

Abstract
Stress-induced activation of p53 is an essential cellular response to prevent aberrant cell proliferation and cancer development. The ubiquitin ligase MDM2 promotes p53 degradation and limits the duration of p53 activation. It remains unclear, however, how p53 persistently escapes MDM2-mediated negative control for making appropriate cell fate decisions. Here we report that TBP-like protein (TLP), a member of the TBP family, is a new regulatory factor for the p53-MDM2 interplay and thus for p53 activation. We found that TLP acts to stabilize p53 protein to ensure long-lasting p53 activation, leading to potentiation of p53-induced apoptosis and senescence after genotoxic stress. Mechanistically, TLP interferes with MDM2 binding and ubiquitination of p53. Moreover, single cell imaging analysis shows that TLP depletion accelerates MDM2-mediated nuclear export of p53. We further show that a cervical cancer-derived TLP mutant has less p53 binding ability and lacks a proliferation-repressive function. Our findings uncover a role of TLP as a competitive MDM2 blocker, proposing a novel mechanism by which p53 escapes the p53-MDM2 negative feedback loop to modulate cell fate decisions.
Keywords: cancer, cell growth, p53, protein stability, transcription factor
Introduction
The tumor suppressor p53 is a potent transcription factor that promotes cell cycle arrest, senescence, and apoptosis in response to various types of stress (1). Although p53 orchestrates more than 100 target genes, it is usually retained in an “off” state because of its rapid turnover (2). This negative regulation is achieved mainly through recruitment of MDM2 to the transactivation domain (TAD)3 in the N terminus of p53 and the E3 ubiquitin ligase activity of MDM2 (3–5). MDM2 mediates both the monoubiquitination and polyubiquitination of p53 and promotes its degradation (6). Monoubiquitination at the DNA binding domain and the C-terminal domain of p53 acts as not only a scaffold of polyubiquitination but also a signal for p53 nuclear export (7–9). In the cytoplasm, other E3 ligases and E4 enzymes such as CBP/p300 can lead polyubiquitination of p53 and 26S proteasomal degradation (10). Additionally, given that p53 principally works as a transcriptional factor in the nucleus, the nuclear export of p53 is a major step that limits p53 function.
The p53-MDM2 interplay forms the basis of p53 dynamics, and several transcription-related factors regulate p53 activity (11). Upon genotoxic stresses, p53 is phosphorylated at Thr-18 and Ser-20, both of which are critical for MDM2 binding, leading to the dissociation of the p53-MDM2 interaction (12, 13). p300/CBP then binds to the p53 TAD and acetylates multiple lysine residues in the C-terminal domain of p53 (14), and p53 escapes from degradation and becomes active as a transcription factor (15). Activated p53 in turn induces the expression of MDM2. High levels of MDM2 associate with HDAC1 and deacetylate p53 to promote its degradation (16). In the late phase of the stress response, TBP-associated factor 1 (TAF1), the largest subunit of the basal transcription factor TFIID, phosphorylates p53 at Thr-55 and dissociates p53 from the p21 promoter by elevating MDM2 binding affinity of p53 (17, 18). In addition, some unknown transcription-related factors may also be involved in p53 regulation, and identification of a novel regulator of the p53-MDM2 interplay is critical for understanding the mechanisms underlying p53 dynamics.
TLP is a member of the TBP family and is also named TBP-related factor 2 (TRF2) and TBP-like 1 (TBPL1) (19, 20). TLP has 38% identity to the C-terminal conserved region of TBP and mimics the function of a basal transcription factor in the regulation of various biological processes (21). TLP, but not TBP, is associated with a number of TATA-less promoters and mediates RNA polymerase II-driven transcription from those promoters including ribosomal protein genes (22). Upon genotoxic stress, TLP represses cell growth via regulation of cell cycle-associated genes such as wee1, Cdkn1a (p21), and Trp63 (23, 24). We have also found that TLP binds to the TAD of p53, as does TBP, and enhances p21 expression in a p53-dependent manner (25–27). However, little is known about the most fundamental question of how TLP regulates p53 target genes or p53 itself.
Here we aimed to investigate the role of TLP in p53 regulation and present evidence that TLP is a new regulatory factor of the p53-MDM2 interplay. In the genotoxic stress response, TLP promotes p53-driven apoptosis and senescence by mediating persistent p53 activation. TLP binds to the p53 TAD and inhibits MDM2 recruitment to p53, which results in suppression of p53 ubiquitination. We also aimed to real time chasing of p53 nuclear export and show that TLP is essential for suppressing MDM2-driven nuclear export of p53. Moreover, a cervical cancer-derived TLP mutant has little p53 binding ability and does not suppress cell growth. Taken together, our findings indicate that TLP disrupts the p53-MDM2 interaction and mediates long-lasting p53 activation in response to genotoxic stress.
Results
TLP Stabilizes p53 Protein and Enhances Its Transcriptional Activity
To explore the role of TLP in p53 function, we first investigated the effects of TLP knockdown on the expression of p53 protein. Both transient and stable knockdown of TLP caused a decrease in p53 protein (Fig. 1, A and B). This decrease was recovered in the short interfering RNAs (siRNA)-resistant TLP (TLPesc)-added HCT116 cells (Fig. 1A). The effect of TLP knockdown was evident based on accelerated cell growth in both TLP siRNA-transfected HCT and HeLa cells (Fig. 1C). To investigate whether TLP regulates p53 at the protein level, we then chased the breakdown of p53 protein using cycloheximide (CHX). Overexpression of TLP increased the stability of p53 in both HCT116 and HeLa cells (Fig. 1D), whereas knockdown of TLP decreased the stability (Fig. 1E). Consistently, TLP-depleted cells contained decreased amounts of p53 targets such as MDM2 and p21 at the protein level (Fig. 1E). The same phenomenon was also observed at the mRNA level, and p53-up-regulated genes and p53-down-regulated genes were decreased and elevated, respectively (Fig. 1F). In contrast, although TLP depletion decreased p53 protein levels, its transcripts were not decreased (Fig. 1F). Additionally, CHX treatment decreased the levels of p53 protein, whereas treatment with the proteasome inhibitor MG132 increased it (Fig. 1G). Overexpressed p53 was also decreased through TLP knockdown in p53-null HCT116 cells (Fig. 1H). Moreover, TLP knockdown in p53-null HCT116 cells did not change the levels of p21 transcript, and the effect of TLP knockdown shown in wild-type HCT116 cells was also observed in the case of p53 overexpression (Fig. 1I). These results suggest that TLP up-regulates p53 at the protein level, which results in inhibition of cell growth through control of p53 target genes.
FIGURE 1.

