

Abstract
Y box protein 1 (YBX1) is a well known oncoprotein that has tumor-promoting functions. YBX1 is widely considered to be an attractive therapeutic target in cancer. To develop novel therapeutics to target YBX1, it is of great importance to understand how YBX1 is finely regulated in cancer. Previously, we have shown that YBX1 could function as a tumor promoter through phosphorylation of its Ser-165 residue, leading to the activation of the NF-κB signaling pathway (1). In this study, using mass spectrometry analysis, we discovered a distinct phosphorylation site, Ser-176, on YBX1. Overexpression of the YBX1-S176A (serine-to-alanine) mutant in either HEK293 cells or colon cancer HT29 cells showed dramatically reduced NF-κB-activating ability compared with that of WT-YBX1, confirming that Ser-176 phosphorylation is critical for the activation of NF-κB by YBX1. Importantly, the mutant of Ser-176 and the previously reported Ser-165 sites regulate distinct groups of NF-κB target genes, suggesting the unique and irreplaceable function of each of these two phosphorylated serine residues. Our important findings could provide a novel cancer therapy strategy by blocking either Ser-176 or Ser-165 phosphorylation or both of YBX1 in colon cancer.
Keywords: colon cancer, NF-κB, phosphorylation, posttranslational modification (PTM), serine, YBX1
Introduction
The Role of YBX1 in Colon Cancer
Y-box binding protein 1 (YBX1) is a multifunctional DNA/RNA-binding protein that regulates transcription and translation. YBX1 specifically interacts with DNA and RNA and regulates many DNA- and mRNA-dependent processes, including DNA transcription, replication, repair, environmental stress, chromatin remodeling, pre-mRNA splicing, etc. (2). High expression of YBX1 is frequently detected in various cancers, including melanoma, osteosarcoma, and prostate, breast, squamous cell, lung, ovarian, thyroid, and colon cancer (3, 4), and is closely related to the progression and poor prognosis of these cancers. For instance, Shibao et al. (5) first demonstrated that YBX1 expression is elevated in colon cancer and positively correlates with DNA topoisomerase IIα and proliferating cell nuclear antigen expression. YBX1 can promote tumorigenesis, cell proliferation, replicative immortality, angiogenesis, invasion, and metastasis, most of which are the “hallmarks of cancer” proposed by Hanahan and Weinberg (6, 7). It is now widely accepted that YBX1 is an oncogene. It has been demonstrated previously that insulin-like growth factor 1 (IGF1)3 activates the PI3K/AKT pathway, leading to the phosphorylation of Ser-102 on YBX1 protein and governs its nuclear translocation in breast cancer cells (8, 9). When this site is disrupted, YBX1 is unable to translocate to the nucleus and activate the target genes, leading to reduced tumor growth in human breast cancer cells (9–11). In contrast, we recently discovered that, in response to IL-1β but not IGF1 treatment, YBX1 can be phosphorylated on Ser-165 (1), leading to colon cancer progression. In this study, we have identified distinct Ser-176 phosphorylation upon treatment with IL-1β. We further prove that Ser-176 phosphorylation of YBX1 is critical for NF-κB activation and its tumor-promoting ability. Ser-176 and the previously reported Ser-165 differentially regulate the expression of different subgroups of NF-κB target genes, offering a potential mechanism underlying the finely tuned YBX1-mediated NF-κB activation in colon cancer.
The Role of NF-κB in Cancer
NF-κB is a family of transcription factors that regulate the expression of genes involved in inflammation, cell proliferation, differentiation, and survival (12). Constitutively active NF-κB has been found in multiple types of cancer (13, 14). There are five proteins in the mammalian NF-κB family: RelA (p65), RelB, c-Rel, p50/p105, and p52/p100. All proteins in the NF-κB family share a Rel homology domain in their N terminus, which results in their classification as NF-κB/Rel proteins. The Rel homology domain is essential for dimerization as well as for binding to cognate DNA elements. The prototypic NF-κB is the heterodimer of p65 and p50. NF-κB activity is primarily regulated by interaction with IκB proteins. In most cells, NF-κB is present as a latent and inactive IκB-bound complex in the cytoplasm (15). When a cell receives an extracellular signal such as stress, cytokines, free radicals, radiation, etc., IκB is degraded, and NF-κB rapidly enters the nucleus and activates target gene expression (16). Constitutive NF-κB has been found in many different types of cancer (13), making NF-κB one of the most popular therapeutic targets in cancer.
Using mass spectrometry analysis, we identified phosphorylation of the novel Ser-176 site on YBX1 upon treatment with IL-1β. We showed that overexpression of the S176A-YBX1 (S176A) mutant led to decreased NF-κB binding ability and reduced cell growth and tumorigenic ability compared with the effect of WT-YBX1 overexpression. We also demonstrated that the phosphorylation of Ser-176 and a site reported previously by our lab, Ser-165 (1), lead to the regulation of quite distinct groups of NF-κB target genes. Therefore, we propose that phosphorylation of Ser-176 on YBX1 is an essential and novel discovery. The functions of phosphorylated Ser-176 or Ser-165 are not interchangeable and replaceable. This study could provide a novel therapeutic strategy for controlling the YBX1:NF-κB axis by blocking phosphorylation of either Ser-176 or Ser-165 or both in colon cancer.
Results
Identification of the Novel Phosphorylation on Ser-176 of YBX1
Previously, YBX1 was shown to be phosphorylated on Ser-102 in response to IGF1 in breast cancer cells (9, 10), and, more recently by us, on Ser-165 upon IL-1β treatment (1). To determine whether any additional serine sites could be phosphorylated by IL-1β stimulation, we conducted further mass spectrometry analysis in greater detail. Interestingly, we identified a novel site of phosphorylation on YBX1, Ser-176, in addition to Ser-165. As shown in Fig. 1, a mass shift of 80 Da was identified on Ser-176 of the FLAG-tagged YBX1 protein purified by FLAG pulldown from IL-1β-treated 293 (Fig. 1A) and HT29 cells (Fig. 1B), indicating the existence of a strong phosphorylation modification signal on Ser-176.
FIGURE 1.
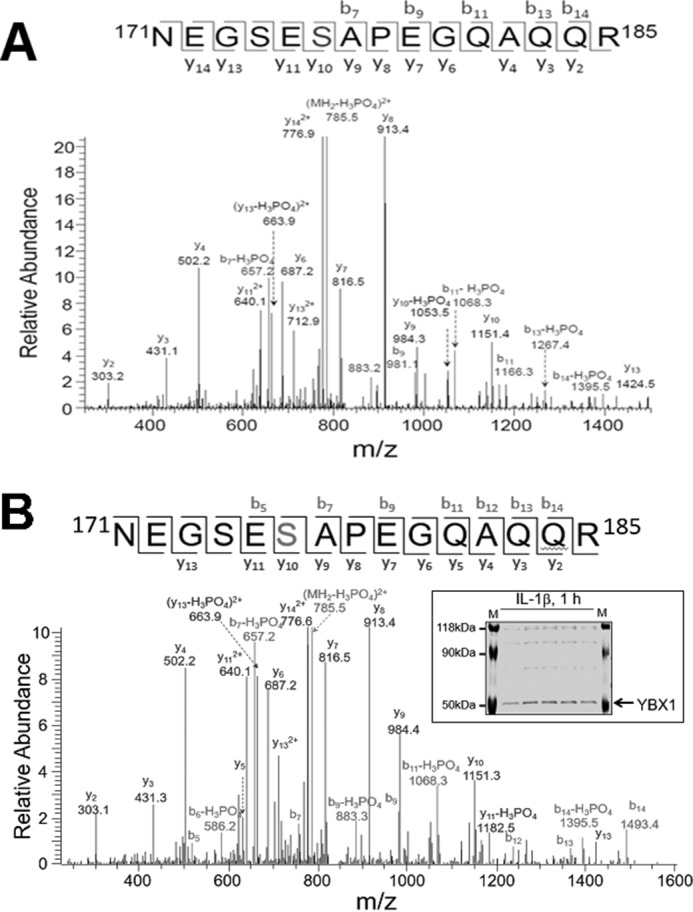
Identification of phosphorylation of Ser-176 on YBX1. A and B, mass spectrometry data for YBX1 showing that, in response to IL-1β treatment, Ser-176 is phosphorylated in 293 cells (A) and in HT29 cells (B). A mass shift of 80 Da was observed, indicating the existence of a phosphorylation modification. Inset, GelCode Blue-stained mass spectrometry gel of HT29 cells with the purified YBX1 band marked.
Phosphorylation of Ser-176 Is Important for the Activation of NF-κB
To determine whether Ser-176 phosphorylation plays an essential role in the activation of NF-κB, we successfully expressed the S176A mutant at a level comparable with WT-YBX1 in 293 cells. Meanwhile, a previously generated shYBX1 293 stable cell line was also included for comparison (1) (Fig. 2A). NF-κB-specific luciferase assay was carried out for the cells described above. As shown in Fig. 2B, upon IL-1β stimulation, NF-κB was greatly induced in control 293 cells. Overexpression of WT-YBX1 dramatically enhanced NF-κB activation, whereas expression of the S176A mutant had considerably weaker NF-κB-activating ability compared with WT-YBX1. These data suggest that phosphorylation of Ser-176 is critical for complete activation of NF-κB by YBX1.
