope for mOC98 (Fig. 6a). We also demonstrate that the absence of antibody reactivity with structures adopted by a FAD mutant Aβ peptide does not necessarily imply the presence of a point mutation in that antibody's apparent linear epitope. For example, mOC116, with an apparent linear epitope composed of residues 3–9 of Aβ (25), is unable to recognize K16N Aβ by dot blot, even though this variant has a mutation well outside of residues 3–9 (Fig. 6b).
FIGURE 6.

Relationship between location of FAD mutation and antibody epitope and reactivity. Dot blotting results for Aβ40 and 11 Aβ variants associated with FAD aggregated under conditions A–C probed with antibody mOC98 (a) and mOC116 (b), along with epitope mapping data for each of the antibodies. Blue depicts the results of a SPOT epitope mapping experiment. Each of the blue panels depicts 40 spots containing peptides C-terminally bound to a cellulose membrane. The peptides are consecutive 10-amino acid-long segments of the Aβ sequence beginning at position −3 (top left corner). The blue color indicates antibody reactivity with a particular spot. The interpretation of the epitope mapping study is to the right of the results, with the apparent epitope highlighted in red. In this assay, a large number of consecutive reactive spots indicates a shorter epitope, as there are a smaller number of amino acids common to the reactive spots. Conversely, a small number of consecutive reactive spots indicates a longer epitope, as the longer epitope would only be contained within a smaller number of spots.
Discussion
The properties associated with FAD mutations in Aβ are a key component of the evidence supporting a causative role for Aβ and APP processing in Alzheimer's pathology. Presenilin mutations and APP mutations localized to the flanking regions of the Aβ peptide increase the production of total Aβ by favoring β-secretase cleavage or increasing the production of the more amyloidogenic Aβ42 peptide relative to the less toxic Aβ40 peptide (36). In contrast, APP FAD mutations that fall within the Aβ sequence seem to have more heterogeneous biochemical properties and pathological effects and lead to the development of diverse clinical presentations (Table 1). Most of the mutations have been reported to increase the rate of aggregation to either fibrillar or oligomeric species that are hypothesized to represent more toxic or pathological species. Our results confirm that most of the mutations do indeed increase fibrillization, as determined by ThT fluorescence. However, A21G exhibited an extended lag time, whereas E22G displayed slower aggregation kinetics. The behavior of E22Δ is unique because it initially displays rapid aggregation with reduced ThT fluorescences slowly dropping to near zero over time. The low ThT fluorescence of E22Δ may be due to an intrinsic difference in its particular fibrillar structure because electron micrographs show that it makes abundant fibrils, and the dot blotting and Western blotting results with aggregation-specific antibodies show that it aggregates rapidly compared with wild type Aβ. These data are in agreement with solid-state nuclear magnetic resonance data indicating that the E22Δ peptide forms structures with distinct dynamics and hydration compared with those formed by WT Aβ or the E22G peptide (37). Although we did not directly assess the levels of soluble Aβ in the ThT reaction by sedimentation of the mixtures after 48 h, our Western blotting data suggest that at least some of the peptides (K16N, E22Δ, and L34V) are still present as soluble lower molecular mass species at the 48-h time point. This is because we observe lower molecular mass species of these peptides that react with antibody mOC87 even at the 10-day time point of condition A, which is the same condition as that used in the ThT experiment (Fig. 5a). Thus, consistent with previous reports (38–40), our results suggest that ThT may represent another probe of fibril structural polymorphism rather than a reliable operational definition of fibril formation. Although the structural basis for ThT binding is not yet known, other dyes like Orange G are known to intercalate and partition into the spaces formed by the interdigitating amino acid side chains that form steric zippers between adjacent β-sheets, suggesting that ThT may prefer particular types of steric zippers over others (41).
