

Abstract
Degradation of the extracellular matrices in the human body is controlled by matrix metalloproteinases (MMPs), a family of more than 20 homologous enzymes. Imbalance in MMP activity can result in many diseases, such as arthritis, cardiovascular diseases, neurological disorders, fibrosis, and cancers. Thus, MMPs present attractive targets for drug design and have been a focus for inhibitor design for as long as 3 decades. Yet, to date, all MMP inhibitors have failed in clinical trials because of their broad activity against numerous MMP family members and the serious side effects of the proposed treatment. In this study, we integrated a computational method and a yeast surface display technique to obtain highly specific inhibitors of MMP-14 by modifying the natural non-specific broad MMP inhibitor protein N-TIMP2 to interact optimally with MMP-14. We identified an N-TIMP2 mutant, with five mutations in its interface, that has an MMP-14 inhibition constant (Ki) of 0.9 pm, the strongest MMP-14 inhibitor reported so far. Compared with wild-type N-TIMP2, this variant displays ∼900-fold improved affinity toward MMP-14 and up to 16,000-fold greater specificity toward MMP-14 relative to other MMPs. In an in vitro and cell-based model of MMP-dependent breast cancer cellular invasiveness, this N-TIMP2 mutant acted as a functional inhibitor. Thus, our study demonstrates the enormous potential of a combined computational/directed evolution approach to protein engineering. Furthermore, it offers fundamental clues into the molecular basis of MMP regulation by N-TIMP2 and identifies a promising MMP-14 inhibitor as a starting point for the development of protein-based anticancer therapeutics.
Keywords: directed evolution, metastasis, protease inhibitor, protein-protein interaction, proteolysis, binding affinity, computational protein design, matrix metalloproteinases, yeast surface display
Introduction
Matrix metalloproteinases (MMPs)5 are a family of enzymes consisting of more than 20 homologous proteins that play a significant role in the degradation of the extracellular matrix, a crucial step in many cellular processes. MMPs are multidomain proteins that differ in domain architecture and substrate preferences (1), but they all share a catalytic domain with a nearly identical active site containing a Zn2+ ion (2). MMPs are synthesized in an inactive form and are subsequently activated by the cleavage of a pro-domain, often by other MMPs or by different proteases (3, 4), thus producing complex protein/protein interaction networks (5). For example, MMP-2 is activated physiologically by MMP-14; MMP-9 is activated by active MMP-2 and active MMP-7; and MMP-1 is also activated by active MMP-7 (4). Some MMPs, such as MMP-3, are “master regulators,” being able to activate themselves as well as other MMPs (6).
Imbalance in MMP activity leads to various diseases, including cancers (7), cardiovascular diseases (8), and arthritis (9). In cancer-related processes, some MMPs play a crucial role in the promotion of metastasis and other aspects of tumor growth through cleavage and activation of a variety of different proteins (10, 11). In particular, there is elevated expression of a membrane-type MMP, MMP-14 (also known as MT1-MMP), in various human carcinomas, including uterine, cervical, stomach, lung, breast, colon, melanoma, head and neck, and brain tumors (12). Elevated MMP-14 expression is also associated with early death of patients with various cancers (13) and is correlated with cancer progression, invasion, lymph node metastases, a poor clinical stage, larger tumor size, and tumor stage progression (14). In the laboratory, MMP-14 has been shown to play a direct role in the multiplication of tumor cells inside a three-dimensional collagen matrix (15).
Because of the obvious importance of MMPs in cancer, many MMP inhibitors have been designed over the past 30 years. Yet, because of their high toxicity, all failed in clinical trials (16, 17). The major drawback of these inhibitors was that they targeted the conserved active site of MMPs and thus reacted with other Zn2+-containing proteins in the body, resulting in high toxicity (2). Moreover, broad inhibition of all MMP family members is not necessarily desirable, because some MMPs, for example, MMP-8 and MMP-12, exhibit anti-tumorigenic functions (18, 19). In addition, it has been shown in animal cancer models that lack of activity of certain MMPs is correlated with more aggressive cancers (17). There is currently an understanding that the correct approach is to target only one MMP family member so as to prevent the undesired effects caused by inhibition or activation of other MMPs. However, such specific MMP inhibitors are difficult to obtain by starting from small molecules.
Protein-based inhibitors, on the contrary, have an inherent potential to be highly specific due to their large interaction surface area, which bind not only highly conserved catalytic sites but also a variety of surrounding residues. In recent years, several antibodies obtained against MMP-2/MMP-9 (20) and MMP-14 (21) have been shown to exhibit high target specificity and therapeutic potential in pre-clinical models. Nonetheless, antibodies have several well known limitations, including high cost of production (22), difficulty in conjugating chemical moieties in a site-specific manner, potential undesired effector functions (23), and considerable intellectual property barriers to their development (24). In addition, the large size of antibodies results in poor tissue perfusion, whereas smaller proteins can permeate a tumor mass far more easily (25). Whereas anti-MMP antibodies remain promising candidates for targeting MMPs, non-antibody scaffolds for MMP inhibition could provide an attractive alternative in MMP drug development. Alternative plausible candidates for MMP inhibitor design are the tissue inhibitors of metalloproteinases (TIMPs), which are broad inhibitors of the MMP family. In nature, there are four such inhibitors (TIMPs 1–4) that show 40–50% sequence identity and exhibit slightly different preferences for various MMPs, but they generally possess low selectivity among MMPs (26). All TIMPs bind to MMPs using the same interaction mode, coordinating the active site but also interacting with a number of additional sites on MMPs (27–33) (Fig. 1). As natural effectors, TIMPs are non-immunogenic and non-toxic to humans. We chose to work only with the N-terminal domain of TIMP-2, N-TIMP2, a tractable domain that is easier to produce than the full-length protein, exhibits a high starting affinity for various MMPs (Ki = 10−10–10−9 m), and has been shown to be necessary and sufficient for MMP inhibition (34, 35). In addition, by not having a C-terminal domain, N-TIMP2 cannot play a role in pro-MMP2 activation by binding to the MMP hemopexin domain and localizing to the cell surface, where pro-MMP2 is activated by MMP-14 (36).
FIGURE 1.
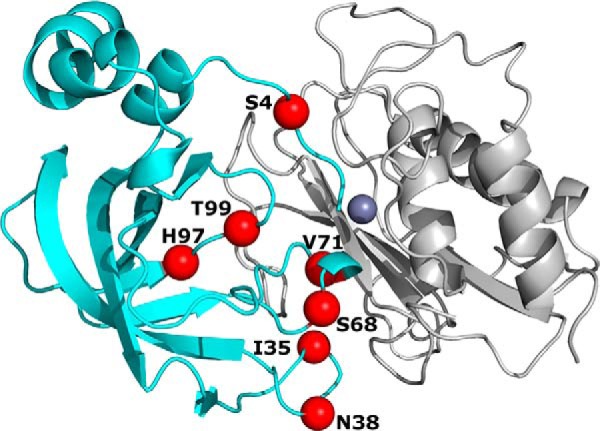
Library design. Structure of N-TIMP2 (shown in cyan) in complex with MMP-14CAT (shown in gray) (adapted from Protein Data Bank code 1BUV (28)) with positions that were chosen for full randomization shown as red spheres. Purple sphere represents the Zn2+ atom found in the active site of MMP-14CAT.
To evolve enhanced selectivity of N-TIMP2, here we employ yeast surface display (YSD), a powerful technique that has been repeatedly utilized for affinity maturation of various biological complexes (37–39), although not previously for TIMP/MMP systems. In the YSD approach, a library of protein mutants is expressed on the surface of the yeast cell and incubated with a fluorescently labeled target protein. The selection for binding can be performed quickly and efficiently using fluorescence-activated cell sorting (FACS). However, due to the limit in transformation efficiency, YSD technology is confined to exploring ∼108 various protein binder sequences, meaning that only 6–7 binder positions can be fully randomized with all 20 amino acids. To overcome this limitation and to increase our chances of success in evolving a potent MMP-14 inhibitor, here we have designed a focused combinatorial library of the most promising N-TIMP2 mutants, based on our previous computational analysis of N-TIMP2/MMP interactions (40).
In our previous study, we computationally explored the effect of various single mutations on N-TIMP2 binding affinity and binding specificity to MMP-14 and MMP-9 and found that N-TIMP2's binding interface is rich in affinity-enhancing mutations (40). Our computational predictions were supported experimentally: out of 13 N-TIMP2 single mutants chosen for expression, purification, and binding measurements, 10 showed enhancement in affinity to MMP-14 and 11 showed enhancement in binding specificity to MMP-14 relative to MMP-9 (40). Yet, the increase in binding affinity and binding specificity due to each single mutation did not exceed a factor of 10, insufficient for obtaining the desired high affinity and high specificity MMP-14 inhibitor. Introduction of multiple mutations into N-TIMP2 should offer the opportunity for more extensive improvements, yet the design of such N-TIMP2 mutants has been limited by our ability to computationally predict the interactive effects of multiple coinciding mutations. In this study, we have taken a novel approach by integrating our computational insights with the power of directed evolution to achieve unprecedented improvements in TIMP selectivity. Our prior computational results serve as a launching-off point for designing a YSD library that very efficiently samples the most relevant areas of sequence space; this formidable combination of computational and YSD methodologies succeeds in producing highly selective N-TIMP2 mutants capable of serving as potent and specific inhibitors of MMP-14 in vitro and in vivo.
Results
Combinatorial Library Design Based on Computational Saturation Mutagenesis Analysis
To maximize our chances of selecting the best MMP-14 inhibitors, we designed a focused library of N-TIMP2 mutants that contained mutations at seven positions with the highest probability of mutations for enhancing affinity and specificity of N-TIMP2 toward the catalytic domain of MMP-14 (MMP-14CAT). The library was designed based on in silico saturation mutagenesis analysis (41, 42) of N-TIMP2 interacting with eight different MMPs performed in our previous study (40). We selected seven N-TIMP2 positions for this study to be randomized in the N-TIMP2 library, namely positions 4, 35, 38, 68, 71, 97, and 99 (Fig. 1). All seven positions lie in the direct binding interface of N-TIMP·MMP complexes, and six of them are coupled in pairs as a result of close proximity (no greater than 5.7 Å) to one another (35 and 38, 68 and 71, and 97 and 99), suggesting that a mutation at one such position is likely to influence the effect of a mutation at another position that is paired with it. Among these chosen positions, positions 4, 35, 38, 68, and 99 were included because they contained a large number of mutations with predicted improvement in the affinity of N-TIMP2 for MMP-14CAT. The other positions were chosen because they have high potential for improving binding specificity, i.e. for facilitating interactions that are mostly neutral for MMP-14 but destabilize complexes with other MMPs. Rather than focusing the library further by restricting the amino acid choices at each position, we allowed full randomization to 20 amino acids at all seven positions, thus producing a library with a theoretical diversity of 1.3 × 109 mutants, which is slightly larger than the limit for YSD. The N-TIMP2 library was then experimentally constructed as detailed under “Experimental Procedures.”