FIGURE 2.
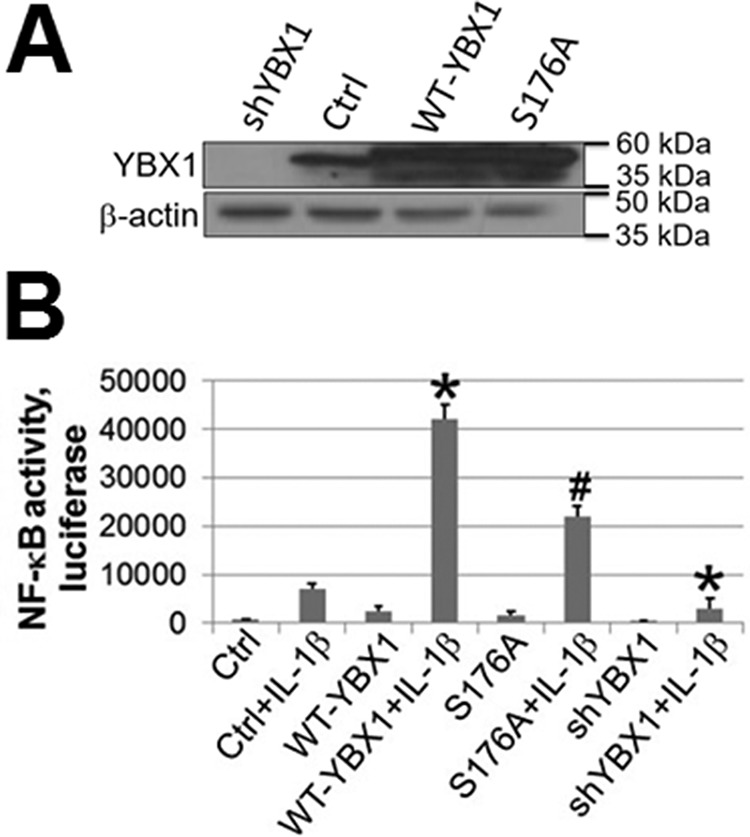
Phosphorylation of Ser-176 on YBX1 is critical for its NF-κB-activating ability. A, Western blot showing that YBX1 was overexpressed at similar levels in WT-YBX1 and the S176A mutant (serine 176-to-alanine mutation) overexpressing 293 cells. shRNA knockdown of YBX1 expression is also shown. B, NF-κB luciferase assay, showing that overexpression of WT-YBX1 could further enhance IL-1β-induced NF-κB activation compared with Ctrl, whereas overexpression of the S176A mutant led to significantly lower NF-κB activation compared with WT-YBX1. The data represent the mean ± S.D. from three independent experiments. *, p < 0.05 versus the Ctrl + IL-1β group; #, p < 0.05 versus the WT-YBX1 + IL-1β group.
Phosphorylation of Ser-176 on YBX1 Differentially Regulates a Subset of NF-κB Target Genes
Posttranslational modification (PTM) of transcription factors may lead to a different gene expression pattern (17). To examine this possibility further, we conducted an Illumina microarray analysis with control 293 cells and WT-YBX1 and S176A overexpression cells. Compared with WT-YBX1 cells (Fig. 3A), about 35% of NF-κB target genes were down-regulated by at least 2-fold in cells expressing S176A mutant protein. About 64% of the genes were not significantly affected, whereas very few genes (∼1%) were up-regulated by at least 2-fold. A representative list of NF-κB target genes that were down-regulated by the S176A mutant is shown in Fig. 3B. These genes can be significantly up-regulated by the overexpression of WT-YBX1 (WT/Ctrl ≥ 2-fold), but their inductions were dramatically reduced by S176A mutant overexpression (S176A/WT ≤ 0.5). Importantly, among these genes are cytokines, chemokines, and signaling components that are involved in tumorigenesis and metastasis, such as IL-17C, interferon regulatory factor 5 (IRF5), and v-myc myelocytomatosis viral oncogene homolog 1, lung carcinoma-derived (MYCL1). Several genes, such as IRF5 and MYCL1, were further confirmed with qPCR analysis in both 293 cells (Fig. 3C, top panels) and HT29 cells (Fig. 3C, bottom panels), showing great reduction in gene expression in the S176A mutant compared with the WT-YBX1 sample under both IL-1β-treated or untreated conditions. Collectively, these data confirm that phosphorylation of YBX1 at Ser-176 is critical for the activation of a subset of NF-κB-inducible genes.
FIGURE 3.

Phosphorylation of Ser-176 differentially regulates the expression of NF-κB target genes. A, comparative analysis of the S176A mutant with WT-YBX1-overexpressing cells on YBX1-inducible NF-κB target gene expression in 293 cells, showing that ∼35% of genes were down-regulated by 2-fold or more (S176A/WT-YBX1 ≤ 0.5) by the S176A mutation. B, list of typical NF-κB-inducible genes that could be up-regulated by WT-YBX1 (WT) but not by S176A. The cutoff -fold for induction is 2-fold, whereas, for reduction, it is 0.5 (decreased by 2-fold). C, qPCR analysis showing confirmation of microarray data. Two genes, IRF5 and MYCL1, showed reduced gene expression in the S176A mutant compared with the WT-YBX1 sample in both 293 (top panel) and HT29 cells (bottom panel). Furthermore, in IL-1β-treated samples, overexpression of WT-YBX1 significantly increased both IRF5 and MYCL expression compared with Ctrl cells. The data represent the mean ± S.D. from three independent experiments. *, p < 0.05 versus the Ctrl group; #, p < 0.05 versus the Ctrl + IL-1β group; §, p < 0.05 versus the WT-YBX1 group; $, p < 0.05 versus the WT-YBX1 + IL-1β group.
Ser-176 Is a Major Phosphorylation Site of YBX1
Previously, we reported that Ser-165 of YBX1 can be phosphorylated upon stimulation by IL-1β (1). We wondered whether both Ser-176 and Ser-165 are phosphorylated to a similar extent or whether one is more dominant than the other after IL-1β treatment. FLAG-tagged WT-YBX1, the S176A single mutant, or the S176A/S165A double mutant were overexpressed at similar levels in 293 cells (Fig. 4A). A co-immunoprecipitation experiment was further conducted using anti-FLAG beads, followed by Western blotting analysis with anti-general phosphorylated serine antibody. As shown in Fig. 4B, upon treatment with IL-1β, FLAG-WT-YBX1 was significantly phosphorylated (Fig. 4B, top panel), whereas the FLAG-S176A mutant exhibited a dramatically reduced phosphorylation ability compared with FLAG-WT-YBX1 protein. Furthermore, the FLAG-S176A/S165A double mutant displayed an even weaker phosphorylation ability than the FLAG-S176A single mutant, suggesting that Ser-176 is a more pronounced phosphorylation site than Ser-165. The intensity of phosphorylation was further quantified and is shown in Fig. 4B, bottom panel.
FIGURE 4.
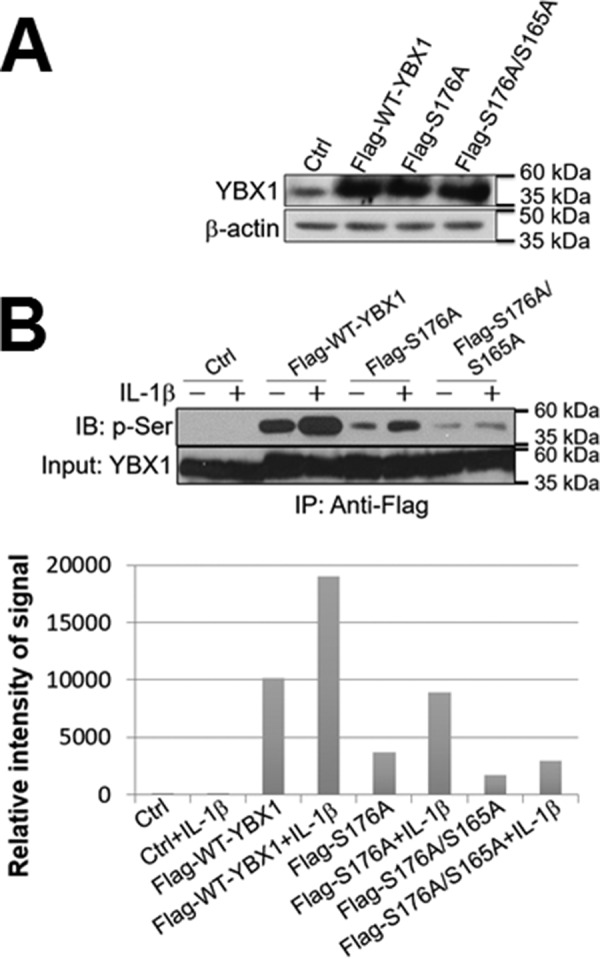
Ser-176 is a major phosphorylation site of YBX1. A, Western blot showing that FLAG-tagged WT-YBX1, S176A, and S176A/S165A were overexpressed at similar levels in 293 cells. Anti-YBX1 antibody was used to detect the total YBX1 expression. β-Actin was probed as a loading control. B, Ser-176 is a major phosphorylation site on YBX1. Top panel, co-immunoprecipitation and Western blot of 293 control cells and cells containing FLAG-tagged WT-YBX1, S176A, or S176A/S165A. These cells were treated with IL-1β for 1 h or left untreated. FLAG-tagged YBX1 was further pulled down with anti-FLAG beads and subjected to Western blotting analysis using an anti-phospho-serine motif antibody. The inputs were probed with an anti-YBX1 antibody. The Western blot shows that FLAG-tagged WT-YBX1 could be significantly phosphorylated upon IL-1β stimulation, whereas FLAG-S176A mutation abolished most of the phosphorylation effect on YBX1, and FLAG-176A/S165A double mutation led to even weaker YBX1 phosphorylation. Bottom panel, quantitative analysis of the phosphorylation signal in the top panel, showing that, in response to IL-1β treatment, Ser-176 is an important phosphorylation site on YBX1. IB, immunoblot.