TLP stabilizes p53 and increases transcription of p53 target genes. A, HCT116 cells were transfected with TLP siRNA (siTLP) together with a FLAG-His-tagged (FH)- and siRNA-resistant TLP expression plasmid (TLPesc). p53, TLP, and GAPDH levels in whole cell lysates were analyzed by Western blot. siCtrl, control siRNA; empty, empty vector. B, proteins in whole cell extracts of two distinct TLP shRNA (shTLP#1 and shTLP#2)-expressing HCT116 clones were analyzed. shCtrl, control shRNA. Because the knockdown effect of the two TLP shRNAs was similar, shTLP#1 and shTLP#2 are described as shTLP in the following figures when either of the TLP shRNAs was used. C, left, HCT116 cells were transfected with the siTLP together with the TLPesc expression vector. Forty-eight hours after transfection the cells were stained with crystal violet. Scale bar, 200 μm. Middle, viable cells were counted at 48 h after transfection. Results are presented as cell number relative to that at the transfection time. Each value is the mean ± S.D. of at least three independent experiments (mean ± S.D.). Right, a repeat experiment as shown in the middle panel of C was performed using HeLa cells. *, p < 0.05; **, p < 0.01 (Tukey's honestly significant difference test). D, HCT116 (left) and HeLa cells (right) were transfected with a FH-TLP expression vector. Twenty-four hours after transfection, the cells were treated with 50 μg/ml CHX and harvested at the indicated times. E, HCT116 (left) and HeLa (right) cells were transfected with siTLP. Forty-eight hours after transfection, the cells were treated with CHX and harvested at the indicated times. F, HCT116 cells (left) and HeLa cells (right) expressing shTLP were analyzed for transcripts of p53-up-regulated and p53-down-regulated genes by RT-qPCR (mean ± S.D.). Results are shown as relative amounts of mRNA to that of TBP. G, HCT116 cells were transfected with siTLP. Forty-eight hours after transfection, the cells were treated with 10 μm of MG132 (M) or/and CHX (C) for 1 h. Cells were harvested, and the indicated proteins in the whole cell lysate were determined. NT, non-treatment. H, two shTLP-expressing p53-null HCT116 clones were transfected with p53 expression vector. Twenty-four hours after transfection, the cell lysates were analyzed for the indicated proteins. I, p53-null HCT116 cells (HCT116 p53−/−) expressing shTLP were transfected with empty or p53 expression vector and incubated for 24 h. Amounts of indicated transcripts were determined for each gene by RT-qPCR (mean ± S.D.).
TLP Is Essential for Persistent Activation of p53 in Response to UV Irradiation
Next, to investigate whether TLP enhances p53 function in response to genotoxic stress, we examined TLP function under UV irradiation. When TLP short hairpin RNA (shRNA)-expressing HCT116 cells were exposed to various doses of UV light and incubated for 36 h, the levels of transcripts of p53 target genes including p21 and MDM2 (Fig. 2A) were less induced compared with control cells. To assess the interplay between p53 and TLP in response to UV irradiation, we chased the expression dynamics of p53 and its target proteins after a high dose (50 J/m2) of UV irradiation.
FIGURE 2.

TLP prolongs the duration of p53 activation upon UV irradiation. A, shTLP-expressing HCT116 cells were exposed to the indicated doses of UV light and incubated for 36 h. Amounts of indicated transcripts were determined for each gene by RT-qPCR (mean ± S.D.). B, left, HCT116 cells expressing shTLP were exposed to 50 J/m2 of UV light and harvested every 4 h. p53, Lys-382-acetylated p53 (Ac-382), MDM2, and p21 in whole cell lysates were detected. Right, p53, p21, and TLP proteins shown in the upper figures of panel B were quantified (means ± S.E.). *, p < 0.05; **, p < 0.01 (p53 and p21, Welch's t test; TLP, Tukey's honestly significant difference test). C, shTLP-expressing HCT116 cells were exposed to UV light and harvested every 8 h. Transcripts were determined by RT-qPCR (mean ± S.E.). D, TLP-depleted HCT116 cells were exposed to UV light and harvested every 16 h. Then chromatin-bound p53 was immunoprecipitated with anti-p53 antibody, and the sequences of the p21, PUMA, and MDM2 promoters were determined by qPCR (mean ± S.E.). E, HCT116 cells were transfected with the FH-TLP expression vector and exposed to UV light for 36 h. Chromatin-bound TLP was immunoprecipitated with anti-FLAG antibody, and the sequences of the p21, PUMA, and MDM2 promoters were determined by qPCR (mean ± S.E.).
The protein levels of both total p53 and Lys-382-acetylated p53, which is an activation marker for p53 transcriptional activity, were elevated within 4 h and decreased around 32–36 h after UV irradiation (Fig. 2B). Notably, TLP was significantly elevated around 32–36 h, and depletion of TLP accelerated the decrease of both total p53 and Lys-382-acetylated p53 during these time points (Fig. 2B, bottom). Consistent with these results, the levels of p21 protein were not elevated in TLP-depleted cells (Fig. 2B, bottom).
We then examined the expression of p53-up-regulated target genes related to cell cycle arrest (p21, 14-3-3σ) and apoptosis (PUMA, Fas) and the expression of p53-down-regulated targets participating in cell proliferation (CDC20, FOXM1) after UV irradiation (Fig. 2C). TLP-depleted cells clearly showed the reduction of p53 transcriptional activity in the late phase (at 32–48 h) of the UV response (Fig. 2C). In contrast, TLP mRNA was not significantly elevated (Fig. 2C; p = 0.14, analysis of variance) although its protein was up-regulated in the late phase of the UV response (Fig. 2B, bottom). This finding suggests that TLP is regulated at the protein level and is not a p53 target.
We also investigated whether p53 and TLP are recruited to the promoters of representative p53 target genes. After UV irradiation, p53 was detected on the p21, PUMA, and MDM2 promoters (Fig. 2D). As expected, the recruitment of p53 to these promoters was considerably reduced in the late phase of UV response of TLP-depleted cells (Fig. 2D). These results indicate that the levels of p53 protein reflect its occupation at these promoters. On the other hand, the recruitment of TLP to those promoters was not obvious (Fig. 2E), suggesting that TLP promotes p53 protein stability but not p53 recruitment to those promoters.
Finally, we investigated the viability and senescent state of the cells after UV irradiation. Under this condition, the viability of TLP knockdown cells was more than three times higher than that of control cells (Fig. 3, A and B). Furthermore, TLP-depleted cells exhibited resistance to senescence after UV irradiation (Fig. 3C). Taken together, these results indicate that TLP elongates the duration of p53 activation to promote apoptosis and senescence in response to the genotoxic stress.
FIGURE 3.