We hypothesized that FAD point mutations may lead to the adoption of alternative amyloid conformations by Aβ, which may be more toxic to cells or more resistant to protein degradation systems. We have previously reported that a panel of anti-Aβ monoclonal antibodies can distinguish alternative polymorphic conformations of amyloid in vitro and in vivo (24, 25). The data presented here, which were obtained using synthetic Aβ40 variants, support the above hypotheses. Our data indicate that multiple alternative conformations are adopted by the aggregates as a consequence of a FAD point mutation at a specific residue and that the specific conformations that are adopted also depend on the aggregation conditions, as reported previously for the E22Δ peptide (37). For example, the three FAD Aβs containing mutations at residue 22 (E22K, E22Q, and E22G) display dramatic differences in their immunological profiles and aggregation kinetics. These results are consistent with the fact that mOC and other anti-amyloid antibodies are generally conformation-specific in nature (25) and the idea that single amino acid substitutions may lead to the formation of alternative amyloid structures (9). These results are also consistent with those obtained using the artificial Aβ mutations E22V, which does not undergo significant aggregation in vitro (42), and E22P, which aggregates aggressively and forms highly stable amyloid structures (43). Again, even though the E22V and E22P mutations both localize to residue 22, they induce vastly different kinetic and structural changes in the Aβ peptide.
A number of the mutations are associated with the deposition of vascular amyloid and CAA (Table 1), so we hypothesized that these mutations might favor the formation of amyloid aggregates that react with the monoclonal antibody mOC31 because it specifically stains a subset of amyloid associated with vascular smooth muscle cells and not parenchymal amyloid deposits in transgenic mouse and human AD brain (25, 27). However, we failed to observe any effect of the mutations on mOC31 immunoreactivity in any of the immunoassays even though mOC31 reacts with some wild type Aβ fibril polymorphs in vitro (24, 25). This suggests that the FAD mutations that increase vascular amyloid do not affect the subtype of vascular amyloid that mOC31 recognizes. In addition, we observed that the existence of a FAD mutation within the apparent linear epitope of an mOC antibody does not abolish its reactivity against structures formed by the Aβ variant. Similarly, we observed that the fact that an mOC antibody is unreactive with the structures formed by an Aβ variant does not indicate that the point mutation for the specific FAD mutant peptide falls within the antibody's apparent linear epitope. These findings suggest that reactivity for many of the antibodies is conformation-specific and sequence-independent and cautions against the sole use of linear epitope mapping data to draw conclusions regarding an antibody's epitope. In summary, these observations demonstrate that FAD mutations produce alterations in the aggregation kinetics of Aβ peptides, and they promote the formation of immunologically and morphologically distinct amyloid structures that may account for some of the heterogeneity of disease phenotypes associated with the mutations.
Experimental Procedures
Antibody Production
mOC antibodies were produced in New Zealand White rabbits using a fibrillar Aβ42 antigen under contract with Epitomics (Burlingame, CA), as described previously (25, 27, 33, 44, 45). The antibodies have been characterized with respect to the epitope of Aβ to which they bind, sequence specificity, the sensitivity of their epitopes to thermal denaturation, dot blotting and Western blotting activities, and reactivity with amyloid deposits in transgenic and human brain (25). Antibodies were produced from hybridoma cell culture at a concentration of 3 μg/ml and used at a final concentration of 60 ng/ml. Antibodies mOC22, mOC31, mOC64, mOC78, mOC87, OC98, and mOC116 are available from Abcam, Cambridge, UK.