Differential and Collaborative Gene Regulation by Phosphorylation of Ser-176 and Ser-165 of YBX1
Because we showed that both Ser-176 and Ser-165 can be phosphorylated upon IL-1β treatment, we wondered whether these two sites could contribute collaboratively or differentially to the NF-κB target gene regulation. A parallel Illumina array experiment for S165A was carried out along with the S176A sample. The data suggest that the expression of ∼33% (185 genes) of NF-κB target genes was reduced by 2-fold or more by S165A mutation (S165A/WT ≤ 0.5) and ∼35% (197 genes) by S176A mutation (S176A/WT ≤ 0.5) (Figs. 3A and 5A, top panel) compared with WT-YBX1. Among the 197 genes that were down-regulated by S176A, 82 genes were solely regulated by S176A but not by S165A (S176A/WT ≤ 0.5, S165A/WT > 0.5); in other words, their gene expression is S176A-dependent (Fig. 5A, bottom panel). On the other hand, among the 185 genes that were down-regulated by S165A, 70 genes were solely regulated by S165A (S165A/WT ≤ 0.5, S176A/YBX1 > 0.5) but not by S176A (Fig. 5A, bottom panel); that is, they are S165A-dependent. Beyond the uniqueness of their gene regulation pattern, both S176A and S165A share the regulation of a common pool of genes (115 genes) (176A/YBX1 ≤ 0.5, S165A/WT ≤ 0.5), indicating that they not only differentially regulate distinct subgroups of genes but also function in a collaborative manner. This interesting phenomenon suggests the sophisticated but elegant gene regulation capacity by the important oncogene YBX1.
FIGURE 5.

Phosphorylation of Ser-176 or Ser-165 results in differential expression of a subgroup of NF-κB target genes. A, comparative analysis of the S176A mutant with the S165A mutant on YBX1-inducible NF-κB target gene expression. The pie chart shows that, although the S176A and S165A mutants shared the regulation of 115 genes, each also had its own pool of solely regulated genes, with S176A regulating 82 unique genes and S165A regulating a completely different set of 70 genes. B, top panel, a short list of representative NF-κB-inducible genes that were down-regulated by both S176A and S165A mutants. Bottom panel, IPA showing that genes commonly regulated by both S176A and S165A are associated with a network of the functions of “protein degradation, synthesis, and cellular function and maintenance.” Importantly, NF-κB is one of the critical nodes in this network. C, top panel, a short list of representative NF-κB-inducible genes that were down-regulated solely by the S176A mutant. Bottom panel, IPA showing that the genes solely regulated by S176A are associated with a network of the functions of “cancer, dermatological diseases and conditions, and gastroenterological diseases.” Importantly, both NF-κB and YBX1 are critical nodes in this network. D, top panel, a short list of representative NF-κB-inducible genes that could be down-regulated solely by the S165A mutant. Bottom panel, IPA showing that genes solely regulated by S165A are associated with a network of the functions of “cell morphology, cell death and survival, and reproductive system development and function.” Importantly, both NF-κB and YBX1 are critical nodes in this network.
Representative genes that are commonly shared by S176A and S165A (Fig. 5B, top panel) or solely by S176A (Fig. 5C) or S165A (Fig. 5D) are shown in Fig. 5. For instance, IRF5 (Fig. 5B) is regulated by either S176A or S165A. This gene is considered to be a novel regulator of C-X-C motif chemokine ligand 13 (CXCL13) expression in cancer (18). In contrast, MYCL1 (Fig. 5C) is solely regulated by S176A. MYCL1 is a member of the MYC gene family, which is well known for its oncogenic potential or correlation with poor prognosis in cancer (19). For S165A-regulated genes, oncostatin-M (OSM) is a very good example. This gene encodes a member of the leukemia inhibitory factor/OSM family of proteins. OSM protein is a secreted cytokine that may regulate the production of other cytokines, including IL6, G-CSF, and GM-CSF in endothelial cells. The OSM receptor is suggested to be a novel therapeutic target in cancer (20). A full list of each group of genes is provided in supplemental Tables S1–S3.
To identify the signature networks of each subgroup of genes, we further conducted an ingenuity pathway analysis (IPA). It is extremely interesting that we observed that each group of genes is associated with quite distinct network functions. For instance, the commonly regulated genes by both S176A and S165A are predominantly associated with network functions of “protein degradation, synthesis, and cellular function and maintenance” (Fig. 5B, bottom panel) and “cellular development, cellular growth and proliferation, and connective tissue development and function” (supplemental Table S4 and Fig. S1). Although S176A solely regulated genes are associated with network functions of “cancer, dermatological diseases and conditions, and gastroenterological diseases” (Fig. 5C, bottom panel) and “cancer, hematological diseases, and hereditary disorders” (supplemental Table S5 and Fig. S2). In great contrast, S165A solely regulated genes are mainly involved in the network functions of “cell morphology, cell death and survival, and reproductive system development and function” (Fig. 5D, bottom panel) and “cell-to-cell signaling and interaction, nervous system development and function, and developmental disorders” (supplemental Table S6 and Fig. S3). Strikingly, NF-κB is one of the most pivotal nodes in each of the key networks (Fig. 5, B–D, bottom panels). Importantly, YBX1 also shows up in two of these networks (Fig. 5, C and D, bottom panels), suggesting an intimate yet sophisticated connection between YBX1 and NF-κB. In short, the above evidence inarguably supports the important and distinct role of differential gene regulation by phosphorylation of Ser-176 and Ser-165 of the YBX1 protein.
Phosphorylation of Ser-176 on YBX1 Plays an Important Role in Cell Proliferation and Anchorage-independent Growth in Colon Cancer Cells
Because NF-κB is well known to be involved in the expression of factors that can promote cancer cell proliferation, we decided to examine the effects of the S176A mutant on NF-κB activity, cell proliferation, and anchorage-independent growth in HT29 colon cancer cells. We established stable HT29 cell lines either with the overexpression of WT-YBX1 or with S176A (Fig. 6A, top panel). By carrying out NF-κB-specific luciferase reporter assays, we further confirmed that overexpression of WT-YBX1 could activate NF-κB, as we observed previously (1) (Fig. 6A, bottom panel). Furthermore, we proved that the S176A overexpression cells had lower NF-κB luciferase activity compared with the WT-YBX1 overexpression cells, confirming that S176A plays an important role in regulating NF-κB activity. Additionally, we determined the effect of S176A on cell proliferation. As shown in Fig. 6B, top panel, overexpression of WT-YBX1 promoted cell growth compared with HT29 control cells, whereas overexpression of S176A showed a significantly reduced cell proliferation ability compared with WT-YBX1 overexpression cells, suggesting that phosphorylation of Ser-176 of YBX1 is important for its cell proliferation ability.
FIGURE 6.

Phosphorylation of Ser-176 of YBX1 plays a crucial role in promoting cell proliferation and anchorage-independent growth in human colon cancer cells. A, top panel, Western blot showing that overexpression of WT-YBX1 or the S176A mutant was at similar levels in HT29 cells. Bottom panel, luciferase assay showing that WT-YBX1 activated NF-κB, whereas the S176A mutation impaired the ability of YBX1 to activate NF-κB. *, p < 0.05 versus the Ctrl group; #, p < 0.05 versus the WT-YBX1 group. B, top panel, cell growth curve showing that overexpression of WT-YBX1 promoted cell growth compared with the Ctrl group, whereas using a pool of shRNA to knockdown YBX1 greatly slowed down cell growth. Moreover, the S176A mutation led to decreased cell growth compared with WT-YBX1 cells. *, p < 0.05 versus the Ctrl group; #, p < 0.05 versus the WT-YBX1 group. Center and bottom panels, anchorage-independent growth (soft agar) assay showing WT-YBX1 cells exhibited increased colony size compared with Ctrl cells. S176A mutant cells showed decreased colony size compared with WT-YBX1 cells. Furthermore, YBX1 shRNA knockdown led to decreased colony size compared with the Ctrl group. *, p < 0.05 versus the Ctrl group; #, p < 0.05 versus the WT-YBX1 group. C and D, first panels, Western blots showing that WT-YBX1 and S176A were expressed at similar levels as the parental HT29 cells (C) or HCT116 cells (D) in put-back cells (into their shYBX1 (3′-UTR) cells, respectively). Second panels, cell growth curves showing that S176A put-back cells displayed a similar cell growth rate as shYBX1 (3′-UTR) cells, whereas WT-YBX1 put-back cells completely rescued cell growth rate and showed similar cell growth rates as their parental HT29 cells (C) or HCT116 cells (D). *, p < 0.05 versus the parental cell group. Third and fourth panels, S176A put-back cells and shYBX1 (3′-UTR cells) had significantly reduced colony formation abilities compared with either parental HT29 cells (C) or HCT116 cells (D) or their WT-YBX1 put-back cells. * p < 0.05 versus the parental cell group.