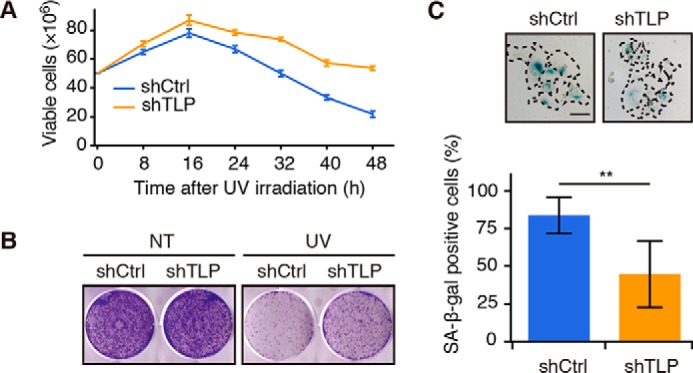
TLP promotes UV-mediated cell death and senescence. A, shTLP-expressing HCT116 cells were exposed to UV light, and viable cells were counted at the indicated times. B, shTLP-expressing HCT116 cells were exposed to UV light, and the cells were re-plated in a 6-well plate. After 7 days, colonies were stained with crystal violet. NT, no treatment. C, top, shTLP-expressing HCT116 cells were exposed to UV light and cultured for 5 days, and then senescence-associated β-galactosidase (SA-β-gal) activity was measured using a SA-β-gal staining method. A single cell is enclosed by a dotted line. Scale bar, 100 μm. Bottom, The SA-β-gal-positive cells were quantified, and results are shown as ratios (mean ± S.D.). **, p < 0.01 (Welch's t test).
TLP Prevents p53 Degradation through Disrupting the p53-MDM2 Interaction
To elucidate the detailed mechanisms by which TLP potentiates p53 activity, we investigated the involvement of MDM2 in the p53-TLP interplay. Overexpression of TLP and MDM2 increased and decreased the levels of p53, respectively (Fig. 4A). In addition, overexpression of both proteins canceled each effect (Fig. 4A). Furthermore, a p53 mutant L22Q/S23S (QS), which is defective in MDM2-binding, did not decrease when TLP was knocked down (Fig. 4B). An in vivo p53 ubiquitination assay showed that the amount of ubiquitinated p53 decreased depending on the TLP expression level (Fig. 4C). This phenomenon became more evident when MDM2 was coexpressed (Fig. 4D). TLP has been shown to bind to the p53 TAD (27); hence, we hypothesized that TLP prevents p53 binding of MDM2.
FIGURE 4.

TLP suppresses p53 ubiquitination and interferes with the p53-MDM2 interaction. A, HCT116 cells were transfected with MDM2 and TLP expression vectors. Left, 35 h after transfection, the amounts of the indicated proteins were determined. Right, relative band intensities of p53 are displayed (mean ± S.D., n = 4). *, p < 0.05; **, p < 0.01 (Tukey's honestly significant difference test). B, HCT116 p53-null cells were transfected with wild-type p53 (p53) or MDM2 binding ability-defective mutant p53 (p53 QS) together with siTLP. p53 protein was detected with polyclonal p53 antibody. C, HCT116 cells were transfected with expression vectors for FH-p53, HA-ubiquitin (Ub), and increased amounts of TLP. Twenty-four hours after transfection the cells were treated with MG132 for 3 h. The whole cell lysate was immunoprecipitated (IP) with anti-FLAG-Sepharose beads. p53-bound HA-tagged ubiquitin (HA-Ub) was detected by Western blot (WB) using anti-HA antibody. D, HCT116 cells were transfected with expression vectors for FH-p53, HA-Ub, MDM2, and increased amounts of TLP. Twenty-four hours after transfection the cells were treated with MG132 for 3 h. Then FH-p53 in whole cell lysate was immunoprecipitated with anti-FLAG-Sepharose beads, and p53-bound ubiquitin was detected. E, Escherichia coli-expressed GST-p53, FH-MDM2 (WT), and FH-MDM2 mutant (G58A) were purified from cell extracts using tag affinity resins. Proteins were stained with Coomassie Brilliant Blue. F, increased amounts of FH-MDM2 or FH-G58A were added to constant amounts of glutathione-Sepharose-bound GST-p53 and FH-TLP. Pull-downed proteins were detected by Western blot with corresponding antibodies. G, expression vectors for FH-p53 and increased amount of HA-TLP were introduced into HCT116 cells. FH-p53 in cell lysates was immunoprecipitated with anti-FLAG-Sepharose beads and eluted with a FLAG-peptide. p53, p53-bound MDM2, and p53-bound TLP were then detected. H, HCT116 cells were transfected with expression vectors for FH-MDM2 or FH-TLP together with HA-p53. Twenty-four hours after transfection, the cells were treated with MG132 for 2 h. FH-MDM2 (upper) and TLP (lower) in whole cell lysate was immunoprecipitated with anti-FLAG-Sepharose beads. I, schematic model of the interplay between p53, MDM2, and TLP. Low levels of TLP resulted in a situation whereby MDM2 promotes p53 ubiquitination and degradation. When TLP levels are elevated (e.g. in response to genotoxic stress), TLP binds to and stabilizes p53 by releasing p53 from MDM2-mediated negative control.
To evaluate the above hypothesis, we performed a competitive pulldown assay using a p53 binding ability-defective MDM2 mutant protein, G58A (Fig. 4E) (28). When wild-type MDM2 was added to the reaction mixture of the GST pulldown assay, the amount of p53-bound TLP was decreased, whereas the G58A mutant interfered with the TLP binding of p53 only slightly (Fig. 4F). We then performed an immunoprecipitation assay for p53 and found that p53-bound MDM2 decreased with increases in p53-bound TLP (Fig. 4G). This suggests that TLP and MDM2 competitively bind to p53 in cells. TLP and MDM2 did not form a complex with p53 simultaneously because TLP and MDM2 were not co-immunoprecipitated (Fig. 4H). These results indicate that TLP stabilizes p53 protein by interfering with MDM2-mediated p53 ubiquitination (Fig. 4I).
TLP Suppresses MDM2-mediated Nuclear Export of p53
Next, to scrutinize the involvement of TLP in MDM2 function in cells, we examined the localization of endogenous p53. Most of the p53 and exogenous TLP were localized in the nucleus of HCT116 cells (Fig. 5A). In contrast, TLP knockdown cells showed increased cytoplasmic localization of p53 (Fig. 5B). These results suggest that TLP retains p53 in the nucleus.
FIGURE 5.