Peptide Production
Side chain protected fluorenylmethyloxycarbonyl (Fmoc) amino acids, Fmoc-PAL-PEG-polystyrene support, and O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU) were purchased from Applied Biosystems, Inc. (Carlsbad, CA), and N,N-diisopropylethylamine (DIEA), dithiothreitol (DTT), thioanisole, ethanedithiol, and anisole were from Aldrich. Trifluoroacetic acid (TFA) was purchased from Advanced Chemical Technology, Inc. (Louisville, KY). WT Aβ40 and Aβ40 containing the FAD mutations were synthesized by the batch-wise method on a CS336X (CS Bio, Inc., Menlo Park, CA) peptide synthesizer using Fmoc/t-butyl chemistry. TBTU/DIEA was used as the coupling reagent for 1 h, and 2% piperidine, 2% 1,8-diazabicyclo[5.4.0]undec-7-ene in dimethylformamide were used as the deprotection reagent for 7 min. Cleavage of the peptide from the resin support and the concomitant deprotection of the amino acid side chains were carried out in reagent R (TFA/thioanisole/ethanediol/anisole at 90:5:3:2) at room temperature for 6 h. This step was followed by removal of the exhausted resin by filtration and precipitation of the peptide product in ice-cold anhydrous ether. The precipitate was allowed to settle overnight at −20 °C and then washed three times with ice-cold water and dried under high vacuum. Preparative reversed phase high performance liquid chromatography was performed using a Waters (Irvine, CA) system (Model 510) with a Vydac C4 (214TP1022) column and a flow rate of 8 ml/min. The crude peptide was loaded after treatment with DTT and eluted using 0.1% TFA/H2O (buffer A) and 0.1% TFA/acetonitrile (buffer B) by gradient (5–95% buffer B). The center cut from the preparative run was frozen in liquid nitrogen immediately after collection and lyophilized under high vacuum. Peptide structure and purity (>90%) were verified by mass spectroscopy.
Aggregation of Aβ40 and Aβ40 FAD Variants
Aβ40, along with 11 Aβ40 variants containing FAD mutations (A2V, H6R, D7N, K16N, A21G, E22Δ, E22Q, E22K, E22G, D23N, and L34V) were aggregated under three different conditions (conditions A–C) over a 10-day time course, as described previously (25). For aggregation under condition A, 0.3 mg of the lyophilized peptide was resuspended in 33 μl of 100 mm NaOH and incubated for 10 min. This solution was then diluted to a 40 μm final concentration by adding 1.5 ml of 10 mm sodium phosphate buffer, pH 7.4. To aggregate the peptides under condition B, 0.5 mg of the lyophilized peptide was resuspended in 333.33 μl of HFIP and incubated for 15 min. This solution was then diluted with 1.33 ml of deionized water, and the tube containing the solution was covered with a punctured cap and placed under a hood; this solution was continuously stirred during the aggregation time course using a stir plate. For aggregation under condition C, 0.3 mg of the peptide was resuspended in 33 μl of 100 mm NaOH and incubated for 10 min. This solution was then diluted to 40 μm by the addition of 1.5 ml of HEPES/NaCl buffer (10 mm HEPES, 100 mm NaCl, pH 6.0). All of the aggregation reactions were carried out in the presence of 0.02% NaN3. All peptide aggregation experiments were performed in duplicate and had similar results. Representative results of one replicate are shown.
ThT Assay
WT Aβ40, along with A2V, H6R, D7N, A21G, E22Δ, E22Q, E22K, E22G, D23N, and L34V Aβ40 peptides were aggregated under condition A as described above. Condition B was not chosen as it is incompatible with the assay because of the presence of HFIP that is evaporating, and condition C was excluded because the rapid kinetics make the differences in aggregation less apparent. 150 μl from each of the reaction mixtures was immediately transferred to a well in a 96-well plate. At the same time, 1 μl of 3 mm ThT was added to each of the wells for a working ThT concentration of 20 μm. The plate was then covered with a Mylar sheet and placed in a 96-well plate reader (Molecular Devices, Sunnyvale, CA). Fluorescence readings from each well were obtained once every 10 min for a total time of 40–48 h. The excitation wavelength was set to 442 nm with the emission wavelength at 482 nm. The plate was maintained at a temperature of 37 °C and was briefly shaken prior to obtaining each reading. All reported values are averages of ThT absorbances taken in triplicate.
Dot Blotting and Western Blotting Assays
These assays were performed as described previously (25). The assays were performed in duplicate, and representative results are displayed.