To test the effect of Ser-176 on anchorage-independent growth, a soft agar assay was performed with the cells described above. As shown in Fig. 6B, center panel, overexpression of WT-YBX1 enhanced colony sizes compared with HT29 control cells, whereas overexpression of S176A showed a much reduced colony-forming ability compared with WT-YBX1. Together, these data suggest that Ser-176 phosphorylation of YBX1 is very important for promoting anchorage-independent growth.
To achieve a cleaner effect of the S176A mutant, we used the shYBX1 (3′-UTR) construct to knock down the endogenous YBX1 in either HT29 or HCT116 colon cancer cells and then reconstituted these cells with similar levels of WT-YBX1 or S176A protein (Fig. 6, C and D, top panels). Cell proliferation and anchorage-independent growth assays were carried out. The data suggested that stable cell lines of shYBX1 (3′-UTR) infected with S176A virus, hereafter referred to as S176A put-back cells, behaved more like the shYBX1 (3′-UTR) cells, showing a slower cell growth rate and weaker colony formation ability in both HT29- and HCT116-derived cells (Fig. 6, C and D). In contrast, stable cell lines of shYBX1 (3′-UTR) infected with WT-YBX1 virus, hereafter referred to as WT-YBX1 put-back cells, showed similar properties as the parental HT29 (Fig. 6C) or HCT116 cells (Fig. 6D). Together, the above data confirm that Ser-176 is an important site to promote colon cancer progression.
Activation of NF-κB by YBX1 Ser-176 Phosphorylation Works Independently of IκBα Degradation
As IκBα degradation is an important step in the activation of NF-κB by cytokines such as IL-1β, we wondered whether the decreased NF-κB-activating ability by S176A is due to its effect on the process of IκBα degradation. Both 293- or HT29-derived cells with WT-YBX1 or S176A overexpression were treated with IL-1β for different times (Fig. 7, A and B). The degradation of IκBα was further determined by Western blotting analysis. The data indicated no significant difference in IκBα degradation among different cell lines, confirming that phosphorylation of Ser-176 had no obvious effect on IκBα degradation. We then tested whether phosphorylation of Ser-176 of YBX1 affected NF-κB DNA binding ability. A κB-specific EMSA was carried out in 293 and HT29 cells with the overexpression of WT-YBX1 or S176A mutant (Fig. 7C). Previously, we and others have verified that prototypical NF-κB (p65/p50 heterodimer) DNA binding ability could be dramatically induced after treatment with IL-1β (1, 12, 13). We now show that overexpression of WT-YBX1 further enhanced IL-1β-induced κB binding activity, whereas S176A mutation had a much lower NF-κB DNA binding ability compared with the WT-YBX1 overexpression cells. These data suggest that Ser-176 phosphorylation is necessary for YBX1 to modulate NF-κB DNA binding activity.
FIGURE 7.
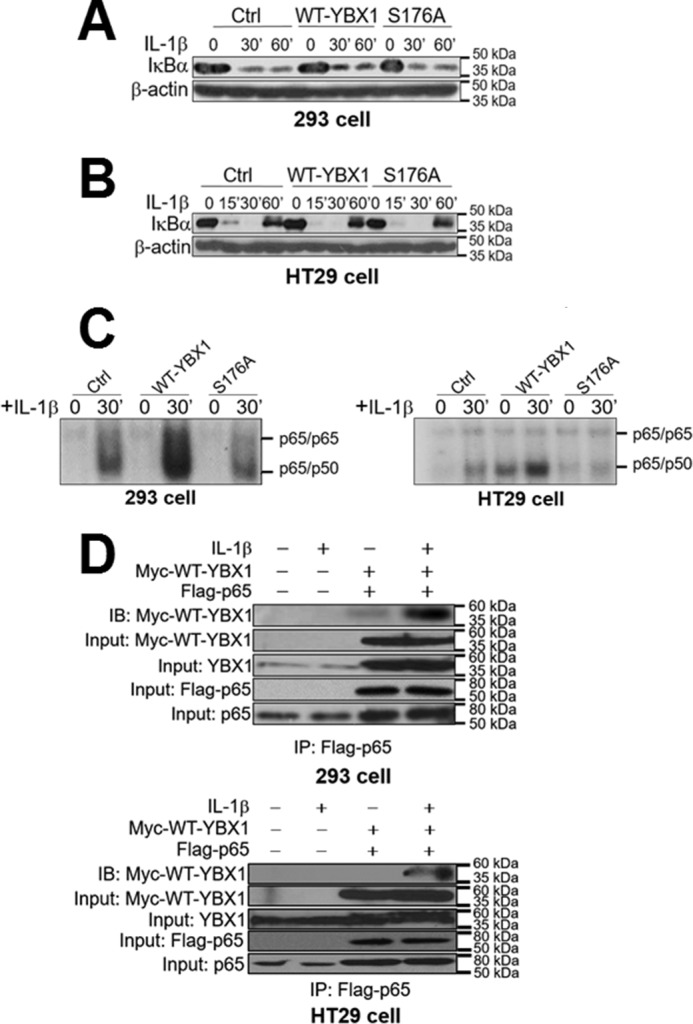
Activation of NF-κB by YBX1 Ser-176 phosphorylation works independently of IκBα degradation. A and B, Western blots in 293 (A) and HT29 cells (B) showing that there is no significant difference for the degradation pattern of IκBα after IL-1β treatment among Ctrl, WT-YBX1, and S176A mutant cells. C, EMSA assay showing that overexpression of WT-YBX1 enhanced NF-κB (mainly p65/p50 heterodimer) DNA binding ability compared with Ctrl in both 293 (left panel) and HT29 cells (right panel), whereas the S176A mutant showed decreased NF-κB DNA binding ability compared with WT-YBX1. IL-1β-induced (30-min treatment) NF-κB binding in Ctrl cells served as the positive control, which has been published previously (1, 12, 13, 36, 37). D, top panel, co-immunoprecipitation (IP) experiments in 293 cells with or without co-expression of Myc-tagged WT-YBX1 and FLAG-tagged p65. These cells were treated with IL-1β for 1 h or left untreated. FLAG-p65 was then pulled down with anti-FLAG beads. Samples were then subjected to Western blotting analysis (immunoblotting, IB) and probed with anti-Myc antibody to detect the co-immunoprecipitation of Myc-WT-YBX1. The data indicated that IL-1β treatment enhanced the interaction between Myc-WT-YBX1 and FLAG-p65 when both Myc-WT-YBX1 and FLAG-p65 were co-expressed. For inputs, anti-Myc antibody was used to detect Myc-WT-YBX1, anti-YBX1 antibody was used to show the input of total YBX1, anti-FLAG antibody was used to detect the input of FLAG-p65, and anti-p65 antibody was used to show the input of total p65. Bottom panel, similar experiments were done in HT29 cells and showed similar results. The conditions and denotations are same as in the top panel.
We also wondered whether IL-1β induces the binding of YBX1 to NF-κB. To examine this possibility, 293 cells or HT29 cells co-expressed with FLAG-p65 (the subunit of NF-κB) (1) and Myc-WT-YBX1 (Fig. 7D) were established and used as the experimental models. Upon treatment with IL-1β for 1 h, we observed increased binding of Myc-WT-YBX1 to FLAG-p65, suggesting that IL-1β may induce the binding of YBX1 to NF-κB, leading to YBX1-enhanced NF-κB activation.
Localization of YBX1 Is Dependent on Ser-176 Phosphorylation
YBX1 nuclear shuttling has been associated with increased tumor promoter activity. We therefore examined whether phosphorylation at Ser-176 is important for nuclear translocation of YBX1. We carried out an immunofluorescence (IF) experiment with a specific monoclonal antibody for FLAG-tagged YBX1 in our FLAG-WT-YBX1 and FLAG-S176A overexpression 293 or HT29 cells. As shown in Fig. 8, upon treatment with IL-1β, FLAG-WT-YBX1 was localized to both the cytoplasm and nucleus, whereas the FLAG-S176A mutant exhibited a different pattern by localizing only to the cytoplasm (Fig. 8). These data suggest that Ser-176 phosphorylation is necessary for nuclear translocation of YBX1.
FIGURE 8.