Sorting of the N-TIMP2 Focused Library
We used a YSD platform to select N-TIMP2 variants that bind with high affinity to MMP-14CAT. For this purpose, we cloned the coding region of N-TIMP2WT into two different YSD plasmids, namely pCTCON or pCHA, for presentation of the proteins on the Saccharomyces cerevisiae yeast surface as a fusion with the Aga2p/Aga1p system (Fig. 2). In the pCTCON plasmid, N-TIMP2 is placed between the Aga2 protein and the c-Myc epitope tag, whereas the pCHA plasmid allows the N terminus of N-TIMP2 to be freely exposed. Expression of N-TIMP2WT in both pCHA and pCTCON was verified by using FACS. Binding of expressed pCHA/N-TIMP2WT but not of the pCTCON/N-TIMP2WT clone to the fluorescently labeled MMP-14CAT was then detected, demonstrating the critical role of a free N terminus for N-TIMP2 binding to MMPs. We therefore continued all our selection experiments using the pCHA construct with the N-TIMP2WT gene being substituted by the designed N-TIMP2 library.
FIGURE 2.

Random mutagenesis library screening. A, schematic representation of N-TIMP2WT expressed using the pCTCON construct, with MMP-14CAT as a soluble target (left panel), and flow cytometry analysis of yeast expressing N-TIMP2WT in the pCTCON construct labeled with both 1 μm MMP-14CAT conjugated to DyLight-488 (emission 525 nm) and with mouse anti-c-Myc antibody followed by sheep anti-mouse secondary antibody conjugated to PE (emission 575 nm) for detection of expression (right panel). B, schematic representation of N-TIMP2WT expressed using the pCHA construct, with MMP-14CAT as its soluble target (left panel), and flow cytometry analysis of the yeast expressing N-TIMP2WT in the pCHA construct under the same conditions as the N-TIMP2WT in the pCTCON construct. C, FACS results showing sorting of focused N-TIMP2 libraries. Sort round 1 (S1) represents a sort for protein expression in which the library is labeled only for detection of expression, and a population with a high expression level is selected from the representative gate. In sort round 2 (S2), 1 μm MMP-14CAT conjugated to DyLight-488 was used to select 1.5% (gated pool) of the high affinity population. In Sort 4 (S4) and Sort 6 (S6), 50 and 5 nm, respectively, of MMP-14CAT conjugated to DyLight-488 were used as targets to select the high affinity pool of clones. The x axis shows c-Myc expression, and the y axis shows receptor binding. Polygons indicate sort gates used to select the desired yeast cell population. D, titration curves of the yeast surface displayed N-TIMP2WT and four selected N-TIMP2 clones (N-TIMP2A–D). The yeast cells expressing four different clones and wild-type N-TIMP2 were labeled with MMP-14CAT conjugated to DyLight-488 at a concentration of 1–1000 nm. The binding signal (DyLight-488, emission 525 nm) was normalized to the expression of each clone (PE, emission 575 nm). E, sequences of the N-TIMP2 variants identified after the sixth sort (S6).
The designed N-TIMP2 library, termed S0, was subjected to an initial round of expression enrichment, based on c-Myc detection, to yield the S1 library (Fig. 2C). Five subsequent rounds of affinity maturation were then performed with decreasing concentrations of MMP-14CAT (ranging from 1 μm in S2 to 5 nm in S6). In sorting rounds 2–6, diagonal sorting gates were used to select cell populations having high target affinity levels relative to protein expression levels, with about 1.5% of the library size being selected each time (Fig. 2C). Such a selection protocol allowed us to dramatically decrease the bias of selecting variants because of their high expression level but possibly low binding level.
Identification of N-TIMP2 Clones with Improved MMP-14CAT Affinity
After each round of selection, we sequenced 15–20 different N-TIMP2 clones and analyzed their sequences with the aim to follow the progression of the selection (Fig. 3). Sequence analysis showed increased amino acid conservation as the selection progressed, indicating convergence upon a small set of high affinity sequences. Computational modeling of the selected sequences in the context of N-TIMP2/MMP-14CAT structure confirmed that the average change in free energy of binding, ΔΔGbind, did indeed decrease as the selection progressed (Fig. 3, right). Sequencing of 15 clones from the final round of selection (S6) led to the identification of four different N-TIMP2 variants, N-TIMP2A–D (Fig. 2E). Clone N-TIMP2B was repeated 12 out of 15 times in S6 and N-TIMP2D was repeated twice. N-TIMP2C included one unexpected mutation, K116E, that is located far from the binding interface. We further confirmed that each of the individual clones identified in S6 bound with higher affinity to MMP-14CAT than to N-TIMP2WT when expressed on the yeast surface (Fig. 2D).
FIGURE 3.
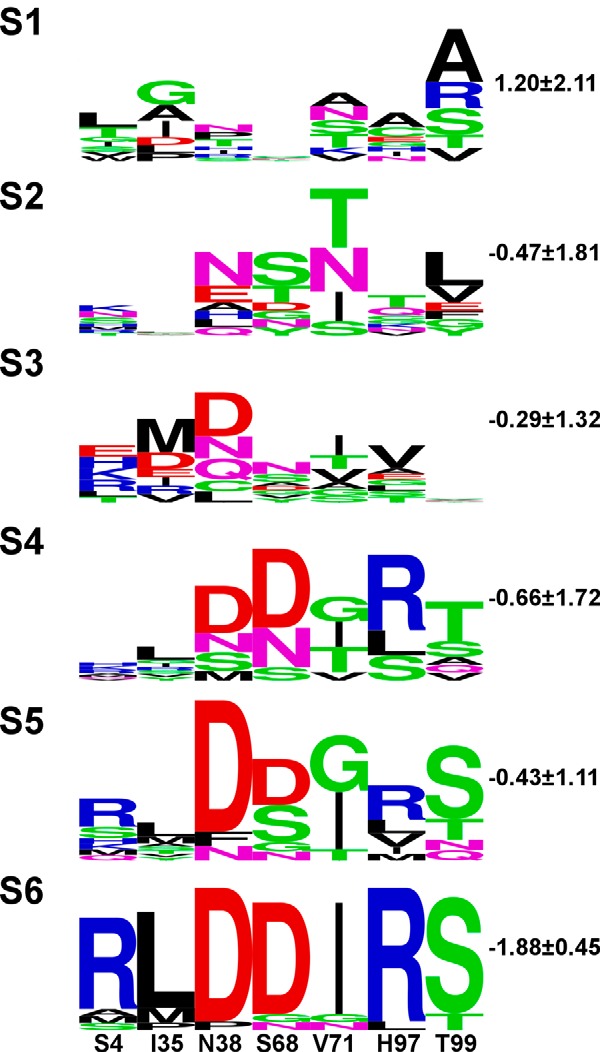
Logo summaries of all N-TIMP2 clones sequenced after each selective round of sorting. The height of each letter is proportional to its frequency at that position. The total height of the stack represents conservation at that position. Green, purple, blue, red, and black letters, respectively, represent polar, neutral, basic, acidic, and hydrophobic amino acids. The identity and position numbers of N-TIMP2WT are denoted at the bottom of the figure. To the top left of each logo, numbers S1–S6 represent the sort number. To the right, the average ΔΔGbind value in kcal/mol is shown (see “Experimental Procedures” for calculation details). The logos were generated by the WebLogo sever.
Affinity-matured N-TIMP2 Variants Show Improved Selective MMP-14 Inhibition in Solution
To assess in vitro N-TIMP2 affinity to MMP-14 and other MMPs, we expressed and purified soluble forms of N-TIMP2WT and the four N-TIMP2 variants (Fig. 4). The expression was performed in Pichia pastoris yeast, which facilitates the correct disulfide bond formation of the N-TIMP2 variants (43) without the need for refolding, as in the case of expression from Escherichia coli (44). For expression, we used the pPICZαA vector that produces the protein with a free N terminus and C-terminal His and c-Myc epitope tags. We purified the proteins using affinity chromatography, followed by size-exclusion chromatography (Fig. 4A). Mass spectrometry analysis confirmed the correct mass for each of the purified variants (Fig. 4B), whereas SDS-PAGE analysis (Fig. 4C) confirmed their high expression level and purity.
FIGURE 4.

Purification, characterization, and MMP-14 inhibitory activity of N-TIMP2 variants. A, size-exclusion chromatography for clone N-TIMP2B. B, mass spectrometry analysis for clone N-TIMP2B after size-exclusion chromatography. C, SDS-PAGE analysis on 15% polyacrylamide gel under reducing conditions for the purified clones N-TIMP2WT, N-TIMP2A, N-TIMP2B, N-TIMP2C, and N-TIMP2D. D, MMP-14 inhibition by N-TIMP2WT. E, MMP-14 inhibition by N-TIMP2 variants. D and E, MMP-14CAT was incubated with N-TIMP2WT and N-TIMP2 variants at various concentrations. The substrate Mca-Lys-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2 was added, and the fluorescent signal upon substrate cleavage by MMP-14CAT was measured. The slopes (cleavage velocities) at each inhibitor concentration were measured, and the curves were fitted by the Morrison equation (Equation 1) to obtain the Ki values.
To determine the binding affinity of each of the purified N-TIMP2 variants to MMP-14 and other MMPs, we utilized an enzyme activity assay. In such an assay, MMP-14CAT was incubated with increasing concentrations of N-TIMP2WT or an N-TIMP2 mutant, and the cleavage of the MMP-14 fluorogenic substrate was determined as a function of time. The slope of each reaction was calculated and fitted to Morrison's tight binding equation (see Equation 1) to determine the Ki value (Fig. 4, D and E, and Table 1). To determine binding specificity of our N-TIMP2 mutants, we also measured Ki values for five additional MMPs belonging to different MMP subgroups, namely gelatinases (MMP-2CAT and MMP-9CAT), collagenases (MMP-1CAT and full-length MMP-8), and stromelysin (MMP-10CAT).
TABLE 1.
Inhibition constants (Ki) of MMP with N-TIMP2 variants
| Clone/MMP |
Kia |
|||||
|---|---|---|---|---|---|---|
| MMP-1CAT | MMP-2CAT | MMP-8 | MMP-9CAT | MMP-10CAT | MMP-14CAT | |
| pm | ||||||
| N-TIMP2WT | 100 ± 7 | 1400 ± 400 | 1300 ± 50 | 100 ± 8 | 2260 ± 150 | 780 ± 80 |
| N-TIMP2A | 10 ± 2 | 1159 ± 10 | 928 ± 50 | 58 ± 10 | 700 ± 20 | 1.6 ± 0.1 |
| N-TIMP2B | 50 ± 2 | 2730 ± 400 | 200 ± 3 | 20 ± 5 | 350 ± 40 | 1.0 ± 0.1 |
| N-TIMP2C | 96 ± 0.8 | 16550 ± 3 | 336 ± 30 | 68 ± 3 | 32600 ± 2755 | 1.5 ± 0.4 |
| N-TIMP2D | 30 ± 9 | 1300 ± 140 | 510 ± 300 | 13 ± 4 | 42150 ± 900 | 0.9 ± 0.1 |
a Values (pm) were obtained from fitting the data shown on Fig. 4 to the Morrison tight binding equation.