TLP suppresses nuclear export of p53. A, HCT116 cells were transfected with FH-TLP, and cells were subjected to immunofluorescence staining with anti-p53 (p53) and anti-FLAG (TLP) antibodies. Scale bar: 10 μm. Nuclei were stained with DAPI. B, TLP-depleted HCT116 cells were treated with 5 μm MG132 for 30 min and subjected to immunofluorescence staining with anti-p53 antibody. Cell membranes were detected with Alexa Fluor 555-conjugated wheat germ agglutinin (WGA). Left, fluorescence was observed using a confocal microscope. Right, histogram of p53 intensity in the cytoplasm/that in the nucleus of each cell (n = 55). **, p < 0.01 (Welch's t test). C, schematic representations of the property of Dronpa. Intense irradiation at 488 nm changed Dronpa to a dark form, and weak irradiation at 405 nm restored it to a bright form. D, HeLa cells were transfected with pEF1α-TET3G and pTRE3G-p53-Dronpa, and the expression of p53-Dronpa under control of the Tet-On system was induced using the indicated amount of Dox. The p53-Dronpa signal in each cell was scanned using a confocal microscope. Three independent HeLa cell batches (cells #1–3) were analyzed. E, shTLP-expressing HeLa cells were transfected with pEF1α-TET3G and pTRE3G-p53-Dronpa, and the expression of p53-Dronpa was induced using 30 ng/ml of Dox. Cells were treated with CHX and MG132, and p53-Dronpa was detected by Western blot. F, shTLP-expressing HeLa cells were transfected with pEF1α-TET3G and pTRE3G-p53-Dronpa, and p53-Dronpa was induced by treatment with Dox. Left, 24 h after transfection the p53-Dronpa signal in each cell was scanned using a confocal microscope. Right, histogram of p53 intensity in the cytoplasm/in the nucleus of each cell (n = 60). Scale bar, 20 μm. *, p = 0.013 (Welch's t test). G, schematic representation of p53-Dronpa detection method for its nuclear export. H, shTLP-expressing HeLa cells were transfected with pEF1α-TET3G and pTRE3G-p53-Dronpa, and p53-Dronpa was induced by treatment with Dox. Cells were treated with CHX and MG132, and the p53-Dronpa signal in each cell was observed using a confocal microscope. Scale bar: 20 μm. I, left, time courses of the nuclear efflux of p53-Dronpa in TLP-depleted HeLa cells. In negative control experiments, leptomycin B (middle) and Nutlin-3a (right) were added to cells in addition to CHX and MG132. The closest time point after the reactivation was defined as time 0, and the increment of the intensity of cytoplasmic p53-Dronpa from time 0 was calculated (mean ± S.E.) and displayed as the relative intensity. At least eight cells were measured per experiment. **, p < 0.01; NS, not significant (two-way repeated measures analysis of variance).
To clarify whether TLP blocks MDM2-mediated nuclear export of p53, we performed a real-time nuclear export assay using the photoswitchable protein, Dronpa (Fig. 5C) (29). Low levels of p53-Dronpa were successfully induced in HeLa cells (Fig. 5, D and E). We confirmed that cytoplasmic localization of p53-Dronpa was emphasized in TLP knockdown cells (Fig. 5F). After treatment with CHX and MG132, fluorescence of p53-Dronpa in a whole cell was erased at 488 nm. Fluorescence in the nucleus was then reactivated through excitation at 405 nm (Fig. 5G). The flux of the florescence of Dronpa was monitored as an indicator of nuclear export. As for control shRNA-expressing cells, <10% of p53-Dronpa was exported to the cytoplasm within 720 s after photoactivation (Fig. 5, H and I, left). In contrast, nuclear export of p53-Dronpa in TLP shRNA-expressing cells was greatly accelerated. We also performed the same experiment with the nuclear export inhibitor leptomycin B and MDM2 antagonist Nutlin-3a to disrupt the p53-MDM2 interaction (7, 30). Leptomycin B-treated cells showed that p53-Dronpa was retained in the nucleus regardless of TLP depletion (Fig. 5I, middle). Similarly, Nutlin-3a blocked the nuclear export of p53-Dronpa in TLP knockdown cells (Fig. 5I, right), indicating that p53 nuclear export observed in TLP knockdown cells is dependent on MDM2. Taken together, these results suggest that TLP retains p53 in the nucleus by suppressing MDM2-driven nuclear export of p53.
TLP Suppresses Tumor Growth
To explore the physiological significance of our findings, we assessed the role of TLP in cancer. We first investigated the involvement of TLP in the growth of xenograft tumors. 25 days after injection the weight of TLP-depleted HCT116-derived tumors was significantly higher than that of control HCT116-derived tumors (Fig. 6A), indicating that depletion of intracellular TLP accelerates tumor growth. We then prepared a human cancer-derived TLP mutant, D99H (31, 32). This mutant is found in cervical cancer that expresses wild-type p53, and the mutation is located in the p53 binding region (Fig. 6B) (27). As expected, the p53 binding ability of D99H was considerably lower than that of wild-type TLP (Fig. 6C). Wild-type TLP-transfected HCT116 cells decreased cell growth in a p53-dependent manner (Fig. 6D). In contrast, overexpression of D99H did not influence cell growth (Fig. 6E). These results suggest that TLP-p53 binding is required for TLP-mediated cell growth repression.
FIGURE 6.

TLP suppresses tumor growth. A, left, a representative photograph of xenograft tumor-bearing mice at 25 days after injection of shCtrl and shTLP-expressing HCT116 cells. Right, weight of each tumor was plotted. **, p < 0.01 (Welch's t test). B, point mutation map of human cancers obtained from The cBio Cancer Genomics Portal. TLP contains two TBP-homology domains (TBP). Mutations at the 99th Glu are scored as D99E and D99H, and the D99H mutant was used in this study. The binding region of p53 (p53 binding: amino acid residue between the 64th and 115th) is depicted as an orange solid line. C, glutathione-Sepharose-bound GST-p53 and FH-TLP (WT) or D99H were mixed, and p53-bound TLP was detected. D, WT and p53-null HCT116 cells were transfected with a mouse TLP expression vector. Forty-eight hours after transfection cell numbers were counted. Results are presented as cell numbers relative to control cells (mean ± S.D.). **, p < 0.01; NS, not significant (Welch's t test). E, HCT116 cells were transfected with wild-type TLP or D99H. Twenty-four hours after transfection the numbers of viable cells were counted at the indicated culture times (mean ± S.D.). Statistical analysis was performed between the values of wild-type TLP and that of D99H. *, p < 0.05; **, p < 0.01 (Welch's t test).
Discussion
Recent studies have proposed that the concentration and duration of activated p53 and its target genes determine the cell fate decision such as cell cycle arrest, apoptosis, and senescence (33, 34). In the present study we showed that TLP stabilizes the p53 protein, thereby enhancing its function. We demonstrated that the TLP function is evident in the late phase of a high dose of UV exposure and is important to mediate the induction of apoptosis and senescence. This indicates that TLP is required for persistent p53 activation and direction of the cell fate decision. We further showed that TLP disrupts the p53-MDM2 interaction and demonstrated that TLP prevented p53 degradation by interfering with MDM2-mediated ubiquitination and nuclear export of p53. Moreover, we found that the expression of TLP is increased at the protein level in the late phase of UV irradiation, suggesting that elevated TLP binds to p53 and releases p53 from the MDM2-mediated negative control. In contrast, the MDM2 function toward p53 becomes stronger when the levels of TLP in cells are low. Notably, MDM2 is one of the targets of p53 and thus is down-regulated by TLP knockdown. Although this result seems to cause p53 stabilization, TLP depletion decreased p53 levels. This situation is because the turnover of p53 became faster in TLP-depleted cells. These phenomena suggest that TLP maintains the protein levels of p53 and MDM2 in cells. Our findings indicate that TLP acts as an MDM2 blocker in the p53-MDM2 negative feedback loop and establishes a novel mechanism for long-lasting p53 activation to direct appropriate cell fate decision (Fig. 7).