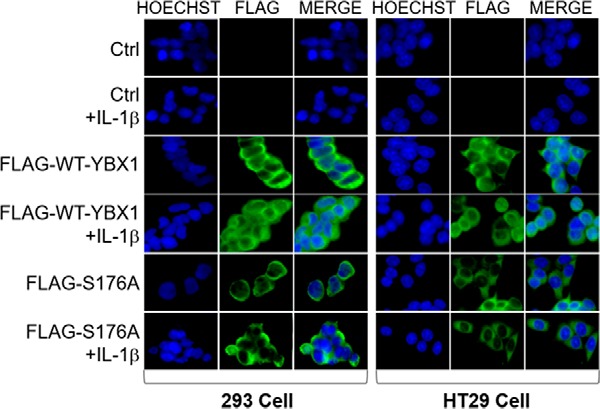
Localization of YBX1 is dependent on Ser-176 phosphorylation. Immunofluorescence experiment showing localization of nuclei stained with Hoechst (blue) and FLAG-tagged YBX1 (green) with an anti-FLAG monoclonal antibody. FLAG-WT-YBX1 exhibited localization to both the cytoplasm and nucleus, with more being localized in the nucleus upon IL-1β treatment in both 293 (left panel) and HT29 cells (right panel) containing FLAG-WT-YBX1 overexpression. On the other hand, the FLAG-S176A mutant exhibited localization mainly to the cytoplasm but not the nucleus, even after treatment with IL-1β. Images were taken at 63× with a fluorescence microscope.
Ser-176 Phosphorylation Is Critical for Cytokine Secretion in Colon Cancer Cells
Activation of NF-κB is associated with the release of many different cytokines from cells. Therefore, we decided to examine the effects of the S176A mutant on cytokine secretion profiles. Conditioned medium was collected from HT29 cells with either WT-YBX1 or S176A mutant overexpression. A cytokine ELISA array was further carried out in triplicate to determine the secretion of different cytokines and growth factors. As shown in Fig. 9, most factors showed reduced secretion in the S176A mutant line compared with the WT-YBX1 sample, whereas there was an overall increase in cytokine expression in the WT-YBX1 line compared with the control. Specifically, secretion of cytokines such as IL-2, VEGF, TNFα, and PDGF was reduced in the S176A mutant compared with the WT-YBX1 sample. Importantly, these cytokines and growth factors are known to be involved in cell proliferation and growth pathways. Overall, our data suggest that Ser-176 phosphorylation is critical for certain cytokines to be secreted into the colon cancer microenvironment.
FIGURE 9.
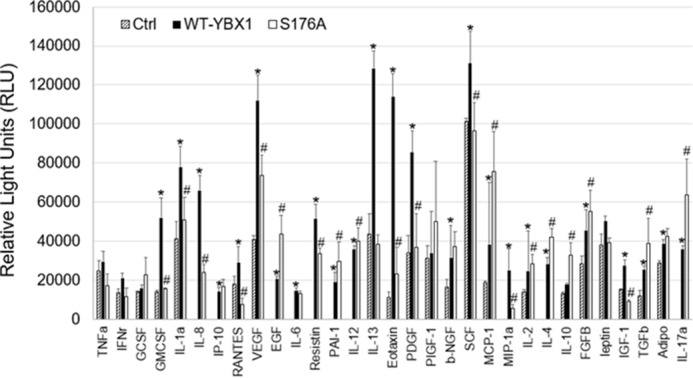
Ser-176 phosphorylation is critical for cytokine secretion. Cytokine array experiment showing that conditioned medium from WT-YBX1 overexpression cells displayed higher secretion of cytokines and growth factors compared with HT29 control cells, whereas medium from S176A-overexpressing cells displayed much lower amounts of secretion of most cytokines than WT-YBX1 cells. Experiments were conducted in triplicate. *, p < 0.05 versus Ctrl; #, p < 0.05 versus WT-YBX1.
Hypothetical Model of Differential Phosphorylation of YBX1 on Ser-176 and Ser-165 and Their Roles in NF-κB Activation
By using the Human Protein Reference Database, we suggest casein kinase I (CKI) as a potential kinase that would phosphorylate Ser-176 on YBX1 (Fig. 10A). To determine the role of CKI in YBX1 phosphorylation, we used a pool of shCKI constructs to knock down the expression of CKI in FLAG-WT-YBX1-overexpressing 293 cells (Fig. 10B, top panel). These cells were further treated with IL-1β, followed by co-immunoprecipitation experiment using anti-FLAG beads. Phosphorylation of YBX1 was further detected by Western blotting analysis using an anti-phospho-serine antibody. The data (Fig. 10B, bottom panel) indicated that, although IL-1β could induce the phosphorylation of YBX1 (this is consistent with what we observed in Fig. 4B), knocking down CKI largely abolished this phosphorylation effect. As we suggested before (Fig. 4B), Ser-176 is the predominant phosphorylation site on YBX1. Thus, based on the data we have presented, we propose that, in the presence of an activating cytokine such as IL-1β, the IκB kinase phosphorylates IκBα, which leads to the degradation of IκBα. While this is occurring, IL-1β can activate CKI, leading to the phosphorylation of YBX1 on Ser-176. Meanwhile, CKII is activated in parallel, which can promote phosphorylation of YBX1 on Ser-165 (1). These two different phosphorylation events on YBX1 can activate NF-κB, leading to the regulation of either some common pool or different subgroups of NF-κB-inducible genes, resulting in finely tuned NF-κB driven functions (Fig. 10C).
FIGURE 10.

Hypothetical model of differential phosphorylation of YBX1 on Ser-176 and Ser-165 and their role in NF-κB activation. A, prediction of the phosphorylation site by CKI on YBX1 at Ser-176 using the Human Protein Reference Database. B, top panel, Western blot showing that CKI protein was successfully knocked down with a pool of shCKI constructs in 293 stable cells containing FLAG-WT-YBX1 (cell line from Fig. 4A). Bottom panel, co-immunoprecipitation (IP) experiment and Western blot showing that, using the above cells, FLAG-tagged WT-YBX1 was pulled down with anti-FLAG beads and then probed with anti-phospho-serine antibody. shCKI cells showed dramatically reduced serine phosphorylation on FLAG-WT-YBX1 upon IL-1β treatment compared with control cells. IB, immunoblot. C, hypothetical model showing that, in the presence of stimuli of the NF-κB pathway such as IL-1β, IκB kinase phosphorylates IκBα, causing degradation of IκB. The free p65/p50 heterodimer then migrates to the nucleus to bind to κB-binding sites on the promoters of specific genes, leading to their activation. While this is occurring, IL-1β can also stimulate YBX1 by activating CKI, which then leads to phosphorylation of YBX1 on Ser-176, and by activating CKII, which can promote phosphorylation of YBX1 on Ser-165 (1). These two different phosphorylation events either regulate some common pool of NF-κB-inducible genes or independently regulates quite distinct subgroups of NF-κB-inducible genes, leading to plasticity of NF-κB-driven biological functions.
Discussion
PTM is one of the key approaches that mammalian cells use to control gene expression and cellular functions in a sophisticated manner (21, 22). One well known example is the PTMs of NF-κB, in which over a dozen different phosphorylation sites were identified either in response to different stimuli or in different cell systems (23–25).
Compared with the study of NF-κB, the understanding of PTMs of the important oncoprotein YBX1 is far less developed. In this study, we provide strong evidence regarding the discovery of the novel phosphorylation of Ser-176 on YBX1. We show that the S176A mutant exhibits decreased activation of NF-κB, reduced secretion of cytokines such as VEGF, IL-12, and TNFα, decreased cell proliferation ability, and compromised anchorage-independent growth capacity. Furthermore, S176A mutant protein is mainly restricted to the cytoplasm, whereas WT-YBX1 is located in both the cytoplasm and nucleus, indicating the importance of Ser-176 phosphorylation for YBX1 translocation to the nucleus. Importantly, the gene regulation levels show that phosphorylation of Ser-176 is critical for the expression of a number of NF-κB-inducible genes. Both Ser-176 and Ser-165 phosphorylation differentially or collaboratively regulate different subgroups of genes that are associated with different networks of functions (Fig. 5, B and D). Collectively, the evidence provides deep insights into how PTMs of YBX1 regulate different cellular functions.
Not surprisingly, the two serine/threonine protein kinases that we proposed in this study, either CKI or II, have been linked to carcinogenesis and its related processes (26–28). It was reported that the highly conserved and ubiquitously expressed pleiotropic CKI family plays critical regulatory roles in many cellular processes, including DNA processing and repair, proliferation, and cell differentiation. Furthermore, CKI is tightly involved in the regulation and degradation of p53, mouse double minute, and β-catenin. In addition, CKI also can phosphorylate and activate Wnt signaling (29). It is important to note that Wnt is known to play an important role in the epithelial-mesenchymal transition (EMT), an important process that contributes to cancer development (30). Interestingly, YBX1 has also been implicated in interacting with promoters of a number of Wnt pathway proteins (31). Mutations and genetic alterations of CKI are often detected in various cancers. Therefore, CKI is considered a highly attractive therapeutic target in cancer (32). On the other hand, although belonging to a different family, CKII is also involved in various important cellular processes, including cell cycle control, apoptosis, and circadian rhythm. It is well known that CKII plays an important role in tumorigenesis and angiogenesis processes (33, 34). In this study, we suggest that Ser-176 is a more predominant phosphorylation site on YBX1 than Ser-165. Therefore, CKI potentially plays a more significant role than CKII in the systems we studied.