N-TIMP2WT bound to MMP-14 with a Ki of 780 ± 80 pm (Table 1), a finding consistent with previous studies (45). All the selected N-TIMP2 variants showed picomolar affinities for MMP-14CAT, exhibiting ∼500- to ∼900-fold improvement in Ki compared with the Ki for N-TIMP2WT (Tables 1 and 2). This outstanding enhancement in binding affinity, by almost 3 orders of magnitude, which was due to only a few mutations, validates the potency of our combined computational/directed evolution methodology.
TABLE 2.
Inhibition specificity of N-TIMP2 variants
| Clone/MMP | MMP-1CAT | MMP-2CAT | MMP-8 | MMP-9CAT | MMP-10CAT | MMP-14CAT |
|---|---|---|---|---|---|---|
| N-TIMP2A | ||||||
| Ki(fold)a | 10 | 1.2 | 1.4 | 1.7 | 3.2 | 487 |
| Specificityb | 48 | 406 | 348 | 286 | 152 | 1 |
| N-TIMP2B | ||||||
| Ki(fold) | 2 | 0.5 | 6.5 | 5 | 6.5 | 780 |
| Specificity | 390 | 1560 | 120 | 156 | 120 | 1 |
| N-TIMP2C | ||||||
| Ki(fold) | 1 | 0.1 | 4 | 1.5 | 0.07 | 520 |
| Specificity | 520 | 5200 | 130 | 346 | 7428 | 1 |
| N-TIMP2D | ||||||
| Ki(fold) | 3 | 1 | 2.5 | 8 | 0.05 | 867 |
| Specificity | 260 | 867 | 340 | 113 | 16164 | 1 |
a Ki(fold) is calculated as the ratio between the Ki of N-TIMP2WT and Ki of N-TIMP2 variant.
b Specificity is calculated as the ratio between the fold of improvement toward MMP-14 in comparison with MMPs. (Ki(fold) for MMP-14CAT/Ki(fold) for MMP-X.)
We found that although our mutants showed greatly improved binding affinity toward MMP-14 vis à vis N-TIMP2WT, affinity improvements toward MMP-9CAT, MMP-2CAT, MMP1CAT, and full-length MMP-8 were relatively small, and for N-TIMP2C and N-TIMP2D, affinity was substantially weakened toward the physiological target MMP-10CAT. Thus, the mutations present in N-TIMP2C and especially N-TIMP2D resulted in enhancement of specificity toward MMP-14CAT over other MMPs by 2–4 orders of magnitude (Table 2). These results demonstrate the high potency and selectivity of all the tested variants toward MMP-14CAT, with N-TIMP2D showing the highest promise as a candidate for in vivo applications targeting MMP-14.
N-TIMP2 Variants Bind with Higher Affinity to MMP-14CAT Because of a Low Off Rate
We next measured the direct binding constants (KD) for N-TIMP2 variants binding to MMP-14CAT by using surface plasmon resonance (SPR). The KD for N-TIMP2WT binding to MMP-14CAT was determined to be 34.9 nm, although for the selected N-TIMP2 variants it was improved to ∼1 nm for N-TIMP2A-C and to 0.6 nm for N-TIMP2D. The SPR sensorgrams (Fig. 5) show that the improvement in KD for the selected N-TIMP2 variants was mostly achieved due to a slower dissociation rate than that for N-TIMP2WT (Table 3). Improvement in the dissociation rate rather than the association rate is expected for variants selected in YSD experiments, and the slow dissociation rate is indeed one of the most important characteristics of a drug candidate molecule.
FIGURE 5.

SPR measurements of MMP-14CAT and N-TIMP2 variants interactions. A, N-TIMP2WT; B, N-TIMP2A; C, N-TIMP2B; D, N-TIMP2C; E, N-TIMP2D. The binding response (y axis) was measured for MMP-14CAT at different concentrations, 20 nm (blue), 10 nm (green), 5 nm (purple), 2.5 nm (yellow), and 0 nm (red).
TABLE 3.
Kinetic rates and binding affinity constants for N-TIMP2 variants measured by SPR
| Clone | kon | koff | KD |
|---|---|---|---|
| 104m−1s−1 | 10−5 s−1 | nm | |
| N-TIMP2WT | 1.41 ± 0.02 | 49.1 ± 0.1 | 34.9 ± 0.6 |
| N-TIMP2A | 5.43 ± 0.01 | 4.65 ± 0.01 | 0.857 ± 0.002 |
| N-TIMP2B | 6.14 ± 0.01 | 6.65 ± 0.01 | 1.08 ± 0.002 |
| N-TIMP2C | 4.72 ± 0.01 | 5.20 ± 0.01 | 1.10 ± 0.01 |
| N-TIMP2D | 6.20 ± 0.01 | 3.74 ± 0.01 | 0.603 ± 0.001 |
N-TIMP2 Variants Inhibit Gelatinolytic Activity of MMP-2 and MMP-9
To test the inhibitory activity of N-TIMP2 variants on the gelatinolytic function of MMP-2 and MMP-9, a gelatin zymography assay was performed with pure full-length MMP-2 and MMP-9 proteins. In this assay, activated MMP-2 and MMP-9 were resolved on gelatin SDS-PAGE incubated with the N-TIMP2 inhibitors, and the gelatinase activity was visualized as clear bands on a dark background (Fig. 6A). Both N-TIMP2WT and the four tested N-TIMP2 variants showed a significant 40–80% inhibition of MMP-2 and MMP-9 activity, as expected (Fig. 6B). The inhibitory activity shown by N-TIMP2WT and all the tested N-TIMP2 variants was similar, in agreement with no significant improvement in Ki values for MMP-9 and MMP-2 of N-TIMP2WT relative to N-TIMP2 variants as shown by in vitro activity assays (Table 2). In addition, inhibition of MMP-9 was stronger than that for MMP-2 for all the proteins, again in agreement with the better Ki values of the N-TIMP2 variants for this enzyme.
FIGURE 6.

Inhibition of gelatin and collagen degradation by N-TIMP2 variants. A, representative gelatin zymography of 1 nm MMP-2 and MMP-9 full-length proteins resolved on SDS-PAGE and treated with 100 nm N-TIMP2 inhibitors. Left lane of each gel represents pro-MMP2 (upper) and active MMP-2 (lower), and the right lane represents active MMP-9. B, quantification of band intensity normalized to the intensity of the control (untreated) gel. The average values for the percentage of inhibition for MMP2 are as follows: N-TIMP2WT, 51%; N-TIMP2A, 57%; N-TIMP2B, 51%; N-TIMP2C, 41%; N-TIMP2D, 55%; and for MMP9 as follows: N-TIMP2WT, 61%; N-TIMP2A, 68%; N-TIMP2B, 68%; N-TIMP2C, 79%; N-TIMP2D, 74%. Error bars represent S.E. Statistical analysis was performed by Student's t test compared with untreated control *, p < 0.05; **, p < 0.01 n = 3. C, representative degradation products of bovine type I collagen. Collagen (1.5 μg) was incubated with (lane 2) or without (lane 1) 2.2 μg of MMP-14CAT, or with 2.2 μg of MMP-14CAT along with 2.5 μm of inhibitors (N-TIMP2WT, N-TIMP2A, N-TIMP2B, N-TIMP2C, and N-TIMP2D, lanes 3–7, respectively). The labels α1 and α2 indicate the α1(I) and α2(I) chains of type I collagen, and the β chains indicate the cross-link between two α1 chains or between α1 chain and α2 chain. The ¾ cleavage product of type I collagen by MMP-14CAT is indicated as TCA (¾) fragment. Note the reduction in collagen degradation in the presence of the N-TIMP2 variants.
N-TIMP2 Variants Inhibit MMP-14 Type I Collagen
To test the inhibitory activity of N-TIMP2 variants on the collagenolytic function of MMP-14, a type I collagen degradation assay was performed with MMP-14CAT. Bovine type I collagen was incubated with MMP-14CAT and N-TIMP2 variants. Samples were loaded on an SDS-polyacrylamide gel, and the cleavage products were detected. All N-TIMP2 variants, but not N-TIMP2WT, showed a significant inhibition of MMP-14CAT (Fig. 6C), with type I collagen cleavage products (TCA fragment) being observed only for the untreated and N-TIMP2WT-treated samples (lanes 2 and 3) but not for the rest of the samples treated with the engineered N-TIMP2 variants (lanes 4–7). This result correlated with the binding affinity (Ki) values.
Binding of Soluble N-TIMP2 Variants to Cancer Cells
To test whether our molecules could potentially inhibit cell invasion by binding to cell surface MMP-14, we measured binding of N-TIMP2WT and the engineered N-TIMP2 variants to MDA-MB-231 breast cancer cells, which endogenously express MMP-14 (46). First, we measured binding of the fluorescently labeled N-TIMP2 variants to the human breast cancer MDA-MB-231 cell line (Fig. 7A). Control experiments showed that MDA-MB-231 cells endogenously expressed MMP-14 (Fig. 7B), and fluorescent labeling of N-TIMP2 variants did not alter the anti-MMP-14 activity of the inhibitors (data not shown). The binding levels were similar for N-TIMP2WT and the engineered variants N-TIMP2A–D. The specific binding of N-TIMP2D to cellular MMP-14 was further validated by performing a competitive assay with LEM-2/63.1, an antibody that selectively binds the MMP-14 catalytic domain and inhibits its activity in vitro and in cells (Fig. 7B) (47). Detection of LEM-2/63.1 with a fluorescently labeled secondary antibody revealed that the binding of LEM-2/63.1 was reduced to a greater extent in the presence of N-TIMP2D versus N-TIMP2WT, supporting the superior specificity of the former toward MMP-14.
FIGURE 7.

Binding of N-TIMP2 variants to MDA-MB-231 cells and inhibition of cancer cell invasion. A, binding of the N-TIMP2 inhibitors, conjugated to DyLight-488 fluorescent dye, to the cells. B, binding of LEM-2/63.1 anti-MMP-14 antibody in the absence and presence of N-TIMP2 inhibitors. *, p < 0.05 for t test comparisons of the indicated condition versus LEM-2/63.1 binding. C, representative micrographs of invading MDA-MB-231 cells treated with 7.5 nm N-TIMP2 inhibitors; the cells were stained with Dipp Kwik Differential Stain and visualized as dark forms on light background by light microscopy using ×40 magnification lenses. D, calculated fold of invasion. The experiment was repeated three times; means and standard error are given. ***, p < 0.001 for t test comparisons of the indicated condition versus N-TIMP2WT.
N-TIMP2D Variant Is the Best Inhibitor of in Vitro Cell Invasion
Previously described Matrigel invasion assays showed that MMP-14, MMP-9, and MMP-2 expression and catalytic activity are essential for the invasiveness of different types of cancer cells (21, 48–50), in particular for MDA-MB-231 cells (21, 51). To test the effect of N-TIMP2 inhibitors on in vitro invasion of MDA-MB-231 cells, a Boyden chamber chemoinvasion assay was employed (47). In such assays, the cells were allowed to migrate through a Matrigel-coated membrane and were visualized by light microscopy (Fig. 7C); the number of invading cells was counted (Fig. 7D). Addition of the N-TIMP2WT, N-TIMP2A, N-TIMP2B, and N-TIMP2C variants resulted in about 40% inhibition of invasion (Fig. 7D). Significantly more effective inhibition of invasion was observed for N-TIMP2D, which inhibits invasion of about 70% of cells, thus making this variant the most promising candidate for drug design. Note that this candidate showed the highest inhibitory activity for both MMP-14 and MMP-9 (but not for MMP-2) among all the tested variants.