Electron Microscopy
5 μl of each sample was deposited onto freshly glow-discharged carbon-Formvar-coated copper TEM grids. After 5 min, the grids were washed twice using deionized water. The peptides were then negative-stained using 1% filtered uranyl acetate for 10 min, and excess uranyl acetate was removed. Several images from each grid were obtained at a magnification of 72,000 using a JEOL 100CX transmission electron microscope operating at 60 kV. The TEM experiment was performed in duplicate, and representative images from three technical replicates per experiment are displayed.
Author Contributions
A. H. and C. G. G. designed the research; A. H., S. Monjazeb, and S. Milton performed the research; A. H. and C. G. G. wrote the paper.
This work was supported by National Institutes of Health Grants AG033069 and AG00538 and a grant from the Cure Alzheimer's fund. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- AD
- Alzheimer's disease
- APP
- amyloid precursor protein
- FAD
- familial AD
- Aβ
- amyloid-β
- TEM
- transmission electron microscopy
- ThT
- thioflavin T
- mOC
- monoclonal antibody cloned from OC serum-producing rabbits
- CAA
- cerebral amyloid angiopathy
- Fmoc
- fluorenylmethyloxycarbonyl
- TBTU
- O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate
- DIEA
- N,N-diisopropylethylamine
- HFIP
- hexafluoroisopropanol.
References
- 1. Weggen S., and Beher D. (2012) Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer's disease. Alzheimers Res. Ther. 4, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bertram L., and Tanzi R. E. (2008) Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nat. Rev. Neurosci. 9, 768–778 [DOI] [PubMed] [Google Scholar]
- 3. Benilova I., Karran E., and De Strooper B. (2012) The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357 [DOI] [PubMed] [Google Scholar]
- 4. Di Fede G., Catania M., Morbin M., Rossi G., Suardi S., Mazzoleni G., Merlin M., Giovagnoli A. R., Prioni S., Erbetta A., Falcone C., Gobbi M., Colombo L., Bastone A., Beeg M., et al. (2009) A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 323, 1473–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pasalar P., Najmabadi H., Noorian A. R., Moghimi B., Jannati A., Soltanzadeh A., Krefft T., Crook R., and Hardy J. (2002) An Iranian family with Alzheimer's disease caused by a novel APP mutation (Thr714Ala). Neurology 58, 1574–1575 [DOI] [PubMed] [Google Scholar]
- 6. Ovchinnikova O. Y., Finder V. H., Vodopivec I., Nitsch R. M., and Glockshuber R. (2011) The Osaka FAD mutation E22Δ leads to the formation of a previously unknown type of amyloid β fibrils and modulates Aβ neurotoxicity. J. Mol. Biol. 408, 780–791 [DOI] [PubMed] [Google Scholar]
- 7. Masuda Y., Nakanishi A., Ohashi R., Takegoshi K., Shimizu T., Shirasawa T., and Irie K. (2008) Verification of the intermolecular parallel β-sheet in E22K-Aβ42 aggregates by solid-state NMR using rotational resonance: implications for the supramolecular arrangement of the toxic conformer of Aβ42. Biosci. Biotechnol. Biochem. 72, 2170–2175 [DOI] [PubMed] [Google Scholar]
- 8. Nilsberth C., Westlind-Danielsson A., Eckman C. B., Condron M. M., Axelman K., Forsell C., Stenh C., Luthman J., Teplow D. B., Younkin S. G., Näslund J., and Lannfelt L. (2001) The ‘Arctic’ APP mutation (E693G) causes Alzheimer's disease by enhanced Aβ protofibril formation. Nat. Neurosci. 4, 887–893 [DOI] [PubMed] [Google Scholar]