FIGURE 7.

Model of the action of TLP on p53-MDM2 interplay. A, regulation of the p53-MDM2 negative feedback loop by TLP. TLP interrupts MDM2-mediated negative regulation toward p53. B, TLP promotes p53 function by disrupting p53 binding of MDM2. TLP binding to p53 interferes with the p53-MDM2 interaction and retains p53 in the nucleus. As a consequence, TLP promotes p53 functions such as growth inhibition and senescence induction.
TBP recognizes the TATA element in promoters of many RNA polymerase II-driven genes, enabling it to constitute the basic transcriptional machinery on those promoters (35). TLP has been identified as a TBP family protein and is considered to be involved in various biological processes (21, 22). TLP binds to some general transcription factors, such as TFIIA, and interferes with the TBP-TFIIA interaction. The TLP-bound TFIIA precursor is protected from Taspase1-mediated cleavage, which leads to inhibition of TBP-mediated transcription (36). Perhaps one role of TLP may be as a blocker of protein-protein interactions. We note that TLP depletion in HeLa cells increased p53 mRNA levels, although p53 protein levels were down-regulated in such a condition. This phenomenon seems to be governed by p53-independent TLP function, suggesting that TLP has multiple roles in both p53-dependent and -independent manners.
The interplay between p53 and transcription-related factors in stress response is highly complicated. It is basically unknown whether basal transcription factors exert their positive or negative effects on p53 function. TFIIH, a multiprotein complex involved in both transcription and DNA repair, and TBP, both, activate p53-driven transcription (37, 38). In contrast, TAF1 phosphorylates p53 at Thr-55 and mediates degradation of p53 (17, 18). TAF1-mediated phosphorylation of p53 increases p53-MDM2 interaction and releases p53 from the p21 promoter in the late phase of UV irradiation. In this study we confirmed that p53 is actually recruited to the promoters of its target genes. However, TLP is not significantly recruited to promoters of representative p53 target genes. These results suggest that TLP affects p53 activity through a mechanism distinct from association of transcription machineries on a promoter. Hence, our findings may help to resolve the mechanisms underlying p53 dynamics and linking basal transcription factors to p53 activation.
The nuclear export of p53 has also been widely discussed because a number of cancers show cytoplasmic localization of p53 (39, 40). It has been speculated that MDM2 induces a conformational change of p53 to expose the nuclear export signal at the C-terminal domain of p53. MDM2 then mediates p53 monoubiquitination and promotes its nuclear export (6–8). We established a method for chasing real-time nuclear export of p53 using Dronpa and showed that TLP depletion increases the nuclear export of p53. However, TLP depletion does not fully export p53 to the cytoplasm, and the remaining p53 still activates transcription. These results suggest that TLP depletion is not sufficient for abolishing p53 activity. Because many factors cooperatively and rapidly regulate the p53 level in normal cells (9), and real-time analysis using Dronpa may help to clarify such a complicated process.
About half of all human cancers have mutations in the p53 gene, whereas the reason why some cancer cells still retain wild-type p53 has not been fully elucidated (41). In these cancer cells several altered cellular processes are likely to suppress p53 function. In this study we used HCT116 and HeLa cells. Interestingly, TLP-elevated p53 stability was clearly observed in HeLa cells. Human papillomavirus E6 protein in HeLa cell leads p53 to degradation (42), and because of these vulnerable conditions, TLP function for p53 may be strengthened. Furthermore, we found that multiple TLP mutations in human cancers were mapped in the p53 binding region of TLP. We used a TLP mutant, D99H, which is found in wild-type p53-expressing cervical cancer. This mutant had a weak p53 binding ability and exhibited a defect in suppression of cell growth. These findings are consistent with the notion that wild-type p53-expressing cancer cells have some p53-suppressive factors. Consequently, TLP is a novel regulator of the p53-MDM2 interplay that provides cells with sensitivity to genotoxic stress by mediating long-lasting p53 activation.
Experimental Procedures
Cell Culture, Transfection, and Cell Counting
Human HCT116 cells and HeLa cells were maintained in Dulbecco's modified minimal essential medium (Sigma) with 10% fetal bovine serum. Transfection of expression plasmids and siRNAs was performed by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendation. Viable cells were counted using trypan blue.
Expression Plasmids, siRNAs, and shRNAs
pCI-neo-FH-mTLP and pCI-neo-HA-mTLP were prepared as described previously (27). The siRNA-resistant TLP mutant that has nucleotide substitutions but retains the native amino acid sequence was constructed by PCR mutagenesis strategy. A p53 expression plasmid supplied by Addgene (Cambridge) was modified to pCI-neo-FH-p53, which contains a FLAG-His (FH) tag at the N terminus. A Dronpa open reading frame (ORF) was subcloned into pCI-neo-FH-p53 in frame with the p53 ORF. In addition, to induce the expression levels of p53 by Tet-On system, p53-Dronpa was subcloned into a Tet promoter-containing vector, pTRE3G (Clontech). pEF1α-TET3G vector, which expresses Tet-On 3G transactivator and activates expression from TRE promoters in the presence of doxycycline (Dox), was transfected with pTRE3G-p53-Dronpa. Sequences of siRNA for human TLP were 5′-UAACAGGGCCCAAUGUAAATT (sense) and 5′-UUUACAUUGGGCCCUAUUATT (antisense). To construct TLP shRNA-expressing vectors, human TLP sequence #1 (5′-GTAACAGGGCCCAATGTAA) and human TLP sequence #2 (5′-GGAGCAAATGTAATTTATA) containing shRNA sequences were cloned into pSilencer 5.1-U6 retro (AM5782).
Antibodies
Antigen-purified TLP antibody was used as described previously (19). To detect total p53 protein, anti-p53 monoclonal antibody (DO-1, Santa Cruz Biotechnology) was used in the most of the experiments. Only in experiments of Fig. 4B, anti-p53 polyclonal antibody (FL-393, Santa Cruz Biotechnology) was used because the DO-1 antibody does not recognize the p53 L22Q/S23S mutant. Antibodies against p53 Ac-382 (Cell Signaling Technology), MDM2 (4B11, Calbiochem), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; FL-335, Santa Cruz Biotechnology), p21 (M-19, Santa Cruz Biotechnology), HA (16B12, BioLegend), and FLAG (2H8, TransGenic Inc.) were used as primary antibodies.
Establishment of Stable Cell Lines
TLP shRNA-expressing HCT116, HCT116 p53−/−, and HeLa cell lines were established through transient transfection of TLP shRNA-containing pSilencer 5.1-U6 Retro (Thermo) followed by puromycin selection. Briefly, cells were transfected with these vectors and were reseeded 24 h after transfection. After 24 h, the cells were treated with 4 μg/ml puromycin and incubated for 1 week. Then puromycin-resistant colonies were selected and expanded to culture scale. Cells in which TLP expression was greatly inhibited were selected by RT-PCR and Western blotting.