Discussion
In this work, we engineered four N-TIMP2 mutants with single-digit picomolar Ki values for binding to MMP-14, improving affinity toward this target by nearly 900-fold and improving binding specificity relative to other MMP family members by 100–16,000-fold. These impressive results demonstrate the enormous potential of the combined computational/directed evolution methodology for protein engineering (39). In this study, we used computational methods to reduce the library size to ∼108 of the most promising N-TIMP2 variants, a number that is tractable by the YSD technique. Our strategy allowed full randomization of seven N-TIMP2 positions most critical to MMP-14 binding affinity and binding specificity, thus greatly increasing our chances of selecting variants with superior affinity and specificity.
Our evolved N-TIMP2 mutants by far surpass all previous MMP inhibitors based on TIMP and other scaffolds in terms of their binding affinities and binding specificity shift toward MMP-14CAT (32, 35, 45, 52). Anti-MMP-14 antibodies previously developed by Sagi and co-workers (53) and by Devy et al. (21) exhibited specific MMP-14 inhibition with nanomolar affinity. Another MMP-14-specific inhibitor, a short peptide (namely peptide G) developed by Suojanen et al. (54), exhibited micromolar affinity toward MMP-14. Previously explored N-TIMP2-based MMP inhibitors usually resulted in 10−10–10−9 m Ki values toward the desired MMP (45, 52, 55) compared with 10−12 Ki value reported here.
Previously reported N-TIMP2-based designs mostly increased its binding specificity by reducing N-TIMP2's affinity to alternative MMPs while maintaining the affinity to the desired MMP at the same or even slightly reduced value (45, 52, 55). We, on the contrary, were able to significantly enhance N-TIMP2's binding affinity toward MMP-14CAT while slightly increasing or decreasing its affinity toward other MMPs, thus making molecules that potentially are much more potent in vivo inhibitors of MMP-14 compared with the previously designed candidates. In addition, N-TIMP2 residues mutated in this study do not include the N-terminal residues 1–3 that have been used in most of the previous N-TIMP2 designs (35, 45, 56), proving that residues that are distant to the catalytic MMP site could be successfully used to modify N-TIMP2's binding specificity.
Our study demonstrates that introduction of a few mutations into the N-TIMP2 sequence is necessary and sufficient to convert this broad MMP inhibitor into a high affinity and high specificity inhibitor of MMP-14. Although the introduction of single mutations in various studies resulted in 10–100-fold specificity enhancements toward the desired MMP (35, 40, 52), introduction of 2–3 mutations in other studies enhanced specificity of N-TIMPs by several hundredfold (45, 52), and introduction of five mutations in this study resulted in up to 16,000-fold specificity enhancement toward MMP-14 relative to other tested MMPs.
Structural modeling of the N-TIMP2 sequences that appeared after the 6th round of YSD selection in complex with MMP-14CAT (Fig. 8) allows us to explain how each of the individual mutations affects binding affinity for MMP-14 (see Figs. 3 and 8). At position 4, an Arg predominates in the selected sequences, although an Ala and a Met are also present. Wild-type Ser at this position has sub-optimal interactions with MMP-14 (40) (Fig. 8A), and single mutations to Arg and Ala at this position were previously shown to result in an ∼2- and 4-fold increase in N-TIMP2 affinity to MMP-14, respectively (40). Here, the Arg is predicted to improve intermolecular interactions by introducing additional hydrogen bonds to Asn-229 and Asn-231 on MMP-14 (Fig. 8A). At position 35, various hydrophobic amino acids (Leu, Met, and Pro) substitute for hydrophobic Ile; such mutations might provide better packing against MMP-14 and against Tyr-36 on N-TIMP2. At position 38, an Asp predominates in the selection; this Asp is predicted to form an intramolecular salt bridge to Lys-41 on N-TIMP2, thus stabilizing the interacting loop in the MMP-14-bound conformation. Another Asp predominates in the selection at position 68. This Asp packs tightly against Phe-204 on MMP-14 and against Ile-71 on N-TIMP2; it is also expected to form a new intramolecular H-bond with the backbone of Ala-70 on N-TIMP2 (Fig. 8B), thus contributing to the stability of the N-TIMP2 loop. At position 71, a Val is substituted by an Ile, improving packing against MMP-14 (Fig. 8B). Mutation H97R, predominating in all the selected sequences, replaces a small residue that does not contact MMP-14 (Fig. 8C) by a larger one, thereby introducing new polar interactions and intermolecular hydrogen bonds with Asp-193 and the backbone of Pro-191 (Fig. 8C). This mutation has also been previously shown to increase N-TIMP2 affinity for MMP-14 by 14-fold and is also likely to increase its affinity to other MMPs (40). Finally, at position 99, either a wild-type Thr or a Ser was observed. The mutation to Ser at this position is predicted to lead to a new hydrogen bond with Asp-193 of MMP-14 (Fig. 8C); despite this Asp residue being present in most MMPs at this position, this interaction may not be possible in other MMPs due to differences in backbone conformation. In summary, there appear to be two scenarios for improvement of binding to and specificity for MMP-14, as have been observed in other studies on TIMP/MMP interactions (32, 45). Some of the acquired mutations are predicted to facilitate favorable intermolecular interactions with MMP-14, although others are expected to stabilize the loops on N-TIMP2 in the MMP-14-bound conformations.
FIGURE 8.

Structural analysis of MMP-14 affinity enhancing mutations. Computationally modeled interactions of mutations that are common after the 6th round of selection for enhancement of affinity of binding of N-TIMP2 for MMP-14. N-TIMP2 is shown in cyan, and MMP-14 is shown in green. A, interactions at position 4 of N-TIMP2; B, interactions at positions 68 and 71 of N-TIMP2; C, interactions at positions 97 and 99 of N-TIMP2. Residues involved in an interaction are shown in stick representation. Mutant residue identities are labeled in red, and WT identities of residues in all panels are labeled in black. Polar interactions are shown as dotted red lines.
Interestingly, some of the mutations appearing after affinity maturation to MMP-14 recapitulate evolutionary trends in TIMP homologues. For example, mutations to Asp at positions 38 and 68 are present in wild-type human TIMP-4 as well as in other mammalian homologues of TIMP-4, as observed in the HOVERGEN database (57), and from a structural alignment with a model of TIMP-4. TIMP-4 is a tissue-specific TIMP, whose expression is normally restricted to the heart, kidney, ovary, pancreas, colon, testes, brain, and adipose tissue, and it is also evident in breast cancer tumors (58, 59). According to Greene et al. (59), the highest expression levels of TIMP-4 are seen in the heart, and this may be biologically significant as cancer metastasis is rare in heart tissue. Zhao et al. (56) reported a Ki value for TIMP-4 binding to MMP-14CAT of 340 pm, compared with 70 pm for TIMP2, indicating that TIMP-4 is a strong inhibitor of MMPs up-regulated in cancers, such as MMP-14. Additionally, Ser at position 99 is observed in vertebrate homologues of TIMP-1 (57). It is known that TIMP-1 exhibits very low affinity for MMP-14 and is not effective in inhibiting its activity (58); therefore, this may indicate that the T99S mutation that we obtained is not effective alone in increasing binding to MMP-14 and may only be significant when coupled with neighboring mutations, such as at position 97 (namely H97R), as we obtained in this work.
Although our results prove that the engineered N-TIMP2 mutants are very potent inhibitors of MMP-14, these molecules might produce undesired effects through interactions with other proteins in the human body. It is known that TIMPs exhibit non-metalloproteinase inhibition-related functions and may act as direct ligands for receptors involved in cell signaling (60–62). For example, TIMP2 was reported to inhibit angiogenesis by binding to α3β1 integrin on human endothelial cells. However, this effect may be strictly limited to the C-terminal domain of TIMP2 (C-TIMP2), which is absent from our constructs, as TIMP2 bound via its C-terminal domain to the hemopexin domain of MMP-2 does not exhibit these inhibitory effects on angiogenesis (4, 60). The importance of the C-terminal domain of TIMP2 in inhibiting angiogenesis and endothelial growth in a non-MMP-dependent manner was further proven by Fernández et al. (43) that showed that C-TIMP2 alone facilitates MMP-2 activation enhancement in cancer cell invasion. In addition to its effects on angiogenesis, TIMP2 is also known to inhibit cell migration and induce arrest of the cell cycle (62). These effects have not been linked exclusively to the either the N- or C-terminal domains of TIMP2, and therefore it is possible that our N-TIMP2 mutants may have unexpected effects on these biological processes and thereby produce adverse effects in cancer treatment. Yet, our constructs lacking the C-terminal domain and containing several mutations in the MMP-binding site are likely to lose at least some of the natural TIMP2 interactions as was discussed in our theoretical study (63) and thus are less likely to participate in undesired interactions compared with WT full-length TIMP2. Nevertheless, further studies are needed to prove this unambiguously.
In conclusion, our best MMP-14 inhibitor, namely N-TIMP2D, possessed 900-fold higher affinity than the parent molecule, making it by far the most potent specific inhibitor of MMP-14 catalytic activity reported to date (21, 53). The mutations present in N-TIMP2 variants resulted in enhancement of specificity toward MMP-14 over other MMPs by 2–5 orders of magnitude in in vitro experiments (Table 2). Importantly, our novel inhibitors, N-TIMP2A–C and particularly N-TIMP2D, also reduced cellular invasion at nanomolar concentrations (Fig. 8). These results suggest that our engineered N-TIMP2 variants may indeed constitute suitable candidates for in vivo applications targeting MMP-14.
Experimental Procedures
TIMP-4 Structure Modeling
A homology model of human TIMP4 was built using the I-TASSER server (64); the input sequence was that deposited in the UNIPROT database (accession no. Q99727) and was modified by removing the signal peptide and truncating the sequence to residue 184. The server produced one output structure.
Preparation of DNA Constructs and Library in Yeast
The gene encoding human N-TIMP2WT (residues 1–127) (40, 65) was expressed in a YSD system in two constructs. For expression in the pCTCON vector (66), N-TIMP2WT was amplified by PCR with Phusion DNA polymerase (New England Biolabs) with primers containing homology regions to pCTCON and recognition sites for the restriction enzymes Nhe and BamHI (forward, 5′-GGTGGTTCTGGTGGTGGTGGTTCTGGTGGTGGTGGTTCTGCTAGCTGCAGCTGCTCCCCGGTG-3′, and reverse, 5′-GCTATTACAAGTCCTCTTCAGAAATAAGCTTTTGTTCAGATGGATCTTGGAAAGCCAATGGTTTATCTGGCAAGGATCCCTCGCAGCCCATCTGGTAC-3′).