- 9. Gessel M. M., Bernstein S., Kemper M., Teplow D. B., and Bowers M. T. (2012) Familial Alzheimer's disease mutations differentially alter amyloid β-protein oligomerization. ACS Chem. Neurosci. 3, 909–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang-Nunes S. X., Maat-Schieman M. L., van Duinen S. G., Roos R. A., Frosch M. P., and Greenberg S. M. (2006) The cerebral β-amyloid angiopathies: hereditary and sporadic. Brain Pathol. 16, 30–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Biffi A., and Greenberg S. M. (2011) Cerebral amyloid angiopathy: a systematic review. J. Clin. Neurol. 7, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Di Fede G., Giaccone G., and Tagliavini F. (2013) Hereditary and sporadic β-amyloidoses. Front. Biosci. 18, 1202–1226 [DOI] [PubMed] [Google Scholar]
- 13. Pezzini A., Del Zotto E., Volonghi I., Giossi A., Costa P., and Padovani A. (2009) Cerebral amyloid angiopathy: a common cause of cerebral hemorrhage. Curr. Med. Chem. 16, 2498–2513 [DOI] [PubMed] [Google Scholar]
- 14. Maia L. F., Mackenzie I. R., and Feldman H. H. (2007) Clinical phenotypes of cerebral amyloid angiopathy. J. Neurol. Sci. 257, 23–30 [DOI] [PubMed] [Google Scholar]
- 15. Grabowski T. J., Cho H. S., Vonsattel J. P., Rebeck G. W., and Greenberg S. M. (2001) Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. (Comment in (2001) Ann. Neurol.49, 691–693) Ann. Neurol. 49, 697–705 [DOI] [PubMed] [Google Scholar]
- 16. Van Nostrand W. E., Melchor J. P., Cho H. S., Greenberg S. M., and Rebeck G. W. (2001) Pathogenic effects of D23N Iowa mutant amyloid β-protein. J. Biol. Chem. 276, 32860–32866 [DOI] [PubMed] [Google Scholar]
- 17. Luyendijk W., Bots G. T., Vegter-van der Vlis M., Went L. N., and Frangione B. (1988) Hereditary cerebral haemorrhage caused by cortical amyloid angiopathy. J. Neurol. Sci. 85, 267–280 [DOI] [PubMed] [Google Scholar]
- 18. Obici L., Demarchi A., de Rosa G., Bellotti V., Marciano S., Donadei S., Arbustini E., Palladini G., Diegoli M., Genovese E., Ferrari G., Coverlizza S., and Merlini G. (2005) A novel AβPP mutation exclusively associated with cerebral amyloid angiopathy. Ann. Neurol. 58, 639–644 [DOI] [PubMed] [Google Scholar]
- 19. Fossati S., Cam J., Meyerson J., Mezhericher E., Romero I. A., Couraud P. O., Weksler B. B., Ghiso J., and Rostagno A. (2010) Differential activation of mitochondrial apoptotic pathways by vasculotropic amyloid-β variants in cells composing the cerebral vessel walls. FASEB J. 24, 229–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Glabe C. G. (2008) Structural classification of toxic amyloid oligomers. J. Biol. Chem. 283, 29639–29643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eisenberg D., and Jucker M. (2012) The amyloid state of proteins in human diseases. Cell 148, 1188–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kayed R., Canto I., Breydo L., Rasool S., Lukacsovich T., Wu J., Albay R. 3rd, Pensalfini A., Yeung S., Head E., Marsh J. L., and Glabe C. (2010) Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Aβ oligomers. Mol. Neurodegener. 5, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pensalfini A., Albay R. 3rd., Rasool S., Wu J. W., Hatami A., Arai H., Margol L., Milton S., Poon W. W., Corrada M. M., Kawas C. H., and Glabe C. G. (2014) Intracellular amyloid and the neuronal origin of Alzheimer neuritic plaques. Neurobiol. Dis. 71, 53–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wälti M. A., Ravotti F., Arai H., Glabe C. G., Wall J. S., Böckmann A., Güntert P., Meier B. H., and Riek R. (2016) Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. U.S.A. 113, E4976–E4984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hatami A., Albay R. 3rd, Monjazeb S., Milton S., and Glabe C. (2014) Monoclonal antibodies against Aβ42 fibrils distinguish multiple aggregation state polymorphisms in vitro and in Alzheimer's disease brain. J. Biol. Chem. 289, 32131–32143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kalimo H., Lalowski M., Bogdanovic N., Philipson O., Bird T. D., Nochlin D., Schellenberg G. D., Brundin R., Olofsson T., Soliymani R., Baumann M., Wirths O., Bayer T. A., Nilsson L. N. G., Basun H., et al. (2013) The Arctic AβPP mutation leads to Alzheimer's disease pathology with highly variable topographic deposition of differentially truncated Aβ. Acta Neuropathol. Commun. 1, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McLean D., Cooke M. J., Albay R. 3rd., Glabe C., and Shoichet M. S. (2013) Positron emission tomography imaging of fibrillar parenchymal and vascular amyloid-β in TgCRND8 mice. ACS Chem. Neurosci. 4, 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Groenning M., Olsen L., van de Weert M., Flink J. M., Frokjaer S., and Jørgensen F. S. (2007) Study on the binding of thioflavin T to β-sheet-rich and non-β-sheet cavities. J. Struct. Biol. 158, 358–369 [DOI] [PubMed] [Google Scholar]
- 29. Cloe A. L., Orgel J. P., Sachleben J. R., Tycko R., and Meredith S. C. (2011) The Japanese mutant Aβ (ΔE22-Aβ1–39) forms fibrils instantaneously, with low-thioflavin T fluorescence: seeding of wild-type Aβ1–40 into atypical fibrils by ΔE22-Aβ1–39. Biochemistry 50, 2026–2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chatani E., and Goto Y. (2005) Structural stability of amyloid fibrils of β(2)-microglobulin in comparison with its native fold. Biochim. Biophys. Acta 1753, 64–75 [DOI] [PubMed] [Google Scholar]
- 31. Shimada H., Ataka S., Tomiyama T., Takechi H., Mori H., and Miki T. (2011) Clinical course of patients with familial early-onset Alzheimer's disease potentially lacking senile plaques bearing the E693Δ mutation in amyloid precursor protein. Dement. Geriatr. Cogn. Disord. 32, 45–54 [DOI] [PubMed] [Google Scholar]
- 32. Kutoku Y., Ohsawa Y., Kuwano R., Ikeuchi T., Inoue H., Ataka S., Shimada H., Mori H., and Sunada Y. (2015) A second pedigree with amyloid-less familial Alzheimer's disease harboring an identical mutation in the amyloid precursor protein gene (E693δ). Intern. Med. 54, 205–208 [DOI] [PubMed] [Google Scholar]
- 33. Kayed R., Head E., Sarsoza F., Saing T., Cotman C. W., Necula M., Margol L., Wu J., Breydo L., Thompson J. L., Rasool S., Gurlo T., Butler P., and Glabe C. G. (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lesné S., Koh M. T., Kotilinek L., Kayed R., Glabe C. G., Yang A., Gallagher M., and Ashe K. H. (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357 [DOI] [PubMed] [Google Scholar]
- 35. Handoko M., Grant M., Kuskowski M., Zahs K. R., Wallin A., Blennow K., and Ashe K. H. (2013) Correlation of specific amyloid-β oligomers with τ in cerebrospinal fluid from cognitively normal older adults. JAMA Neurol. 70, 594–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wolfe M. S. (2007) When loss is gain: reduced presenilin proteolytic function leads to increased Aβ42/Aβ40. Talking point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 8, 136–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Elkins M. R., Wang T., Nick M., Jo H., Lemmin T., Prusiner S. B., DeGrado W. F., Stöhr J., and Hong M. (2016) Structural polymorphism of Alzheimer's β-amyloid fibrils as controlled by an E22 switch: a solid-state NMR study. J. Am. Chem. Soc. 138, 9840–9852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reinke A. A., and Gestwicki J. E. (2011) Insight into amyloid structure using chemical probes. Chem. Biol. Drug Des. 77, 399–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Biancalana M., and Koide S. (2010) Molecular mechanism of thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 1804, 1405–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Duboisset J., Ferrand P., He W., Wang X., Rigneault H., and Brasselet S. (2013) Thioflavine-T and Congo Red reveal the polymorphism of insulin amyloid fibrils when probed by polarization-resolved fluorescence microscopy. J. Phys. Chem. B 117, 784–788 [DOI] [PubMed] [Google Scholar]