As proposed in our model, both Ser-176 and Ser-165 (1) are critical for the nuclear translocation of YBX1 and its ability to enhance NF-κB binding to DNA. However, details are still unknown regarding the exact order of these phosphorylation events. In the future, it would be important to further study the role of CKI in Ser-176 and CKII in Ser-165 phosphorylation. This endeavor may provide a better understanding of the intricacies of communication between the YBX1 and NF-κB signaling pathways that are linked through Ser-176 and Ser-165 phosphorylation on YBX1. Additionally, how exactly YBX1 may enhance NF-κB binding to DNA is still unknown. Based on our EMSA data (Fig. 7C), we speculate that at least the phosphorylation of Ser-176 and Ser-165 (1) is critical to this process. Furthermore, the IF data (Fig. 8) also affirmed that lack of phosphorylation of Ser-165 (1) and Ser-176 could lead to the retention of most YBX1 into the cytoplasm and prevent it from translocating into the nucleus. In the future, studies such as chromatin immunoprecipitation experiments and in silico crystal structure prediction of the binding between YBX1 and NF-κB may help us to better understand this important aspect of YBX1-mediated NF-κB activation.
Furthermore, the discovery of Ser-176 phosphorylation of YBX1 and its link to NF-κB activation is significant. Although both NF-κB and YBX1 are known to play critical roles in cancer progression, the interaction between YBX1 and NF-κB is still understudied. Interestingly, NF-κB is one of the very few critical nodes in the three networks of functions we proposed (Fig. 5, B–D). More importantly, YBX1 also serves as a critical node in two of these three networks, suggesting that the crosstalk between YBX1 and NF-κB is critical and complicated. Phosphorylation sites such as Ser-176 and Ser-165 of YBX1 and their different regulatory abilities on NF-κB target genes are therefore of particular importance in terms of increasing our knowledge of the connection between the YBX1 and NF-κB signaling pathways.
Notably, YBX1 is an excellent example of how a novel NF-κB modulator may regulate NF-κB and its downstream target gene expression. The concept of protein modulators of RelA, i.e. the p65 subunit of NF-κB, has been developed previously by Li et al. (35) with computational approaches (35). In their report, the authors provided a repertoire of near 600 modulators for experimental validation. They further hypothesized that these RelA modulators might influence the expression of “certain groups” of NF-κB-dependent genes. Remarkably, one of their predicted novel modulators was YBX1. Although a complete mystery at that time, this study has inarguably validated the role of YBX1 as a novel NF-κB modulator and unveiled the underlying mechanism of how YBX1 may regulate NF-κB and lead to the activation of certain groups of its target genes. Therefore, this study is of great novelty and significance.
In a broader scope, a fascinating aspect regarding the novel phosphorylation of Ser-176 and Ser-165 of YBX1 could be its potential influence on the overall YBX1 functions. For instance, YBX1 is well known for playing an important role in translation control. Evdokimova et al. (36) demonstrated that YBX-1 regulated EMT by inducing cap-independent translation of EMT-promoting factors and suppressing cap-dependent translation of growth-promoting factors, confirming YBX-1 as a restriction point of EMT. Interestingly, NF-κB is famous for being a key player in EMT gene regulation (37). In the future, it would be of immense interest to explore whether phosphorylation of Ser-176 and Ser-165 on YBX1 may play any role in translational control of EMT genes as well as other important functions of YBX1.
Finally, this research may provide a potential therapeutic approach in colon cancer treatment. Targeting YBX1 phosphorylation may lead to the inhibition of NF-κB activity in cancer, providing a more specific therapeutic approach than targeting NF-κB itself in general. In the future, it would be very interesting to explore whether phosphorylation of Ser-176 and Ser-165 of YBX1 could be generalized to some other types of cancer beyond colon cancer, therefore providing novel therapeutic approaches to other types of cancer as well.
Experimental Procedures
Cell Lines and Antibodies
The 293 cell line has been described previously (38). The HT29 and HCT116 colon cancer cell lines were purchased from the ATCC (Manassas, VA) and cultivated in RPMI 1640 medium with 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% fetal calf serum. The following antibodies were obtained from commercial sources: anti-YBX1 (Abcam, Cambridge, MA), anti-IκBα and anti-CKI (Santa Cruz Biotechnology Inc., Dallas, TX), anti-β-actin (Sigma-Aldrich, St. Louis, MO), and anti-phosphorylated serine and anti-Myc (Cell Signaling Technology, Danvers, MA).
Construction of Stable Cell lines
FLAG-tagged WT-YBX1 cDNA was amplified by reverse transcription from total mRNA derived from 293 cells. The sequence was confirmed and then cloned into the pLVX-IRES-puro vector (1). Mutations of the Ser-176 residue to Ala (S176A) or S176A/S165A were generated using the QuikChange II XL site-directed mutagenesis kit following the protocol of the manufacturer (Agilent Technologies, Inc., Santa Clara, CA). The mutated site was then confirmed by sequencing. The shRNA pool against YBX1 was purchased from Sigma-Aldrich. All cell lines were generated using either 293, HT29, or HCT116 colon cancer cell lines. To generate stable cell lines, the lentiviral plasmid containing the DNA of interest or shRNAs targeting either exon or 3′-UTR (for put-back expression) of YBX1 or CKI were transfected into a 293T packaging cell line to produce viruses. 293, HT29, or HCT116 cells were then infected with these viruses and further selected with 1 μg/ml of puromycin, as the lentiviral vector construct is comprised of a puromycin resistance gene. Expression of the respective constructs was confirmed using Western blotting with specific antibodies. For stable YBX1 put-back cells, stable shYBX1–3′-UTR cells were first established. The same numbers of these cells under the same conditions were then infected with the same titers of viruses for either WT-YBX1 or the S176A mutant. Cells were then selected under either 0.5 or 1 μg/ml puromycin. Two stable pools per cell line were collected. Samples were then examined by Western blotting analysis, and cells with comparable YBX1 expression (parent, WT-YBX1, and S176A mutant cells) were used for both cell growth and soft agar experiments.
Transfections and Luciferase Assays
Constructs were transfected into cell lines using the Lipofectamine and PLUS reagents (Life Technologies/Invitrogen). For NF-κB luciferase assays, the κB-luciferase construct p5XIP10 κB (39) was transfected transiently into the cells, and luciferase activity was determined 48 h later. A β-galactosidase construct was co-transfected to normalize for transfection efficiency. Transfections and luciferase assays were carried out as described previously (39).
Western Analyses
Cells were cultured to ∼95% confluence, and samples were collected and assayed by Western blotting as described previously (39). Different antibodies were used based on different experiments as described in the text.
Co-immunoprecipitations
Cells cultured in 10-cm plates to 95% confluency were lysed in co-immunoprecipitation buffer (1% Triton X-100 (v/v), 50 mm Tris·HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, 1 mm sodium orthovanadate, 20 μm aprotinin, 1 mm phenylmethanesulfonyl fluoride, and 1 mm pepstatin A). Prewashed anti-FLAG-M2 antibody, EZView beads were (Sigma) mixed with cell lysates with equivalent amounts of protein at 4 °C overnight. Gel beads were washed four times with co-immunoprecipitation buffer with rotation at 4 °C for 5 min each time. For the last step, FLAG peptide was added to elute the FLAG-tagged protein. Samples were added to SDS sample loading buffer (6% (v/v) glycerol, 1% (v/v) β-mercaptoethanol, 2% (w/v) SDS, 50 mm Tris·HCl (pH 6.7), 0.004% (w/v) bromphenol blue), and we further proceeded with Western blotting analysis as described above.
EMSA
The oligomer used for NF-κB binding site was 5′AGTTGAGGGGACTTTCCCAGGC-3′ (Santa Cruz Biotechnology, Inc.). It was labeled with [γ-32P]ATP by the polynucleotide kinase method, following the protocol provided by Promega Corp. (Madison, WI). Whole cell lysates were prepared and analyzed as described previously (1).
Cell Growth and Soft Agar Assays
HT29 cells overexpressing WT-YBX1, S176A, and shRNA-YBX1 knockdown cell lines were plated at 2 × 104 cells/well in a 6-well plate with 3 ml of RPMI 1640 medium. Cells were seeded in triplicate and counted on different days using a cell counting chamber. For soft agar assays, type VII agarose (Sigma) was autoclaved and mixed with RPMI 1640 cell growth medium. Cell culture dishes were coated with 1.2% type VII agarose as the bottom layer. Cells were resuspended in 0.6% of type VII agarose and plated on top of the bottom layer. Cells were cultured for 2–3 weeks before being checked under a microscope, measured, and quantified with the aid of ImageJ software (http://imagej.nih.gov/ij/).