For the construction of yeast-displayed N-TIMP2WT with a free N terminus, the pCHA-VRC01-scFv vector was used (obtained from Dane Wittrup, Massachusetts Institute of Technology) (67). In this construct, the C terminus of the gene of interest is fused to the N terminus of Aga-2 (68), leaving the N terminus of the displayed protein exposed to the solvent. The gene was amplified with a forward primer (5′-TACCGTCGGTTCCGCTGCAGAAGGCTCTTTGGACAAGAGATGCAGCTGCTCCCCGGTG-3′) containing the KR sequence at the N terminus of N-TIMP2, which results in a free N terminus after post-modification cleavage by the Kex-2 protease. The reverse primer (5′-GTCTTCTTCGGAGATAAGCTTTTGTTCTGCACGCGTGGATCCCTCGCAGCCCATCTGGTACC-3′) contains homologous regions to the vector and the BamHI restriction site at the 3′ end of the reverse primer. The pCT and pCHA vectors were linearized with BamHI-HF and Nhe-HF enzymes (New England Biolabs) and, together with the respective N-TIMP2WT insert, were transformed by homologous recombination into a competent EBY100 S. cerevisiae yeast strain using a MicroPulser electroporator (Bio-Rad), as described previously (69). The transformed yeast was first grown on selective SDCAA plates (0.54% Na2HPO4, 0.856% Na2HPO4H2O, 18.2% sorbitol, 1.5% agar, 2% dextrose, 0.67% yeast nitrogen base, 0.5% BactoTM casamino acids) at 30 °C. A single colony was then grown in selective SDCAA culture medium (2% dextrose, 0.67% yeast nitrogen base, 0.5% BactoTM casamino acids, 1.47% sodium citrate, 0.429% citric acid monohydrate, pH 4.5) at 30 °C, and the correct sequence was confirmed by DNA sequencing (sequencing laboratory of the National Institute for Biotechnology in the Negev (NIBN), Ben-Gurion University of the Negev, Israel).
The YSD-focused N-TIMP2 library was designed on the basis of positions that were previously shown to be tolerant to mutation by Sharabi et al. (40) and purchased from GenScript. Briefly, the focused library was prepared using NNS (where N represents A, C, T, or G nucleotides, and S represents C and G) degenerate codons at positions 4, 35, 38, 68, 71, 97, and 99 of the N-TIMP2WT gene. The focused library was amplified with forward and reverse primers (forward, 5′-CCGGTTATTTCTACTACCGTCGGTTC-3′, and reverse-5′-GTCAGTTCCTGCAAGTCTTCTTCG-3′) and transformed into competent EBY100 S. cerevisiae along with the linear pCHA vector. The transformed yeast was grown in selective SDCAA culture medium (2% dextrose, 0.67% yeast nitrogen base, 0.5% BactoTM casamino acids, 1.47% sodium citrate, 0.429% citric acid monohydrate, pH 4.5). A library size of 8 × 106 clones was confirmed by performing serial dilutions of the transformed yeast on SDCAA plates and counting after 48 h of growth.
Flow Cytometry Analysis and Cell Screening
The yeast containing the gene for N-TIMP2WT was grown in the selective and protein expression-inducing SGCAA medium (2% galactose, 0.67% yeast nitrogen base, 0.5% BactoTM casamino acids, 1.47% sodium citrate, 0.429% citric acid monohydrate) overnight at 30 °C to a density of 108 cells/ml (OD600 nm = 10.0). The yeast cells (106 cells) were then collected and washed with a buffer containing 50 mm Tris, pH 7.5, 100 mm NaCl, 5 mm CaCl2, and 1% bovine serum albumin (BSA). For the detection of protein expression, the yeast was incubated with mouse anti-c-Myc antibody (Abcam, Cambridge, UK) at a ratio of 1:50 at room temperature for 1 h. Thereafter, the cells were washed and incubated with sheep anti-mouse secondary antibody conjugated to phycoerythrin (PE) (Sigma) at a 1:50 ratio for 30 min on ice. The catalytic domain of MMP-14 (MMP-14CAT) was purified (see details below) and labeled for 1 h at room temperature with DyLight-488 Amine Reactive Dye, which contains a hydroxysuccinimide ester that forms a covalent bond with primary amines (Thermo Fisher Scientific, Waltham, MA), at a ratio of 1:5 (protein/dye). For the detection of binding to MMP-14CAT, the cells were incubated with 1 μm MMP-14CAT labeled with DyLight-488 for 30 min on ice. The yeast was then washed and analyzed using an Accuri C6 flow cytometer (BD Biosciences). In the flow cytometry plots, the horizontal axis provides an indication of the expression of the displayed protein (PE signal), and the vertical axis shows the binding of the displayed protein to the soluble target MMP-14CAT (DyLight-488 signal).
A similar labeling process was performed for yeast expressing the N-TIMP2 focused library for enrichment of the high affinity cell population. For the first sort (c-Myc-clear, S1), 108 cells (10 times more than the library diversity size) were labeled, and for the following sorts, 2–20 × 106 cells were labeled, depending on the library size. In the S1 sort, the high-expressing cells were selected. In the subsequent sorts (S2–S6), the cells were labeled for both detection of expression and binding to MMP-14CAT conjugated to DyLight-488, with decreasing concentrations of labeled MMP-14CAT for each round of sorting, beginning with 1 μm of labeled MMP-14CAT for S2 and ending with 5 nm in the final sort (S6). The affinity maturation screens were performed on an iCyt Synergy FACS apparatus (Sony Biotechnology, San Jose, CA). For each sort, about 1.5% of the binding population normalized to expression was selected using a polygonal gate.
To obtain the YSD-based affinity titration curves, the same labeling process was implemented, and the levels of binding of N-TIMP2WT and the selected N-TIMP2 variants were measured by incubating the yeast cells with different concentrations (1, 5, 10, 20, 50, 100, 500, 750, and 1000 nm) of labeled MMP-14CAT for 30 min at room temperature.
DNA Sequencing
To sequence the pre- and post-sorted N-TIMP2 libraries, the plasmid DNA was purified from the yeast using the ZymoprepTM yeast plasmid miniprep I kit (ZymoResearch, Irvine, CA). The plasmid was then transformed into electro-competent E. coli bacteria and grown on ampicillin (100 μg/ml) LB agar plates. Thereafter, about 20 colonies were transferred to liquid ampicillin/LB culture medium and grown overnight at 37 °C. The plasmid was purified from the bacteria according to the manufacturer's protocol with the HiYield plasmid mini kit (RBC Bioscience, Taiwan). The plasmids were sequenced using the Sanger sequencing method (Genetics Unit, NIBN, Ben-Gurion University of the Negev, Israel), and the sequences were analyzed using Geneious R7 software (Biomatters, New Zealand).
In Silico Thermodynamic and Structural Analysis of Affinity-enhanced Variants
ΔΔGbind was calculated for each variant for which DNA sequencing was performed after each sorting round. Multiple mutations were computationally inserted into the structure, and a ΔΔGbind value was determined as described above for single mutations (saturation mutagenesis). This calculation provided both ΔΔGbind values and structural models for these variants. The average ΔΔGbind value for each sorting round was determined by multiplying the ΔΔGbind value of each variant present in the sorting round by the number of times it appeared in the sorting round. The sum of these values was then divided by the total number of sequences obtained for each sorting round. The standard deviations of those values were calculated in a likewise manner. Variants containing a proline residue at position 35 were excluded, because they exhibited unusually high energy values, indicative of artifacts resulting from inaccuracies of our energy function or the need to account for backbone flexibility.
Protein Expression and Purification
The human MMP-14 catalytic domain gene (MMP-14CAT, residues 112–292) in a pET3a vector with a C-terminal His6 tag was expressed in Bl21 (DE3) E. coli bacteria and purified as described previously (32, 70). The human MMP-9 catalytic domain (MMP-9CAT) lacking the fibronectin-like domain (residues 107–215, 391–443) was purified as described previously (20), with the following modifications: the gene was expressed in Bl21 (DE3) pLysS bacteria cells in a pet28 vector (with an N-terminal His6 tag) and induced with 1 mm isopropyl β-d-1-thiogalactopyranoside (IPTG) overnight at 30 °C.
The MMP-2 catalytic domain (MMP2CAT) without the fibronectin-like domain (residues 110–226 and 395–446) was cloned into the pET3a vector with a C-terminal His6 tag and purified from the Rosetta E. coli strain. Induction was performed with 1 mm IPTG for 18 h at 18 °C. The cells were then harvested, and three rounds of sonication and centrifugation at 12,000 × g were performed. Thereafter, the protein was loaded onto a nickel column and eluted with 20 mm Tris, pH 7.5, 150 mm NaCl, and 500 mm imidazole. The protein was then loaded on a Mono Q ion-exchange column (GE Healthcare) and washed with low-salt buffer (50 mm Tris, pH 7.5, 50 mm NaCl, 10% glycerol, 5 μm β-mercaptoethanol, and 0.1% EDTA). The protein was eluted using a gradient up to 1 m NaCl. The protein was then washed with 20 mm Tris, pH 7.5, and 150 mm NaCl buffer and purified on a gel filtration Superdex 200 followed by Superdex 75 column (GE Healthcare) with 20 mm Tris, pH 7.5, and 150 mm NaCl buffer. The catalytic domains of MMP-1 and MMP-10 (MMP-1CAT and MMP-10CAT, respectively) were purified as described previously (33, 71). Pro-MMP-2, pro-MMP-9, and pro-MMP-8 (residues 21–467) were purchased from R&D Systems (Minneapolis, MN) and were activated before use according to the manufacturer's instructions. The concentrations of MMP-14CAT, MMP-2CAT, MMP-9CAT, MMP-1CAT, MMP-10CAT, and MMP-8 were determined by UV-visible absorbance at 280 nm, with extinction coefficients (ϵ280) of 34,500, 33,920, 18,934, 17,928, and 79,885 m−1 cm−1, respectively.