- 41. Colletier J.-P., Laganowsky A., Landau M., Zhao M., Soriaga A. B., Goldschmidt L., Flot D., Cascio D., Sawaya M. R., and Eisenberg D. (2011) Molecular basis for amyloid-β polymorphism. Proc. Natl. Acad. Sci. U.S.A. 108, 16938–16943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Murakami K., Irie K., Morimoto A., Ohigashi H., Shindo M., Nagao M., Shimizu T., and Shirasawa T. (2003) Neurotoxicity and physicochemical properties of Aβ mutant peptides from cerebral amyloid angiopathy: implication for the pathogenesis of cerebral amyloid angiopathy and Alzheimer's disease. J. Biol. Chem. 278, 46179–46187 [DOI] [PubMed] [Google Scholar]
- 43. Masuda Y., Uemura S., Ohashi R., Nakanishi A., Takegoshi K., Shimizu T., Shirasawa T., and Irie K. (2009) Identification of physiological and toxic conformations in Aβ42 aggregates. ChemBioChem 10, 287–295 [DOI] [PubMed] [Google Scholar]
- 44. Nussbaum J. M., Schilling S., Cynis H., Silva A., Swanson E., Wangsanut T., Tayler K., Wiltgen B., Hatami A., Rönicke R., Reymann K., Hutter-Paier B., Alexandru A., Jagla W., Graubner S., et al. (2012) Prion-like behaviour and τ-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature 485, 651–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nussbaum J. M., Seward M. E., and Bloom G. S. (2013) Alzheimer disease: a tale of two prions. Prion 7, 14–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tycko R., Sciarretta K. L., Orgel J. P., and Meredith S. C. (2009) Evidence for novel β-sheet structures in Iowa mutant β-amyloid fibrils. Biochemistry 48, 6072–6084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Benilova I., Gallardo R., Ungureanu A.-A., Castillo Cano V., Snellinx A., Ramakers M., Bartic C., Rousseau F., Schymkowitz J., and De Strooper B. (2014) The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of amyloid-β (Aβ) aggregation. J. Biol. Chem. 289, 30977–30989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Giaccone G., Morbin M., Moda F., Botta M., Mazzoleni G., Uggetti A., Catania M., Moro M. L., Redaelli V., Spagnoli A., Rossi R. S., Salmona M., Di Fede G., and Tagliavini F. (2010) Neuropathology of the recessive A673V APP mutation: Alzheimer disease with distinctive features. Acta Neuropathol. 120, 803–812 [DOI] [PubMed] [Google Scholar]
- 49. Hori Y., Hashimoto T., Wakutani Y., Urakami K., Nakashima K., Condron M. M., Tsubuki S., Saido T. C., Teplow D. B., and Iwatsubo T. (2007) The Tottori (D7N) and English (H6R) familial Alzheimer disease mutations accelerate Aβ fibril formation without increasing protofibril formation. J. Biol. Chem. 282, 4916–4923 [DOI] [PubMed] [Google Scholar]
- 50. Ono K., Condron M. M., and Teplow D. B. (2010) Effects of the English (H6R) and Tottori (D7N) familial Alzheimer disease mutations on amyloid β-protein assembly and toxicity. J. Biol. Chem. 285, 23186–23197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kaden D., Harmeier A., Weise C., Munter L. M., Althoff V., Rost B. R., Hildebrand P. W., Schmitz D., Schaefer M., Lurz R., Skodda S., Yamamoto R., Arlt S., Finckh U., and Multhaup G. (2012) Novel APP/Aβ mutation K16N produces highly toxic heteromeric Aβ oligomers. EMBO Mol. Med. 4, 647–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tang T.