Preparation of Samples for MS Experiments
Ten 15-cm plates of 293 or HT29 cells with the stably expressed FLAG-tagged WT-YBX1 protein were cultured to 80% confluence. Five were used as controls, and the other five plates were treated with IL-1β for 1 h. Cells were then lysed with co-immunoprecipitation buffer (see “Co-immunoprecipitations”). After spinning the debris for 10 min at 4 °C, the supernatant solution was incubated with EZview Red anti-FLAG M2 affinity gel overnight at 4 °C. Gel beads were washed with 20 volumes of co-immunoprecipitation buffer with rotation at 4 °C for 5 min each time. Protein was eluted with FLAG peptide (Sigma) following the standard protocol of the manufacturer. The supernatant solution was mixed with 5× SDS sample loading buffer, boiled for 5 min, and separated in a 10% Tris-HCl SDS/PAGE gel (40). The gel was then treated with fixing buffer (50% ethanol and 10% acetic acid) for 20 min, and washed with distilled water for 1 h before being stained with GelCode Blue stain (Pierce) overnight. The gel was destained with distilled water for 2 h before analysis by MS.
Protein In-gel Digestion
Pieces cut from SDS/PAGE gels were excised and subjected to in-gel tryptic digestion. The excised bands were washed twice with 100 mm ammonium bicarbonate containing 50% acetonitrile for 1 h and twice with acetonitrile for 10 min. The proteins in the gels were then treated with 20 mm DTT at room temperature for 30 min, followed by 50 mm iodoacetamide for 30 min in 100 mm ammonium bicarbonate. After the treatment, the reagents were removed, and the gel pieces were washed with 100 mm ammonium bicarbonate and then dehydrated in acetonitrile. The dried gel pieces were then reswollen in 50 mm ammonium bicarbonate containing sequencing-grade modified trypsin for overnight digestion. Tryptic peptides were extracted from the gel with 50% acetonitrile in 5% formic acid.
MS Analysis
The proteolytic digests were analyzed using an LTQ Orbitrap XL linear ion trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA) coupled with an Ultimate 3000 HPLC system (Dionex, Sunnyvale, CA). The reverse-phase C18 column (0.075 × 150 mm, Dionex) was equilibrated with 0.1% formic acid/4% acetonitrile (v/v), and the proteolytic digests were injected on it. A linear gradient of acetonitrile from 4% to 40% in water in the presence of 0.1% formic acid over a period of 45 min was used at a flow rate of 300 nl/min. The spectra were acquired by data-dependent methods consisting of a full scan (m/z 400–2000) and then tandem MS on the five most abundant precursor ions. The previously selected precursor ions were scanned once for 30 s and then excluded for 30 s. The obtained data were submitted to Mascot software (Matrix Science, Inc., Boston, MA) to search for phosphorylated residues on the YBX1 protein. The tandem mass spectra of the possibly modified peptides were further interpreted manually.
Illumina Microarrays
RNA (250 ng) was reverse-transcribed into cDNA and labeled with biotin-UTP using the Illumina TotalPrep RNA amplification kit (Ambion/Applied Biosystems, Foster City, CA). The amount of cDNA was determined using a NanoDrop spectrophotometer, and the cDNA quality (size distribution) was further analyzed in a 1% (w/v) agarose gel. cDNA was hybridized to Illumina Human Ref-v3 v1 Expression BeadChips and scanned in a BeadArray reader using standard protocols (Illumina, San Diego, CA). Illumina's BeadStudio software was used for data analysis.
IF Experiment
Coverslips were coated with 0.1% sterile gelatin for 2 h and dried for 30 min at room temperature. 1 × 105 cells/well were then seeded onto coverslips in a 24-well plate and left overnight. Cells were then treated with or without IL-1β for 1 h to continue with IF experiments. Cells were fixed with 4% formaldehyde for 30 min and then blocked with blocking buffer for 10 min at room temperature. Coverslips were further probed with anti-FLAG antibody for FLAG-tagged WT-YBX1 or S176A and Alexa Fluor 488 (green) goat anti-mouse IgG. Before sealing the coverslips, mounting medium with Hoechst was used to stain the nucleus. The slides were examined under a Leica DMI6000B series fluorescent microscope with ×63 magnification.
Conditioned Medium Collection and Human Cytokine ELISA Arrays
Human cytokine ELISA arrays were purchased from Signosis, Inc. (Santa Clara, CA). Experiments were carried out according to the protocol of the manufacturer. HT29 control, WT-YBX1, and S176A overexpression stable cell lines were seeded and allowed to grow to 90% confluence and then cultured for 3 days. This medium was then collected by centrifugation and added to specific cytokine capture antibody-precoated wells for 2 h at room temperature. After incubation, the wells were washed to remove unbound labeled antibodies. The plate was further detected with HRP luminescent substrate. The level of expression for each specific cytokine was directly proportional to the luminescence that was emitted.
qPCR Analyses
RNA samples were purified with TRIzol (Thermo Fisher Scientific) reagent as described previously (41). cDNA was made by reverse PCR from total RNA by using the SuperScript III first-strand synthesis system (Thermo Fisher Scientific). FastStart Universal SYBR Green Master ROX (Roche Diagnostics) was used for the qPCR reactions. Primers were designed by Primer Express 3.0 software.
IPA
Three groups of genes, i.e. S176A or S165A solely regulated or commonly regulated genes, were analyzed with IPA analysis. The setting and filter were as follows: reference set: Ingenuity Knowledge Base (Genes + Endogenous Chemicals); Relationship to include: Direct and Indirect; Includes Endogenous Chemicals; Filter Summary: Consider only molecules where species = Human OR Rat OR Mouse. The p values for the enrichment test were calculated using Fisher's exact test, right-tailed. Usually the −log10(p) is visualized to the left of the p value. p < 0.05 was considered significant.
Statistical Analysis
Statistical analysis was performed using Prism 6 software (GraphPad, San Diego, CA). The data represent the mean ± S.D. from three independent experiments. A two-tailed Student's t test was used when comparing two means and to test for significant differences between relative luciferase activity and relative gene expression in different groups. All statistics were calculated on triplicate experiments. For all statistics, p < 0.05 was considered statistically significant.
Author Contributions
M. M., L. H., B. W., and H. W. carried out the major experimental work and data analysis. L. P., G. J., and A. V. H. contributed to part of the experimental work or data analysis. T. L. and M. M. contributed to the experimental design. M. M. and T. L. wrote the manuscript. Y. L. provided valuable feedback.
Supplementary Material
Acknowledgments
We thank Lisa King (Department of Pharmacology and Toxicology, Indiana University School of Medicine) for help with editing this manuscript.
This work was supported by Grants 4186265 (to T. L.) from the American Cancer Society, 068058-00002B (to T. L.) from the V Foundation Kay Yow Cancer Fund, and 4486233 (to T. L.) from the Showalter Trust Fund, as well as Biomedical Research Grant 2286229 (to T. L.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1–S3 and Tables S1–S6.
- IGF
- insulin-like growth factor
- PTM
- posttranslational modification
- qPCR
- quantitative PCR
- IPA
- ingenuity pathway analysis
- IF
- immunofluorescence
- CKI
- casein kinase I
- EMT
- epithelial-mesenchymal transition
- Ctrl
- control.