Preparation of Cell Lysate and Western Blotting
To prepare cell lysates, cells were suspended in a lysis buffer (Tris-HCl, pH 7.6, 150 mm NaCl, 1 mm EDTA, 10% glycerol, 0.1% Nonidet P-40, and protease inhibitor (PI) mixture) and underwent three cycles of freeze and thaw. After centrifugation at 13,000 rpm for 20 min, the supernatant fraction was collected as a whole cell extract. Proteins were separated by 12.5% SDS-polyacrylamide gel electrophoresis and transferred to an Immobilon-P PVDF membrane (Millipore). Proteins on the membrane were detected by ImmunoStar Zeta (Wako).
RT-qPCR
Total cellular RNAs were prepared using an RNase Mini kit (Qiagen), and reverse transcription-PCR (RT-PCR) was performed. Briefly, cDNA was synthesized from 500 ng of total RNA using Prime Script II (Takara). Quantitative determination of the PCR products (qPCR) was performed using a Thunderbird qPCR Mix (Toyobo) and CFX384 Touch Real-Time PCR Detection System (Bio-Rad). All reactions were performed in triplicate. Primer sets to detect transcripts are described in Table 1.
TABLE 1.
Primers for RT-qPCR
| Gene | Forward (5′-) | Reverse (5′-) | Size | Reference |
|---|---|---|---|---|
| bp | ||||
| 14–3–3σ | ACTACGAGATCGCCAACAGC | CAGTGTCAGGTTGTCTCGCA | 149 | |
| CDC20 | GCAAGGAGAACCAGCCTGAA | ACCGTTCAGGTTCAAAGCCC | 80 | |
| Cyclin B1 | CGCCTGAGCCTATTTTGGTTG | CAGAGAAAGCCTGACACAGGT | 96 | |
| Fas | CTTCCCATCCTCCTGACCAC | TCGTAAACCGCTTCCCTCAC | 113 | |
| FoxM1 | TGCCCAGATGTGCGCTATTA | TCAATGCCAGTCTCCCTGGTA | 69 | 46 |
| GADD45A | ACGATCACTGTCGGGGTGTA | CCACATCTCTGTCGTCGTCC | 106 | |
| MDM2 | CAGCAGGAATCATCGGACTCA | AGGTCCTTTTGATCACTCCCAC | 80 | |
| NOXA | CTGGAAGTCGAGTGTGCTAC | GTTCCTGAGCAGAAGAGTTTGG | 101 | |
| p21 | CTGGGGATGTCCGTCAGAAC | CATTAGCGCATCACAGTCGC | 101 | |
| p53 | GTTCCGAGAGCTGAATGAGG | TTATGGCGGGAGGTAGACTG | 123 | 47 |
| PIG3 | AAATTCACCAAAGGTGCTGGAGT | TCCGCCTATGCAGTCTAGAATAAGA | 51 | 48 |
| PTEN | GACAGCCATCATCAAAGAGATCG | TCTGCAGGAAATCCCATAGCAA | 117 | |
| PUMA | CTCGGTGCTCCTTCACTCTG | AGGCTAGTGGTCACGTTTGG | 108 | |
| TLP | TGCAGACAGTGATGTTGCAT | GCTCCTTCCAAAGCAATCTTCC | 102 |
ChIP Assay
ChIP assay was performed as described previously with a few modifications (26). Sonicated chromatin of HCT116 cells was incubated with a specific antibody. The sample was then immunoprecipitated with protein G-Sepharose (GE Healthcare), and the presence of target DNA sequences in the precipitates was analyzed by qPCR. Primer sets used are described previously (p21 and PUMA; Ref. 43; MDM2; Ref. 44).
Colony Formation Assay
HCT116 cells were exposed to 50 J/m2 of UV light. Then the cells were trypsinized and re-plated into a 6-well plate. Seven days after re-plating, the cells were fixed and stained by a staining solution (PBS, crystal violet, 1% formaldehyde, and 1% methanol). Numbers of colonies with >100 cells were counted.
Senescence-associated β-Galactosidase Staining
Senescence states of UV light-irradiated HCT116 cells were detected using Cellular Senescence Assay kits (Chemicon). β-Galactosidase-positive cells was counted using ImageJ.
Immunoprecipitation
Cells transfected with a FLAG-tagged protein were suspended in IP buffer (20 mm Hepes-KOH, pH 7.8, 150 mm KCl, 1 mm EDTA, 10% glycerol, 0.1% Nonidet P-40, and PI mixture), disrupted by sonication, and centrifuged at 13,000 rpm for 20 min. The supernatant fraction was then collected. FH proteins in the extracts were precipitated by anti-FLAG M2 Affinity Gel (Sigma) at 4 ºC for 2 h. IgG-Sepharose 6 Fast Flow (GE Healthcare) was used as a control antibody. Bound proteins were eluted with FLAG peptides (0.5 mg/ml) at 4 ºC for 30 min, boiled for 5 min in SDS sample buffer, and analyzed by Western blotting.
In Vivo Ubiquitination Assay
To detect in vivo ubiquitination of p53, HCT116 cells were transfected with various combinations of plasmids as described in the figure legends. Twenty-four hours after transfection, the cells were treated with 10 μm MG132 for 3 h, and an in vivo ubiquitination assay was performed in two ways. One method for the assay, for which results are shown in Fig. 4C, was performed as below. Cells were lysed with 500 μl of radioimmune precipitation assay buffer (50 mm Tris-HCl pH 7.6, 150 mm NaCl, 1 mm EDTA, 10% glycerol, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 20 mm N-ethylmaleimide (NEM), and PI mixture) and subsequently immunoprecipitated as described above. As for another method, for which results are shown in Fig. 4D, cells were lysed with 200 μl of SDS lysis buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1 mm EDTA, 10% glycerol, 1% SDS, 20 mm NEM, and PI mixture) and then boiled for 10 min. The lysate was homogenized and subsequently diluted with 1.8 ml of dilution buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1 mm EDTA, 10% glycerol, 1% Triton X-100, 20 mm NEM, and PI mixture). FLAG-tagged p53 in the lysate was immunoprecipitated.
Immunofluorescence
Cells were washed with PBS, fixed with fix solution (PBS, 4% paraformaldehyde, 20 mm MgCl2, and 5 mm EGTA) for 10 min, washed again with PBS, and permeabilized with 0.5% Triton X-100 for 10 min. The cells were then incubated with Blocking One Histo (Nacalai Tesque Inc.) at room temperature for 15 min and subsequently incubated with anti-p53 or FLAG antibody at 37 ºC for 1 h. After washing with PBS, the cells were incubated with Alexa Fluor 488-conjugated anti-mouse IgG antibody and Alexa Fluor 555-conjugated wheat germ agglutinin at 37 ºC for 1 h. After washing again, the cells were mounted with Fluoro-KEEPER Antifade Reagent with DAPI (Nacalai Tesque Inc.) on a slide glass.