-C., Hu Y., Kienlen-Campard P., El Haylani L., Decock M., Van Hees J., Fu Z., Octave J.-N., Constantinescu Stefan N., and Smith S. O. (2014) Conformational changes induced by the A21G flemish mutation in the amyloid precursor protein lead to increased Aβ production. Structure 22, 387–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang Z., Natté R., Berliner J. A., van Duinen S. G., and Vinters H. V. (2000) Toxicity of Dutch (E22Q) and Flemish (A21G) mutant amyloid β proteins to human cerebral microvessel and aortic smooth muscle cells. Stroke 31, 534–538 [DOI] [PubMed] [Google Scholar]
- 54. Tsubuki S., Takaki Y., and Saido T. C. (2003) Dutch, Flemish, Italian, and Arctic mutations of APP and resistance of Aβ to physiologically relevant proteolytic degradation. Lancet 361, 1957–1958 [DOI] [PubMed] [Google Scholar]
- 55. Betts V., Leissring M. A., Dolios G., Wang R., Selkoe D. J., and Walsh D. M. (2008) Aggregation and catabolism of disease-associated intra-Aβ mutations: reduced proteolysis of AβA21G by neprilysin. Neurobiol. Dis. 31, 442–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nishitsuji K., Tomiyama T., Ishibashi K., Ito K., Teraoka R., Lambert M. P., Klein W. L., and Mori H. (2009) The E693Δ mutation in amyloid precursor protein increases intracellular accumulation of amyloid β oligomers and causes endoplasmic reticulum stress-induced apoptosis in cultured cells. Am. J. Pathol. 174, 957–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tomiyama T., Nagata T., Shimada H., Teraoka R., Fukushima A., Kanemitsu H., Takuma H., Kuwano R., Imagawa M., Ataka S., Wada Y., Yoshioka E., Nishizaki T., Watanabe Y., and Mori H. (2008) A new amyloid β variant favoring oligomerization in Alzheimer's-type dementia. Ann. Neurol. 63, 377–387 [DOI] [PubMed] [Google Scholar]
- 58. Kassler K., Horn A. H., and Sticht H. (2010) Effect of pathogenic mutations on the structure and dynamics of Alzheimer's Aβ42-amyloid oligomers. J. Mol. Model. 16, 1011–1020 [DOI] [PubMed] [Google Scholar]
- 59. Hernandez-Guillamon M., Mawhirt S., Fossati S., Blais S., Pares M., Penalba A., Boada M., Couraud P.-O., Neubert T. A., Montaner J., Ghiso J., and Rostagno A. (2010) Matrix metalloproteinase 2 (MMP-2) degrades soluble vasculotropic amyloid-β E22Q and L34V mutants, delaying their toxicity for human brain microvascular endothelial cells. J. Biol. Chem. 285, 27144–27158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Van Nostrand W. E., Melchor J. P., and Ruffini L. (1998) Pathologic amyloid β-protein cell surface fibril assembly on cultured human cerebrovascular smooth muscle cells. J. Neurochem. 70, 216–223 [DOI] [PubMed] [Google Scholar]
- 61. Van Nostrand W. E., Melchor J. P., Romanov G., Zeigler K., and Davis J. (2002) Pathogenic effects of cerebral amyloid angiopathy mutations in the amyloid β-protein precursor. Ann. N.Y. Acad. Sci. 977, 258–265 [DOI] [PubMed] [Google Scholar]
- 62. Revesz T., Holton J. L., Lashley T., Plant G., Frangione B., Rostagno A., and Ghiso J. (2009) Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol. 118, 115–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Perálvarez-Marín A., Mateos L., Zhang C., Singh S., Cedazo-Mínguez A., Visa N., Morozova-Roche L., Gräslund A., and Barth A. (2009) Influence of residue 22 on the folding, aggregation profile, and toxicity of the Alzheimer's amyloid β peptide. Biophys. J. 97, 277–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fawzi N. L., Kohlstedt K. L., Okabe Y., and Head-Gordon T. (2008) Protofibril assemblies of the Arctic, Dutch, and Flemish mutants of the Alzheimer's Aβ40 peptide. Biophys. J. 94, 2007–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]