References
- 1. Prabhu L., Mundade R., Wang B., Wei H., Hartley A. V., Martin M., McElyea K., Temm C. J., Sandusky G., Liu Y., and Lu T. (2015) Critical role of phosphorylation of serine 165 of YBX1 on the activation of NF-κB in colon cancer. Oncotarget 6, 29396–29412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Eliseeva I. A., Kim E. R., Guryanov S. G., Ovchinnikov L. P., and Lyabin D. N. (2011) Y-box-binding protein 1 (YB-1) and its functions. Biochemistry 76, 1402–1433 [DOI] [PubMed] [Google Scholar]
- 3. Kohno K., Izumi H., Uchiumi T., Ashizuka M., and Kuwano M. (2003) The pleiotropic functions of the Y-box-binding protein, YB-1. BioEssays 25, 691–698 [DOI] [PubMed] [Google Scholar]
- 4. Kuwano M., Oda Y., Izumi H., Yang S. J., Uchiumi T., Iwamoto Y., Toi M., Fujii T., Yamana H., Kinoshita H., Kamura T., Tsuneyoshi M., Yasumoto K., and Kohno K. (2004) The role of nuclear Y-box binding protein 1 as a global marker in drug resistance. Mol. Cancer Ther. 3, 1485–1492 [PubMed] [Google Scholar]
- 5. Shibao K., Takano H., Nakayama Y., Okazaki K., Nagata N., Izumi H., Uchiumi T., Kuwano M., Kohno K., and Itoh H. (1999) Enhanced coexpression of YB-1 and DNA topoisomerase II α genes in human colorectal carcinomas. Int. J. Cancer 83, 732–737 [DOI] [PubMed] [Google Scholar]
- 6. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 7. Lasham A., Print C. G., Woolley A. G., Dunn S. E., and Braithwaite A. W. (2013) YB-1: oncoprotein, prognostic marker and therapeutic target? Biochem. J. 449, 11–23 [DOI] [PubMed] [Google Scholar]
- 8. Evdokimova V., Ruzanov P., Anglesio M. S., Sorokin A. V., Ovchinnikov L. P., Buckley J., Triche T. J., Sonenberg N., and Sorensen P. H. (2006) Akt-mediated YB-1 phosphorylation activates translation of silent mRNA species. Mol. Cell Biol. 26, 277–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sutherland B. W., Kucab J., Wu J., Lee C., Cheang M. C., Yorida E., Turbin D., Dedhar S., Nelson C., Pollak M., Leighton Grimes H., Miller K., Badve S., Huntsman D., Blake-Gilks C., et al. (2005) Akt phosphorylates the Y-box binding protein 1 at Ser102 located in the cold shock domain and affects the anchorage-independent growth of breast cancer cells. Oncogene 24, 4281–4292 [DOI] [PubMed] [Google Scholar]
- 10. Wu J., Lee C., Yokom D., Jiang H., Cheang M. C., Yorida E., Turbin D., Berquin I. M., Mertens P. R., Iftner T., Gilks C. B., and Dunn S. E. (2006) Disruption of the Y-box binding protein-1 results in suppression of the epidermal growth factor receptor and HER-2. Cancer Res. 66, 4872–4879 [DOI] [PubMed] [Google Scholar]
- 11. Basaki Y., Hosoi F., Oda Y., Fotovati A., Maruyama Y., Oie S., Ono M., Izumi H., Kohno K., Sakai K., Shimoyama T., Nishio K., and Kuwano M. (2007) Akt-dependent nuclear localization of Y-box-binding protein 1 in acquisition of malignant characteristics by human ovarian cancer cells. Oncogene 26, 2736–2746 [DOI] [PubMed] [Google Scholar]
- 12. Oeckinghaus A., and Ghosh S. (2009) The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 1, a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu T., and Stark G. R. (2004) Cytokine overexpression and constitutive NF-κB in cancer. Cell Cycle 3, 1114–1117 [PubMed] [Google Scholar]
- 14. Prasad S., Ravindran J., and Aggarwal B. B. (2010) NF-κB and cancer: how intimate is this relationship. Mol. Cell Biochem. 336, 25–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baeuerle P. A., and Baltimore D. (1988) Activation of DNA-binding activity in an apparently cytoplasmic precursor of the NF-κB transcription factor. Cell 53, 211–217 [DOI] [PubMed] [Google Scholar]
- 16. Hayden M. S., West A. P., and Ghosh S. (2006) NF-κB and the immune response. Oncogene 25, 6758–6780 [DOI] [PubMed] [Google Scholar]
- 17. Zheng Y., McFarland B. C., Drygin D., Yu H., Bellis S. L., Kim H., Bredel M., and Benveniste E. N. (2013) Targeting protein kinase CK2 suppresses pro-survival signaling pathways and growth of glioblastoma. Clin. Cancer Res. 19, 6484–6494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garaud S., and Willard-Gallo K. (2015) IRF5: a rheostat for tumor-infiltrating lymphocyte trafficking in breast cancer? Immunol. Cell Biol. 93, 425–426 [DOI] [PubMed] [Google Scholar]
- 19. Ryan S. L., Schwalbe E. C., Cole M., Lu Y., Lusher M. E., Megahed H., O'Toole K., Nicholson S. L., Bognar L., Garami M., Hauser P., Korshunov A., Pfister S. M., Williamson D., Taylor R. E., et al. (2012) MYC family amplification and clinical risk-factors interact to predict an extremely poor prognosis in childhood medulloblastoma. Acta Neuropathol. 123, 501–513 [DOI] [PubMed] [Google Scholar]
- 20. Caffarel M. M., and Coleman N. (2014) Oncostatin M receptor is a novel therapeutic target in cervical squamous cell carcinoma. J. Pathol. 232, 386–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Prabhu L., Hartley A. V., Martin M., Warsame F., Sun E., and Lu T. (2015) Role of post-translational modification of the Y box binding protein 1 in human cancers. Genes Dis. 2, 240–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Narayan S., Bader G. D., and Reimand J. (2016) Frequent mutations in acetylation and ubiquitination sites suggest novel driver mechanisms of cancer. Genome Med. 8, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu T., and Stark G. R. (2015) NF-κB: regulation by methylation. Cancer Res. 75, 3692–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang B., Wei H., Prabhu L., Zhao W., Martin M., Hartley A. V., and Lu T. (2015) Role of novel serine 316 phosphorylation of the p65 subunit of NF-κB in differential gene regulation. J. Biol. Chem. 290, 20336–20347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perkins N. D. (2006) Post-translational modifications regulating the activity and function of the NF-κB pathway. Oncogene 25, 6717–6730 [DOI] [PubMed] [Google Scholar]
- 26. Hirner H., Günes C., Bischof J., Wolff S., Grothey A., Kühl M., Oswald F., Wegwitz F., Bösl M. R., Trauzold A., Henne-Bruns D., Peifer C., Leithäuser F., Deppert W., and Knippschild U. (2012) Impaired CK1 δ activity attenuates SV40-induced cellular transformation in vitro and mouse mammary carcinogenesis in vivo. PLoS ONE 7, e29709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hagan C. R., Knutson T. P., and Lange C. A. (2013) A common docking domain in progesterone receptor-B links DUSP6 and CK2 signaling to proliferative transcriptional programs in breast cancer cells. Nucleic Acids Res. 41, 8926–8942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bidère N., Ngo V. N., Lee J., Collins C., Zheng L., Wan F., Davis R. E., Lenz G., Anderson D. E., Arnoult D., Vazquez A., Sakai K., Zhang J., Meng Z., Veenstra T. D., et al. (2009) Casein kinase 1α governs antigen-receptor-induced NF-κB activation and human lymphoma cell survival. Nature 458, 92–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bryja V., Schulte G., Rawal N., Grahn A., and Arenas E. (2007) Wnt-5a induces Dishevelled phosphorylation and dopaminergic differentiation via a CK1-dependent mechanism. J. Cell Sci. 120, 586–595 [DOI] [PubMed] [Google Scholar]
- 30. Fodde R., and Brabletz T. (2007) Wnt/β-catenin signaling in cancer stemness and malignant behavior. Curr. Opin. Cell Biol. 19, 150–158 [DOI] [PubMed] [Google Scholar]
- 31. Gopal S. K., Greening D. W., Mathias R. A., Ji H., Rai A., Chen M., Zhu H. J., and Simpson R. J. (2015) YBX1/YB-1 induces partial EMT and tumourigenicity through secretion of angiogenic factors into the extracellular microenvironment. Oncotarget 6, 13718–13730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Knippschild U., Krüger M., Richter J., Xu P., García-Reyes B., Peifer C., Halekotte J., Bakulev V., and Bischof J. (2014) The CK1 family: contribution to cellular stress response and its role in carcinogenesis. Front. Oncol. 4, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Filhol O., Giacosa S., Wallez Y., and Cochet C. (2015) Protein kinase CK2 in breast cancer: the CK2β regulatory subunit takes center stage in epithelial plasticity. Cell Mol. Life Sci. 72, 3305–3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Montenarh M. (2014) Protein kinase CK2 and angiogenesis. Adv. Clin. Exp. Med. 23, 153–158 [DOI] [PubMed] [Google Scholar]
- 35. Li X., Zhao Y., Tian B., Jamaluddin M., Mitra A., Yang J., Rowicka M., Brasier A. R., and Kudlicki A. (2014) Modulation of gene expression regulated by the transcription factor NF-κB/RelA. J. Biol. Chem. 289, 11927–11944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Evdokimova V., Tognon C., Ng T., Ruzanov P., Melnyk N., Fink D., Sorokin A., Ovchinnikov L. P., Davicioni E., Triche T. J., and Sorensen P. H. (2009) Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell 15, 402–415 [DOI] [PubMed] [Google Scholar]
- 37. Min C., Eddy S. F., Sherr D. H., and Sonenshein G. E. (2008) NF-κB and epithelial to mesenchymal transition of cancer. J. Cell Biochem. 104, 733–744 [DOI] [PubMed] [Google Scholar]
- 38. Mattioli I., Geng H., Sebald A., Hodel M., Bucher C., Kracht M., and Schmitz M. L. (2006) Inducible phosphorylation of NF-κB p65 at serine 468 by T cell costimulation is mediated by IKKϵ. J. Biol. Chem. 281, 6175–6183 [DOI] [PubMed] [Google Scholar]
- 39. Wei H., Wang B., Miyagi M., She Y., Gopalan B., Huang D. B., Ghosh G., Stark G. R., and Lu T. (2013) PRMT5 dimethylates R30 of the p65 subunit to activate NF-κB. Proc. Natl. Acad. Sci. U.S.A. 110, 13516–13521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lu T., Jackson M. W., Wang B., Yang M., Chance M. R., Miyagi M., Gudkov A.V., and Stark G. R. (2010) Regulation of NF-κB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc. Natl. Acad. Sci. U.S.A. 107, 46–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lu T., Jackson M. W., Singhi A. D., Kandel E. S., Yang M., Zhang Y., Gudkov A. V., and Stark G. R. (2009) Validation-based insertional mutagenesis identifies lysine demethylase FBXL11 as a negative regulator of NF-κB. Proc. Natl. Acad. Sci. U.S.A. 106, 16339–16344 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.