Real-time Nuclear Export Assay
Nucleocytoplasmic shuttling of p53-Dronpa was analyzed as described previously (29). HeLa cells were transfected with pTRE3G-p53-Dronpa and pEF1α-TET3G and then treated with 30 ng/ml Dox for an experiment at 24 h post transfection. 50 μg/μl cycloheximide, 10 μm MG132, 10 nm leptomycin B, and 5 μm Nutlin-3a were added when necessary. p53-Dronpa-expressing cells were observed under a FV1200 confocal microscope (Olympus) equipped with a 60× objective lens and two scanning units. Fluorescence of Dronpa was erased at 488 nm for 5 s followed by its photoactivation at the predetermined region by excitation at 405 nm for 10 ms. The flux of the fluorescence of Dronpa was monitored through weak excitation. Background noise subtraction and measurement of fluorescence signal intensities were executed using MetaMorph software (Molecular Devices Co.). The nuclear export rate was calculated using the equation Export = (Ct/C0) × (T0/Tt), where Ct is the average signal intensity in the cytoplasm at time t after photoactivation. To calculate the nuclear export of p53-Dronpa, the average signal intensity of reactivating states in the cytoplasm was determined as C0. Tt is the average signal intensity of the entire cell at time t, and T0 is the average signal intensity of the entire cell after reactivation during the measurement of nuclear export. The ratio of T0/Tt serves as a correction factor to control and eliminate loss of fluorescence signal during the image acquisition phase (45). All values were derived from at least 10 cells.
Tumor Xenograft Assay
Five-week-old BALB/c nu/nu male athymic nude mice (Japan SLC) were used. All animal experiments described adhered to policies and practices approved by Chiba University. TLP-depleted HCT116 cells (1 × 106 cells in 20 μl) were injected subcutaneously into nude mice. Tumor weight was measured 25 days after the injection.
Statistical Analysis
Welch's t test, Tukey's honestly significant difference test, and two-way repeated measures analysis of variance (analysis of variance) with replicate data sets were performed using R software.
Author Contributions
R. M., N. A., K. U., T. E., and T. T. conceived and designed the experiments. R. M., H. Tamashiro, K. T., H. S., S. S., and W. K. performed the experiments. R. M. and H. Takahashi analyzed the data. R. M., H. S., and T. T. contributed reagents, materials, and analysis tools. R. M. and T. T. wrote the paper.
This work was supported by the Japan Society for the Promotion of Science (272670 (to R. M.), 26114703 (to H. Tamashiro), 254583 (to H. S.), and 24570193 (to T. T.)). The authors declare that they have no conflicts of interest with the contents of this article.
- TAD
- transactivation domain
- CBP
- CREB (cAMP-response element-binding protein)-binding protein
- TAF1
- TBP-associated factor 1
- TLP
- TBP-like protein
- CHX
- cycloheximide
- FH
- FLAG-His
- Dox
- doxycycline
- PI
- protease inhibitor
- qPCR
- quantitative PCR
- NEM
- N-ethylmaleimide.
References
- 1. Vogelstein B., Lane D., and Levine A. J. (2000) Surfing the p53 network. Nature 408, 307–310 [DOI] [PubMed] [Google Scholar]
- 2. Wei C. L., Wu Q., Vega V. B., Chiu K. P., Ng P., Zhang T., Shahab A., Yong H. C., Fu Y., Weng Z., Liu J., Zhao X. D., Chew J. L., Lee Y. L., Kuznetsov V. A., et al. (2006) A global map of p53 transcription-factor binding sites in the human genome. Cell 124, 207–219 [DOI] [PubMed] [Google Scholar]
- 3. Kussie P. H., Gorina S., Marechal V., Elenbaas B., Moreau J., Levine A. J., and Pavletich N. P. (1996) Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274, 948–953 [DOI] [PubMed] [Google Scholar]
- 4. Haupt Y., Maya R., Kazaz A., and Oren M. (1997) Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299 [DOI] [PubMed] [Google Scholar]
- 5. Kubbutat M. H., Jones S. N., and Vousden K. H. (1997) Regulation of p53 stability by Mdm2. Nature 387, 299–303 [DOI] [PubMed] [Google Scholar]
- 6. Li M., Brooks C. L., Wu-Baer F., Chen D., Baer R., and Gu W. (2003) Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science 302, 1972–1975 [DOI] [PubMed] [Google Scholar]
- 7. Geyer R. K., Yu Z. K., and Maki C. G. (2000) The MDM2 RING-finger domain is required to promote p53 nuclear export. Nat. Cell Biol. 2, 569–573 [DOI] [PubMed] [Google Scholar]
- 8. Nie L., Sasaki M., and Maki C. G. (2007) Regulation of p53 nuclear export through sequential changes in conformation and ubiquitination. J. Biol. Chem. 282, 14616–14625 [DOI] [PubMed] [Google Scholar]
- 9. Carter S., Bischof O., Dejean A., and Vousden K. H. (2007) C-terminal modifications regulate MDM2 dissociation and nuclear export of p53. Nat. Cell Biol. 9, 428–435 [DOI] [PubMed] [Google Scholar]
- 10. Shi D., Pop M. S., Kulikov R., Love I. M., Kung A. L., Kung A., and Grossman S. R. (2009) CBP and p300 are cytoplasmic E4 polyubiquitin ligases for p53. Proc. Natl. Acad. Sci. U.S.A. 106, 16275–16280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beckerman R., and Prives C. (2010) Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2, a000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Unger T., Juven-Gershon T., Moallem E., Berger M., Vogt Sionov R., Lozano G., Oren M., and Haupt Y. (1999) Critical role for Ser-20 of human p53 in the negative regulation of p53 by Mdm2. EMBO J. 18, 1805–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sakaguchi K., Saito S., Higashimoto Y., Roy S., Anderson C. W., and Appella E. (2000) Damage-mediated phosphorylation of human p53 threonine 18 through a cascade mediated by a casein 1-like kinase: effect on MDM2 binding. J. Biol. Chem. 275, 9278–9283 [DOI] [PubMed] [Google Scholar]
- 14. Teufel D. P., Bycroft M., and Fersht A. R. (2009) Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 28, 2112–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tang Y., Zhao W., Chen Y., Zhao Y., and Gu W. (2008) Acetylation is indispensable for p53 activation. Cell 133, 612–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ito A., Kawaguchi Y., Lai C. H., Kovacs J. J., Higashimoto Y., Appella E., and Yao T. P. (2002) MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 21, 6236–6245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li H. H., Li A. G., Sheppard H. M., and Liu X. (2004) Phosphorylation on Thr-55 by TAF1 mediates degradation of p53: a role for TAF1 in cell G1 progression. Mol. Cell 13, 867–878 [DOI] [PubMed] [Google Scholar]