N-TIMP2WT and its variants were expressed and produced in the methylotrophic X33 P. pastoris yeast strain. The genes were first amplified with forward primers for N-TIMP2WT (5′-GGTATCTCTCGAGAAAAGATGCAGCTGCTCCCCG-3′) and the N-TIMP2A–D variants (5′-GGTATCTCTCGAGAAAAGATGCAGCTGCGCGCCG-3′, 5′-GGTATCTCTCGAGAAAAGATGCAGCTGCAGGCCG-3′, 5′-GGTATCTCTCGAGAAAAGATGCAGCTGCATGCCG-3′, and 5′-GGTATCTCTCGAGAAAAGATGCAGCTGCTCGCCG-3′, respectively), and the reverse primer (5′-GCTGGCGGCCGCCTCGCAGCCCATCTGGTA-3′). The pPICZαA vector, encoding for the Zeocin resistance gene, containing the AOX1 promoter at its N terminus and a His6 tag at the construct's C terminus, and the amplified N-TIMP2 insert clones were digested with XhoI and NotI restriction enzymes (New England Biolabs). Thereafter, the inserts and the vector were ligated and transformed into E. coli electro-competent cells. The transformed bacteria were plated on LB agar plates containing 50 μg/ml Zeocin (Invitrogen). The plasmid was extracted from several colonies, and the correct sequence was verified (Genetics Unit, NIBN, Ben-Gurion University of the Negev, Israel). Thereafter, 100 μg of plasmids with the correct sequence were linearized by treatment with the SacI restriction enzyme (New England Biolabs). The N-TIMP2 variants containing the plasmids were transformed into electro-competent X33 P. pastoris according to the pPICZα protocol (Invitrogen). The transformed yeast was grown on YPDS plates (2% peptone, 1% yeast extract, 2% d-glucose, 1 m sorbitol, 2% agar) for 72 h at 30 °C. Thereafter, expression levels of several colonies from each N-TIMP2 variant transformation were determined; for this purpose, four colonies from each variant were taken from the plates and grown in 5 ml of BMGY medium (2% peptone, 1% yeast extract, 0.23% K2H(PO4), 1.1812% KH2(PO4), 1.34% yeast nitrogen base, 4 × 10−5% biotin, 1% glycerol). After overnight growth at 30 °C, the cultures were grown in inductive BMMY medium (2% peptone, 1% yeast extract, 0.23% K2H(PO4), 1.1812% KH2 (PO4), 1.34% yeast nitrogen base, 4 × 10−5% biotin, 0.5% methanol) for 72 h at 30 °C, with the addition of 1% methanol each day. Overexpression of the secreted proteins was determined by Western blotting, using a 1:3000 dilution of mouse anti-His6 antibody (Abcam, Cambridge, UK) primary antibody, followed by a 1:5000 dilution of anti-mouse secondary antibody conjugated to alkaline phosphatase (Jackson ImmunoResearch, West Grove, PA) and detection by incubation in 2 ml of 5-bromo-4-chloro-3-indolyl phosphate reagent (Sigma). Large scale production of the proteins was performed by growth of the N-TIMP2-expressing yeast exhibiting the highest protein overexpression in 50 ml of BMGY medium overnight, followed by 72 h of growth in BMMY medium, with daily additions of 1% methanol. The proteins were purified by centrifugation of the yeast cell suspension at 3800 × g for 10 min and filtration of the supernatant, followed by addition of 500 mm NaCl and 10 mm imidazole, pH 8.0. The supernatant was incubated for 1 h at 4 °C and then loaded onto nickel-nitrilotriacetic acid-Sepharose beads (Invitrogen), washed with 50 mm Tris, pH 7.5, 100 mm NaCl, and 10 mm imidazole, eluted with 20 ml of 50 mm Tris, pH 7.5, 100 mm NaCl, 300 mm imidazole, and 5 mm CaCl2, and concentrated using a Vivaspin centrifugal concentrator with a 3-kDa cutoff (GE Healthcare). The proteins were further purified using a Superdex 75 column with elution buffer (50 mm Tris, pH 7.5, 100 mm NaCl, and 5 mm CaCl2) in an ÄKTA pure instrument (GE Healthcare). SDS-PAGE analysis on a 15% polyacrylamide gel under reducing conditions for the purified proteins was then performed. Bands were visualized by staining with Instant Blue (CBS Scientific). Protein samples were concentrated using a Vivaspin centrifugal concentrator with a 3-kDa cutoff (GE Healthcare) and subjected to mass spectrometry analysis (Ilse Katz Institute for Nanoscale Science and Technology, Ben-Gurion University of the Negev, Israel). Protein concentrations were determined by UV-visible absorbance at 280 nm, using a NanoDrop spectrophotometer (Thermo Fisher Scientific), with an extinction coefficient (ϵ280) of 13,500 m−1 cm−1 for N-TIMP2WT and all its variants. The production yielded an average of about 1 mg of protein for all variants.
MMP Inhibition Studies
N-TIMP2WT and its variants were tested for inhibitory activity against 0.0075 nm MMP-14CAT, 0.0075 nm MMP-9CAT, 6.25 nm MMP-2CAT, 0.2 nm MMP-1CAT, 0.2 nm MMP-10CAT, and 0.075 nm full-length MMP-8 enzymes. MMP-14CAT was incubated with 0.4–25 nm N-TIMP2WT and with 0.015–0.125 nm N-TIMP2 variants in TCNB buffer (50 mm Tris, pH 7.5, 100 mm NaCl, 5 mm CaCl2, and 0.05% Brij) for 1 h at 37 °C. MMP-9CAT was incubated with 0.03–1.25 nm N-TIMP2 inhibitors for 1 h at 37 °C in TCNB buffer. MMP-2CAT was incubated with 50–500 nm of inhibitors in 50 mm Tris, pH 7.5, and 10 mm CaCl2 for 1 h at 25 °C. MMP-1CAT, MMP-10CAT, and MMP-8 were incubated in TCNB buffer with inhibitor concentrations of 0.2–12.5 nm for 1 h at 37 °C. Thereafter, the fluorogenic substrate Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2·TFA (where Mca is (7-methoxycoumarin-4-yl)acetyl and Dpa is N-3-(2,4-dinitrophenyl)-l-2,3-diaminopropionyl and TFA is trifluoroacetic acid) (Merck Millipore), at a final concentration of 7.5 μm for MMP-14 or 12.5 μm for the other MMPs, was added to the reaction, and the fluorescence was monitored (with 340/30 excitation and 400/30 emission filters) using a Synergy 2 plate reader (BioTek, Winooski, VT) at 37 °C. Reactions were followed spectroscopically for 120 min, and initial rates were determined from the linear portion of the increase in the fluorescence signal caused by the cleavage of the fluorescent substrate Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2·TFA. Data were globally fitted by multiple regression to Morrison's tight binding inhibition equation (72) (see Equation 1) using Prism (GraphPad Software, San Diego). Ki values were calculated by plotting the initial velocities against different concentrations of the inhibitors. Reported inhibition constants are average values obtained from three independent experiments. Calculations were performed using Km values of 1.31 ± 0.2 μm for MMP-14CAT, 2 ± 0.57 μm for MMP-9CAT, 14 ± 0.7 μm for MMP-2CAT, 19 ± 4.3 μm for MMP-1CAT, 13.8 ± 1.9 μm for MMP-10CAT, and 38 ± 7 μm for MMP-8, as determined from at least three Michaelis-Menten kinetic experiments in our laboratory.
| (Eq. 1) |
where Vi is enzyme velocity in the presence of inhibitor; V0 is enzyme velocity in the absence of inhibitor; E is enzyme concentration; I is inhibitor concentration; S is substrate concentration; Km is Michaelis-Menten constant; and Kiapp is apparent inhibition constant, which is given by Equation 2.
| (Eq. 2) |
Binding Measurements between MMP-14CAT and N-TIMP2 Variants Using SPR
Binding affinity between the N-TIMP2 variants and MMP-14CAT was detected using surface plasmon resonance (SPR) spectroscopy on a ProteOn XPR36 system (Bio-Rad) (73). An HTG chip (Bio-Rad) was loaded with 150 μl of 10 mm NiSO4, pH 6.3, at a rate of 30 μl/min for 5 min. Then, 150 μl of 0.033 μg/μl N-TIMP2 variants (tagged with His6) were immobilized at flow rate of 30 μl/min for 5 min via polyHis-Ni2+ interactions; an empty channel was used as control. MMP-14CAT was diluted in elution buffer to 2.5, 5, 10, and 20 nm and was circulated at a flow rate of 25 μl/min for 200 s, followed by 20 min of dissociation. The response was monitored as a function of time at 25 °C. The rate binding constants, kon and koff, and the equilibrium affinity constant KD between N-TIMP2WT and MMP-14CAT were determined using the Langmuir model.
Gelatinase Zymography Assay
The gelatinolytic inhibitory activity of the N-TIMP2 variants was tested in a gelatinase zymography assay, as was described previously (47), with the following modifications. Inhibition of gelatinolytic activity of MMP-2 and MMP-9 full-length proteins was performed on 8% SDS-PAGE with embedded 1% gelatin. The proteins were first activated according to the manufacturer's protocol and then resolved on SDS-PAGE at a concentration of 1 nm. Afterward, the gels were rinsed for 1 h with gentle agitation in 2.5% Triton X-100 at room temperature and then incubated overnight with 100 nm N-TIMP2 inhibitors in 50 mm Tris-HCl, pH 7.5, 10 mm CaCl2, and 200 mm NaCl at 37 °C. After incubation, the gels were stained with Simply-Blue SafeStain (Thermo Fisher Scientific), and the gelatinolytic activity was visualized as clear bands. The signal was quantified using ImageJ software.
Type I Collagen Degradation Assay
Inhibition of MMP-14CAT activity on type I collagen was performed as described previously (74), with the following modifications. Briefly, bovine type I collagen (1.5 mg) (Sigma) was incubated with 2.2 μg of MMP-14CAT with or without 2.5 μm of each inhibitor in 100 μl of 50 mm Tris-HCl, pH 7.5, 0.15 m NaCl, 10 mm CaCl2, 0.05% Brij 35, and 0.02% NaN3 for 24 h at 27 °C. Each reaction was loaded onto 8% SDS-polyacrylamide gel, and the bands were visualized by Instant Blue staining (CBS Scientific). The activity of MMP-14CAT was detected by the characteristic ¾ fragments (TCA) of type I collagen, which represent the degradation products of α1 and α2 collagen type I chains.
Cell Culture
MDA-MB-231 human breast cancer cell line (kindly provided by Smadar Cohen, Ben-Gurion University of the Negev, Israel) was maintained in RPMI 1640 (Roswell Park Memorial Institute) medium (Biological Industries Israel Beit-Haemek Ltd., Israel) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific), 1% l-glutamine (Biological Industries Israel Beit-Haemek Ltd., Israel), and 1% penicillin/streptomycin (Biological Industries Beit-Haemek Ltd., Israel). Murine fibroblast NIH-3T3 cells (American Type Culture Collection, Manassas, VA) were maintained in DMEM (Biological Industries Israel Beit-Haemek Ltd., Israel) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, MA), 1% l-glutamine (Biological Industries, Beit-Haemek Ltd., Israel), and 1% penicillin/streptomycin (Biological Industries, Beit-Haemek Ltd., Israel).
Cell Binding Assay
N-TIMP2 proteins were labeled with DyLight-488 (Thermo Fisher Scientific) at a ratio of 1:5 (protein/dye) for 1 h at room temperature and washed three times with 50 mm Tris-HCl, pH 7.5, 5 mm CaCl2, and 100 mm NaCl using Vivaspin centrifugal concentrators with a 3-kDa cutoff (GE Healthcare). The inhibitory activity of the labeled N-TIMP2 inhibitors was determined as described above. Briefly, MMP-14CAT was incubated with the labeled and non-labeled inhibitors for 1 h at 37 °C. Thereafter, the cleavage of the fluorescent substrate Mca-Lys-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2 was measured. For the binding assays, various concentrations of N-TIMP2 proteins (10 nm to 1 μm) were added to 1 × 105 cells in volumes that were appropriate to prevent ligand depletion. Cells were incubated at room temperature for 2 h with gentle agitation, centrifuged at 150 × g at 4 °C for 5 min, washed twice with 50 mm Tris-HCl, pH 7.5, 5 mm CaCl2, 100 mm NaCl, and 0.1% BSA, and analyzed by flow cytometry using an Accuri flow cytometer (BD Biosciences). Mean fluorescence values were measured using the Accuri C6 software (BD Biosciences) and were plotted against the log of the concentration of the inhibitors. Error is reported as the standard deviation of triplicate measurements. MMP-14 expression on the membrane of MDA-MB-231 cells and N-TIMP2D binding specificity for cellular MMP-14 were determined by incubation of 1 × 105 MDA-MB-231 cells with 10 μg/ml of Ms-anti-MMP-14 antibody LEM-2/63.1 (Abcam, Cambridge, UK) (47). These mixtures were in the presence or absence of 1 μm N-TIMP2 variant for 1 h at room temperature with gentle agitation. Thereafter, the cells were washed and incubated with goat anti-mouse secondary antibody conjugated to FITC (Sigma) to detect the LEM-2/63.1 antibody and analyzed using Accuri C6 as above.
Invasion Assay
The Boyden chamber chemoinvasion assay was performed as described previously (75) with the following modifications. The experiment was performed with polycarbonate 8-μm filters (Neuoro Probe, Inc., Gaithersburg, MD) coated with Matrigel inside Transwell chambers (Neuoro Probe, Inc.). The bottom part of each chamber was filled with conditioned medium containing DMEM collected from NIH-3T3 fibroblast cells after 24 h of culture and supplemented with 0.1% of BSA. The upper part of the chamber was filled with 200 μl of MDA-MB-231 (1 × 105). Then, 600 μl of DMEM without serum was added to the control chamber, or 7.5 nm of inhibitors was added to the sample chambers. The cells were incubated for 4 h at 37 °C. Thereafter, the invasive cells were fixed and stained using the Dipp Kwik Differential Stain kit (American Mastertech Scientific). The cells that had migrated were counted using EVOS FL Cell Imaging System (Thermo Fisher Scientific), at ×40 magnification in 2.2-mm2 fields. The experiment was performed in triplicate, and 10 fields were counted for each sample.
Author Contributions
V. A., J. S., J. M. S., and N. P. designed the research. V. A. performed and analyzed the experiments shown in Figs. 2, 4, 6, and 7 and Tables 1 and 2. G. Y. performed and analyzed the experiments shown in Figs. 5 and 6 and Table 3. J. S. generated the computational data shown in Figs. 1, 3, and 8. I. C. designed the N-TIMP2 constructs and performed the experiments shown in Fig. 2, A and B. Y. H. performed and analyzed the experiments shown in Fig. 4 and Tables 1 and 2. E. S. R., I. S., and M. G. provided recombinant MMPs and consulted on kinetics studies. V. A., G. Y., J. S., J. M. S., and N. P. wrote the paper, and all authors reviewed and approved the final version of the manuscript.
Acknowledgments
We thank Prof. Reuven Reich (Hebrew University of Jerusalem) for providing Matrigel and for consultation on the invasion studies. We thank Dr. Alon Zilka for technical assistance. FACS and Proteon experiments were performed at the NIBN proteomics unit.
This work was supported in part by European Research Council “Ideas Program” ERC-2013-StG by Contract Grant 336041 (to N. P.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- MMP
- matrix metalloproteinase
- YSD
- yeast surface display
- TIMP
- tissue inhibitor of metalloproteinase
- FACS
- fluorescence-activated cell sorting
- SPR
- surface plasmon resonance
- PE
- phycoerythrin
- Mca
- (7-methoxycoumarin-4-yl)acetyl
- Dpa
- N-3-(2,4-dinitrophenyl)-l-2,3-diaminopropionyl
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- NIBN
- National Institute for Biotechnology in the Negev.
References
- 1. Brinckerhoff C. E., and Matrisian L. M. (2002) Matrix metalloproteinases: a tail of a frog that became a prince. Nat. Rev. Mol. Cell Biol. 3, 207–214 [DOI] [PubMed] [Google Scholar]
- 2. Vandenbroucke R. E., and Libert C. (2014) Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 13, 904–927 [DOI] [PubMed] [Google Scholar]
- 3. Seiki M. (1999) Membrane-type matrix metalloproteinases. APMIS 107, 137–143 [DOI] [PubMed] [Google Scholar]
- 4. Dollery C. M., and Libby P. (2006) Atherosclerosis and proteinase activation. Cardiovasc. Res. 69, 625–635 [DOI] [PubMed] [Google Scholar]
- 5. Ra H. J., and Parks W. C. (2007) Control of matrix metalloproteinase catalytic activity. Matrix Biol. 26, 587–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reddy N. R., Roopa D., Babu D. S., Kumar P. M., Raju C. M., and Kumar N. S. (2012) Estimation of matrix metalloproteinase-3 levels in gingival crevicular fluid in periodontal disease, health and after scaling and root planing. J. Indian Soc. Periodontol. 16, 549–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Murphy G., and Nagase H. (2008) Reappraising metalloproteinases in rheumatoid arthritis and osteoarthritis: destruction or repair? Nat. Clin. Pract. Rheumatol. 4, 128–135 [DOI] [PubMed] [Google Scholar]
- 8. Raffetto J. D., and Khalil R. A. (2008) Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem. Pharmacol. 75, 346–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burrage P. S., Mix K. S., and Brinckerhoff C. E. (2006) Matrix metalloproteinases: role in arthritis. Front. Biosci. 11, 529–543 [DOI] [PubMed] [Google Scholar]
- 10. Egeblad M., and Werb Z. (2002) New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2, 161–174 [DOI] [PubMed] [Google Scholar]
- 11. Coussens L. M., Fingleton B., and Matrisian L. M. (2002) Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 295, 2387–2392 [DOI] [PubMed] [Google Scholar]
- 12. Bartolomé R. A., Ferreiro S., Miquilena-Colina M. E., Martínez-Prats L., Soto-Montenegro M. L., García-Bernal D., Vaquero J. J., Agami R., Delgado R., Desco M., Sánchez-Mateos P., and Teixidó J. (2009) The chemokine receptor CXCR4 and the metalloproteinase MT1-MMP are mutually required during melanoma metastasis to lungs. Am. J. Pathol. 174, 602–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang H., Liu M., Sun Y., and Lu J. (2009) MMP-14 can serve as a prognostic marker in patients with supraglottic cancer. Eur. Arch. Otorhinolaryngol. 266, 1427–1434 [DOI] [PubMed] [Google Scholar]
- 14. Têtu B., Brisson J., Wang C. S., Lapointe H., Beaudry G., Blanchette C., and Trudel D. (2006) The influence of MMP-14, TIMP-2 and MMP-2 expression on breast cancer prognosis. Breast Cancer Res. 8, R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hotary K. B., Allen E. D., Brooks P. C., Datta N. S., Long M. W., and Weiss S. J. (2003) Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell 114, 33–45 [DOI] [PubMed] [Google Scholar]
- 16. Overall C. M., and Kleifeld O. (2006) Tumour microenvironment–opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 6, 227–239 [DOI] [PubMed] [Google Scholar]
- 17. López-Otín C., and Matrisian L. M. (2007) Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 7, 800–808 [DOI] [PubMed] [Google Scholar]
- 18. Folgueras A. R., Pendás A. M., Sánchez L. M., and López-Otín C. (2004) Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int. J. Dev. Biol. 48, 411–424 [DOI] [PubMed] [Google Scholar]
- 19. Decock J., Thirkettle S., Wagstaff L., and Edwards D. R. (2011) Matrix metalloproteinases: protective roles in cancer. J. Cell. Mol. Med. 15, 1254–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sela-Passwell N., Kikkeri R., Dym O., Rozenberg H., Margalit R., Arad-Yellin R., Eisenstein M., Brenner O., Shoham T., Danon T., Shanzer A., and Sagi I. (2011) Antibodies targeting the catalytic zinc complex of activated matrix metalloproteinases show therapeutic potential. Nat. Med. 18, 143–147 [DOI] [PubMed] [Google Scholar]
- 21. Devy L., Huang L., Naa L., Yanamandra N., Pieters H., Frans N., Chang E., Tao Q., Vanhove M., Lejeune A., van Gool R., Sexton D. J., Kuang G., Rank D., Hogan S., et al. (2009) Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 69, 1517–1526 [DOI] [PubMed] [Google Scholar]
- 22. Roque A. C., Lowe C. R., and Taipa M. A. (2004) Antibodies and genetically engineered related molecules: production and purification. Biotechnol. Prog. 20, 639–654 [DOI] [PubMed] [Google Scholar]
- 23. Romer T., Leonhardt H., and Rothbauer U. (2011) Engineering antibodies and proteins for molecular in vivo imaging. Curr. Opin. Biotechnol. 22, 882–887 [DOI] [PubMed] [Google Scholar]
- 24. McCamish M., and Woollett G. (2011) Worldwide experience with biosimilar development. MAbs 3, 209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thurber G. M., Schmidt M. M., and Wittrup K. D. (2008) Factors determining antibody distribution in tumors. Trends Pharmacol. Sci. 29, 57–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brew K., and Nagase H. (2010) The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim. Biophys. Acta 1803, 55–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gomis-Rüth F. X., Maskos K., Betz M., Bergner A., Huber R., Suzuki K., Yoshida N., Nagase H., Brew K., Bourenkov G. P., Bartunik H., and Bode W. (1997) Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by TIMP-1. Nature 389, 77–81 [DOI] [PubMed] [Google Scholar]
- 28. Fernandez-Catalan C., Bode W., Huber R., Turk D., Calvete J. J., Lichte A., Tschesche H., and Maskos K. (1998) Crystal structure of the complex formed by the membrane type 1-matrix metalloproteinase with the tissue inhibitor of metalloproteinases-2, the soluble progelatinase A receptor. EMBO J. 17, 5238–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maskos K., Lang R., Tschesche H., and Bode W. (2007) Flexibility and variability of TIMP binding: X-ray structure of the complex between collagenase-3/MMP-13 and TIMP-2. J. Mol. Biol. 366, 1222–1231 [DOI] [PubMed] [Google Scholar]
- 30. Wisniewska M., Goettig P., Maskos K., Belouski E., Winters D., Hecht R., Black R., and Bode W. (2008) Structural determinants of the ADAM inhibition by TIMP-3: crystal structure of the TACE-N-TIMP-3 complex. J. Mol. Biol. 381, 1307–1319 [DOI] [PubMed] [Google Scholar]
- 31. Iyer S., Wei S., Brew K., and Acharya K. R. (2007) Crystal structure of the catalytic domain of matrix metalloproteinase-1 in complex with the inhibitory domain of tissue inhibitor of metalloproteinase-1. J. Biol. Chem. 282, 364–371 [DOI] [PubMed] [Google Scholar]
- 32. Grossman M., Tworowski D., Dym O., Lee M. H., Levy Y., Murphy G., and Sagi I. (2010) The intrinsic protein flexibility of endogenous protease inhibitor TIMP-1 controls its binding interface and affects its function. Biochemistry 49, 6184–6192 [DOI] [PubMed] [Google Scholar]
- 33. Batra J., Soares A. S., Mehner C., and Radisky E. S. (2013) Matrix metalloproteinase-10/TIMP-2 structure and analyses define conserved core interactions and diverse exosite interactions in MMP/TIMP complexes. PLoS one 8, e75836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang W., Suzuki K., Nagase H., Arumugam S., Van Doren S. R., and Brew K. (1996) Folding and characterization of the amino-terminal domain of human tissue inhibitor of metalloproteinases-1 (TIMP-1) expressed at high yield in E. coli. FEBS Lett. 384, 155–161 [DOI] [PubMed] [Google Scholar]