- 18. Wu Y., Lin J. C., Piluso L. G., Dhahbi J. M., Bobadilla S., Spindler S. R., and Liu X. (2014) Phosphorylation of p53 by TAF1 inactivates p53-dependent transcription in the DNA damage response. Mol. Cell 53, 63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ohbayashi T., Makino Y., and Tamura T. (1999) Identification of a mouse TBP-like protein (TLP) distantly related to the Drosophila TBP-related factor. Nucleic Acids Res. 27, 750–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rabenstein M. D., Zhou S., Lis J. T., and Tjian R. (1999) TATA box-binding protein (TBP)-related factor 2 (TRF2), a third member of the TBP family. Proc. Natl. Acad. Sci. U.S.A. 96, 4791–4796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zehavi Y., Kedmi A., Ideses D., and Juven-Gershon T. (2015) TRF2: TRansForming the view of general transcription factors. Transcription 6, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Y. L., Duttke S. H., Chen K., Johnston J., Kassavetis G. A., Zeitlinger J., and Kadonaga J. T. (2014) TRF2, but not TBP, mediates the transcription of ribosomal protein genes. Genes Dev. 28, 1550–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tanaka Y., Nanba Y. A., Park K. A., Mabuchi T., Suenaga Y., Shiraishi S., Shimada M., Nakadai T., and Tamura T. (2007) Transcriptional repression of the mouse wee1 gene by TBP-related factor 2. Biochem. Biophys. Res. Commun. 352, 21–28 [DOI] [PubMed] [Google Scholar]
- 24. Suenaga Y., Ozaki T., Tanaka Y., Bu Y., Kamijo T., Tokuhisa T., Nakagawara A., and Tamura T. (2009) TATA-binding protein (TBP)-like protein is engaged in etoposide-induced apoptosis through transcriptional activation of human TAp63 gene. J. Biol. Chem. 284, 35433–35440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Horikoshi N., Usheva A., Chen J., Levine A. J., Weinmann R., and Shenk T. (1995) Two domains of p53 interact with the TATA-binding protein, and the adenovirus 13S E1A protein disrupts the association, relieving p53-mediated transcriptional repression. Mol. Cell. Biol. 15, 227–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Suzuki H., Ito R., Ikeda K., and Tamura T. (2012) TATA-binding Protein (TBP)-like protein is required for p53-dependent transcriptional activation of upstream promoter of p21Waf1/Cip1 gene. J. Biol. Chem. 287, 19792–19803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maeda R., Suzuki H., Tanaka Y., and Tamura T. (2014) Interaction between transactivation domain of p53 and middle part of TBP-like protein (TLP) is involved in TLP-stimulated and p53-activated transcription from the p21 upstream promoter. PLoS ONE 9, e90190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Freedman D. A., Epstein C. B., Roth J. C., and Levine A. J. (1997) A genetic approach to mapping the p53 binding site in the MDM2 protein. Mol. Med. 3, 248–259 [PMC free article] [PubMed] [Google Scholar]
- 29. Ando R., Mizuno H., and Miyawaki A. (2004) Regulated fast nucleocytoplasmic shuttling observed by reversible protein highlighting. Science 306, 1370–1373 [DOI] [PubMed] [Google Scholar]
- 30. Vassilev L. T., Vu B. T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C., Fotouhi N., and Liu E. A. (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 [DOI] [PubMed] [Google Scholar]
- 31. Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., and Schultz N. (2012) The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., and Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leontieva O. V., Gudkov A. V., and Blagosklonny M. V. (2010) Weak p53 permits senescence during cell cycle arrest. Cell Cycle 9, 4323–4327 [DOI] [PubMed] [Google Scholar]
- 34. Kracikova M., Akiri G., George A., Sachidanandam R., and Aaronson S. A. (2013) A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ. 20, 576–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hernandez N. (1993) TBP, a universal eukaryotic transcription factor? Genes Dev. 7, 1291–1308 [DOI] [PubMed] [Google Scholar]
- 36. Suzuki H., Isogai M., Maeda R., Ura K., and Tamura T. (2015) TBP-like protein (TLP) interferes with Taspase1-mediated processing of TFIIA and represses TATA box gene expression. Nucleic Acids Res. 43, 6285–6298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen X., Farmer G., Zhu H., Prywes R., and Prives C. (1993) Cooperative DNA binding of p53 with TFIID (TBP): a possible mechanism for transcriptional activation. Genes Dev. 7, 1837–1849 [DOI] [PubMed] [Google Scholar]
- 38. Xiao H., Pearson A., Coulombe B., Truant R., Zhang S., Regier J. L., Triezenberg S. J., Reinberg D., Flores O., and Ingles C. J. (1994) Binding of basal transcription factor TFIIH to the acidic activation domains of VP16 and p53. Mol. Cell. Biol. 14, 7013–7024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moll U. M., Riou G., and Levine A. J. (1992) Two distinct mechanisms alter p53 in breast cancer: mutation and nuclear exclusion. Proc. Natl. Acad. Sci. U.S.A. 89, 7262–7266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goldman S. C., Chen C. Y., Lansing T. J., Gilmer T. M., and Kastan M. B. (1996) The p53 signal transduction pathway is intact in human neuroblastoma despite cytoplasmic localization. Am. J. Pathol. 148, 1381–1385 [PMC free article] [PubMed] [Google Scholar]
- 41. Freed-Pastor W. A., and Prives C. (2012) Mutant p53: one name, many proteins. Genes Dev. 26, 1268–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Freedman D. A., and Levine A. J. (1998) Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol. Cell. Biol. 18, 7288–7293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cross B., Chen L., Cheng Q., Li B., Yuan Z. M., and Chen J. (2011) Inhibition of p53 DNA binding function by the MDM2 protein acidic domain. J. Biol. Chem. 286, 16018–16029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fischer M., Steiner L., and Engeland K. (2014) The transcription factor p53: not a repressor, solely an activator. Cell Cycle 13, 3037–3058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Trotman L. C., Wang X., Alimonti A., Chen Z., Teruya-Feldstein J., Yang H., Pavletich N. P., Carver B. S., Cordon-Cardo C., Erdjument-Bromage H., Tempst P., Chi S. G., Kim H. J., et al. (2007) Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128, 141–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barsotti A. M., and Prives C. (2009) Pro-proliferative FoxM1 is a target of p53-mediated repression. Oncogene 28, 4295–4305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mizuno S., Bogaard H. J., Voelkel N. F., Umeda Y., Kadowaki M., Ameshima S., Miyamori I., and Ishizaki T. (2009) Hypoxia regulates human lung fibroblast proliferation via p53-dependent and -independent pathways. Respir. Res. 10, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Towers C. G., Guarnieri A. L., Micalizzi D. S., Harrell J. C., Gillen A. E., Kim J., Wang C. A., Oliphant M. U., Drasin D. J., Guney M. A., Kabos P., Sartorius C. A., Tan A. C., Perou C. M., Espinosa J. M., and Ford H. L. (2015) The Six1 oncoprotein down-regulates p53 via concomitant regulation of RPL26 and microRNA-27a-3p. Nat. Commun. 6, 10077. [DOI] [PMC free article] [PubMed] [Google Scholar]