- 35. Butler G. S., Hutton M., Wattam B. A., Williamson R. A., Knäuper V., Willenbrock F., and Murphy G. (1999) The specificity of TIMP-2 for matrix metalloproteinases can be modified by single amino acid mutations. J. Biol. Chem. 274, 20391–20396 [DOI] [PubMed] [Google Scholar]
- 36. Morgunova E., Tuuttila A., Bergmann U., and Tryggvason K. (2002) Structural insight into the complex formation of latent matrix metalloproteinase 2 with tissue inhibitor of metalloproteinase 2. Proc. Natl. Acad. Sci. U.S.A. 99, 7414–7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boder E. T., Midelfort K. S., and Wittrup K. D. (2000) Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc. Natl. Acad. Sci. U.S.A. 97, 10701–10705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Graff C. P., Chester K., Begent R., and Wittrup K. D. (2004) Directed evolution of an anti-carcinoembryonic antigen scFv with a 4-day monovalent dissociation half-time at 37°C. Protein Eng. Des. Sel. 17, 293–304 [DOI] [PubMed] [Google Scholar]
- 39. Rosenfeld L., Heyne M., Shifman J. M., and Papo N. (2016) Protein engineering by combined computational and in vitro evolution approaches. Trends Biochem. Sci. 41, 421–433 [DOI] [PubMed] [Google Scholar]
- 40. Sharabi O., Shirian J., Grossman M., Lebendiker M., Sagi I., and Shifman J. (2014) Affinity- and specificity-enhancing mutations are frequent in multispecific interactions between TIMP2 and MMPs. PLoS ONE 9, e93712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sharabi O., Shirian J., and Shifman J. M. (2013) Predicting affinity- and specificity-enhancing mutations at protein-protein interfaces. Biochem. Soc. Trans. 41, 1166–1169 [DOI] [PubMed] [Google Scholar]
- 42. Sharabi O., Erijman A., and Shifman J. M. (2013) Computational methods for controlling binding specificity. Methods Enzymol. 523, 41–59 [DOI] [PubMed] [Google Scholar]
- 43. Fernández C. A., Butterfield C., Jackson G., and Moses M. A. (2003) Structural and functional uncoupling of the enzymatic and angiogenic inhibitory activities of tissue inhibitor of metalloproteinase-2 (TIMP-2): loop 6 is a novel angiogenesis inhibitor. J. Biol. Chem. 278, 40989–40995 [DOI] [PubMed] [Google Scholar]
- 44. Williamson R. A., Natalia D., Gee C. K., Murphy G., Carr M. D., and Freedman R. B. (1996) Chemically and conformationally authentic active domain of human tissue inhibitor of metalloproteinases-2 refolded from bacterial inclusion bodies. Eur. J. Biochem. 241, 476–483 [DOI] [PubMed] [Google Scholar]
- 45. Bahudhanapati H., Zhang Y., Sidhu S. S., and Brew K. (2011) Phage display of tissue inhibitor of metalloproteinases-2 (TIMP-2): identification of selective inhibitors of collagenase-1 (metalloproteinase 1 (MMP-1)). J. Biol. Chem. 286, 31761–31770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hegedüs L., Cho H., Xie X., and Eliceiri G. L. (2008) Additional MDA-MB-231 breast cancer cell matrix metalloproteinases promote invasiveness. J. Cell. Physiol. 216, 480–485 [DOI] [PubMed] [Google Scholar]
- 47. Gálvez B. G., Matías-Román S., Albar J. P., Sánchez-Madrid F., and Arroyo A. G. (2001) Membrane type 1-matrix metalloproteinase is activated during migration of human endothelial cells and modulates endothelial motility and matrix remodeling. J. Biol. Chem. 276, 37491–37500 [DOI] [PubMed] [Google Scholar]
- 48. Yan T., Lin Z., Jiang J., Lu S., Chen M., Que H., He X., Que G., Mao J., Xiao J., and Zheng Q. (2015) MMP14 regulates cell migration and invasion through epithelial-mesenchymal transition in nasopharyngeal carcinoma. Am. J. Transl. Res. 7, 950–958 [PMC free article] [PubMed] [Google Scholar]
- 49. Yu Q., and Stamenkovic I. (2000) Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes Dev. 14, 163–176 [PMC free article] [PubMed] [Google Scholar]
- 50. Mehner C., Hockla A., Miller E., Ran S., Radisky D. C., and Radisky E. S. (2014) Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer. Oncotarget 5, 2736–2749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jiang W. G., Davies G., Martin T. A., Parr C., Watkins G., Mason M. D., and Mansel R. E. (2006) Expression of membrane type-1 matrix metalloproteinase, MT1-MMP in human breast cancer and its impact on invasiveness of breast cancer cells. Int. J. Mol. Med. 17, 583–590 [PubMed] [Google Scholar]
- 52. Wei S., Chen Y., Chung L., Nagase H., and Brew K. (2003) Protein engineering of the tissue inhibitor of metalloproteinase 1 (TIMP-1) inhibitory domain. In search of selective matrix metalloproteinase inhibitors. J. Biol. Chem. 278, 9831–9834 [DOI] [PubMed] [Google Scholar]
- 53. Udi Y., Grossman M., Solomonov I., Dym O., Rozenberg H., Moreno V., Cuniasse P., Dive V., Arroyo A. G., and Sagi I. (2015) Inhibition mechanism of membrane metalloprotease by an exosite-swiveling conformational antibody. Structure 23, 104–115 [DOI] [PubMed] [Google Scholar]
- 54. Suojanen J., Salo T., Koivunen E., Sorsa T., and Pirilä E. (2009) A novel and selective membrane type-1 matrix metalloproteinase (MT1-MMP) inhibitor reduces cancer cell motility and tumor growth. Cancer Biol. Ther. 8, 2362–2370 [DOI] [PubMed] [Google Scholar]
- 55. Kveiborg M., Jacobsen J., Lee M. H., Nagase H., Wewer U. M., and Murphy G. (2010) Selective inhibition of ADAM12 catalytic activity through engineering of tissue inhibitor of metalloproteinase 2 (TIMP-2). Biochem. J. 430, 79–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhao H., Bernardo M. M., Osenkowski P., Sohail A., Pei D., Nagase H., Kashiwagi M., Soloway P. D., DeClerck Y. A., and Fridman R. (2004) Differential inhibition of membrane type 3 (MT3)-matrix metalloproteinase (MMP) and MT1-MMP by tissue inhibitor of metalloproteinase (TIMP)-2 and TIMP-3 regulates pro-MMP-2 activation. J. Biol. Chem. 279, 8592–8601 [DOI] [PubMed] [Google Scholar]
- 57. Dufayard J.-F., Duret L., Penel S., Gouy M., Rechenmann F., and Perrière G. (2005) Tree pattern matching in phylogenetic trees: automatic search for orthologs or paralogs in homologous gene sequence databases. Bioinformatics 21, 2596–2603 [DOI] [PubMed] [Google Scholar]
- 58. Murphy G. (2011) Tissue inhibitors of metalloproteinases. Genome Biol. 12, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Greene J., Wang M., Liu Y. E., Raymond L. A., Rosen C., and Shi Y. E. (1996) Molecular cloning and characterization of human tissue inhibitor of metalloproteinase 4. J. Biol. Chem. 271, 30375–30380 [DOI] [PubMed] [Google Scholar]
- 60. Vanhoutte D., and Heymans S. (2010) TIMPs and cardiac remodeling: ‘Embracing the MMP-independent-side of the family’. J. Mol. Cell. Cardiol. 48, 445–453 [DOI] [PubMed] [Google Scholar]
- 61. Moore C. S., and Crocker S. J. (2012) An alternate perspective on the roles of TIMPs and MMPs in pathology. Am. J. Pathol. 180, 12–16 [DOI] [PubMed] [Google Scholar]
- 62. Stetler-Stevenson W. G. (2008) Tissue inhibitors of metalloproteinases in cell signaling: metalloproteinase-independent biological activities. Sci. Signal. 1, re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fromer M., and Shifman J. M. (2009) Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Comput. Biol. 5, e1000627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yang J., and Zhang Y. (2015) Protein structure and function prediction using I-TASSER. Curr. Protoc. Bioinformatics 52, 5.8.1–5.815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tam B. Y., Wei K., Rudge J. S., Hoffman J., Holash J., Park S. K., Yuan J., Hefner C., Chartier C., Lee J. S., Jiang S., Nayak N. R., Niyak N. R., Kuypers F. A., Ma L., et al. (2006) VEGF modulates erythropoiesis through regulation of adult hepatic erythropoietin synthesis. Nat. Med. 12, 793–800 [DOI] [PubMed] [Google Scholar]
- 66. Boder E. T., and Wittrup K. D. (1997) Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 15, 553–557 [DOI] [PubMed] [Google Scholar]
- 67. Mata-Fink J., Kriegsman B., Yu H. X., Zhu H., Hanson M. C., Irvine D. J., and Wittrup K. D. (2013) Rapid conformational epitope mapping of anti-gp120 antibodies with a designed mutant panel displayed on yeast. J. Mol. Biol. 425, 444–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Angelini A., Chen T. F., de Picciotto S., Yang N. J., Tzeng A., Santos M. S., Van Deventer J. A., Traxlmayr M. W., and Wittrup K. D. (2015) Protein engineering and selection using yeast surface display. Methods Mol. Biol. 1319, 3–36 [DOI] [PubMed] [Google Scholar]
- 69. Chao G., Lau W. L., Hackel B. J., Sazinsky S. L., Lippow S. M., and Wittrup K. D. (2006) Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 1, 755–768 [DOI] [PubMed] [Google Scholar]
- 70. Ogata H., Decaneto E., Grossman M., Havenith M., Sagi I., Lubitz W., and Knipp M. (2014) Crystallization and preliminary X-ray crystallographic analysis of the catalytic domain of membrane type 1 matrix metalloproteinase. Acta Crystallogr. F Struct. Biol. Commun. 70, 232–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Batra J., Robinson J., Soares A. S., Fields A. P., Radisky D. C., and Radisky E. S. (2012) Matrix metalloproteinase-10 (MMP-10) interaction with tissue inhibitors of metalloproteinases TIMP-1 and TIMP-2: binding studies and crystal structure. J. Biol. Chem. 287, 15935–15946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Morrison J. F. (1969) Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta 185, 269–286 [DOI] [PubMed] [Google Scholar]
- 73. Anderson G. P., Glaven R. H., Algar W. R., Susumu K., Stewart M. H., Medintz I. L., and Goldman E. R. (2013) Single domain antibody-quantum dot conjugates for ricin detection by both fluoroimmunoassay and surface plasmon resonance. Anal. Chim. Acta 786, 132–138 [DOI] [PubMed] [Google Scholar]
- 74. Ohuchi E., Imai K., Fujii Y., Sato H., Seiki M., and Okada Y. (1997) Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J. Biol. Chem. 272, 2446–2451 [DOI] [PubMed] [Google Scholar]
- 75. Reich R., Katz Y., Hadar R., and Breuer E. (2005) Carbamoylphosphonate matrix metalloproteinase inhibitors 3: in vivo evaluation of cyclopentylcarbamoylphosphonic acid in experimental metastasis and angiogenesis. Clin. Cancer Res. 11, 3925–3929 [DOI] [PubMed] [Google Scholar]