

Abstract
Macrocycles have attracted significant attention in drug discovery recently. In fact, a few de novo designed macrocyclic kinases inhibitors are currently in clinical trials with good potency and selectivity for their intended target. In this paper, we have successfully engaged a structure-based drug design approach to discover macrocyclic pyrimidines as potent Mer tyrosine kinase (MerTK)-specific inhibitors. An enzyme-linked immunosorbent assay (ELISA) in a 384-well format was employed to evaluate the inhibitory activity of macrocycles in a cell-based assay assessing tyrosine phosphorylation of MerTK. Through SAR studies, analogue 11 (UNC2541) was identified as a potent and MerTK-specific inhibitor and exhibits sub-micro molar inhibitory activity in the cell-based ELISA assay. In addition, an X-ray structure of MerTK protein in complex with 11 was resolved to show that these macro-cycles bind in the MerTK ATP pocket.
Keywords: MerTK, MerTK-specific inhibitors, macrocycles, pyrimidines, intramolecular hydrogen bonds
Graphical abstract

A structure-based drug design approach led to macrocyclic pyrimidines as potent Mer tyrosine kinase (MerTK)-specific inhibitors. Through SAR studies, analogue 11 (UNC2541) was identified as a key compound and exhibits sub-micro molar inhibitory activity in the cell-based enzyme-linked immunosorbent assay (ELISA). In addition, an X-ray structure of MerTK protein in complex with 11 was resolved to show that these macrocycles bind in the MerTK ATP pocket.
Macrocycles, especially de novo designed macrocycles, have recently gained attention in drug discovery due to the new physicochemical properties and broader intellectual property (IP) that they may provide.1-3 In macrocycles, cyclization leads to a structural preorganization which can increase both binding affinity and selectivity by engaging targets through numerous and spatially distributed binding interactions.1,4 A few macrocyclic kinase inhibitors are currently in clinical trials with good potency and selectivity for their intended target.5-7 We have been interested in Mer tyrosine kinase (MerTK) as a therapeutic target8,9 and have developed several MerTK inhibitors with varying selectivity profiles.10-14 The most advanced compound among these inhibitors is UNC2025 which is a potent and highly orally bioavailable MerTK inhibitor.14 It is also equally potent against FMS-like tyrosine kinase (Flt3). This dual inhibitory activity of UNC2025 is desirable for certain diseases such as AML, however, inhibition of Flt3 has been associated with hematopoietic toxicity15,16 and is therefore inadvisable for other applications of MerTK inhibitors. The new pyrrolopyrimidine macrocycles that we have developed recently share the same undesired selectivity profile.17 In this paper, we will present a new type of MerTK-specific inhibitor—macrocyclic pyrimidines.
We have recently discovered substituted-pyrimidines as novel MerTK specific inhibitors via a structure-based drug design approach.12 Based on our published X-ray crystal structure of MerTK in complex with 1 (Figures 1a and 1b), the butyl side chain and the cyclohexyl alcohol are close to each other and well-positioned to form a macrocycle. One example of this design is compound 2 (Figure 1c), which has a hydrogen donor, an amino group, at the same position as the hydroxyl group in 1. The macrocycle is connected by an amide bond and the cyclohexyl ring has been opened to eliminate the chance to introduce new stereogenic centers. Compound 2 fits the MerTK docking model and is predicted to retain three key hydrogen bonds with MerTK protein (Figure 1d, two with the hinge area (F673 and P672) and one with either D741, R727, or N728). Since substituted-pyrimidines show some selectivity for MerTK over Flt3 (e.g. compound 1 is 14-fold more active against MerTK versus Flt3),12 we were interested to see if macrocycles based on this scaffold could improve MerTK inhibitory activity and/or the selectivity profile over other TAM family members and Flt3.
Figure 1.
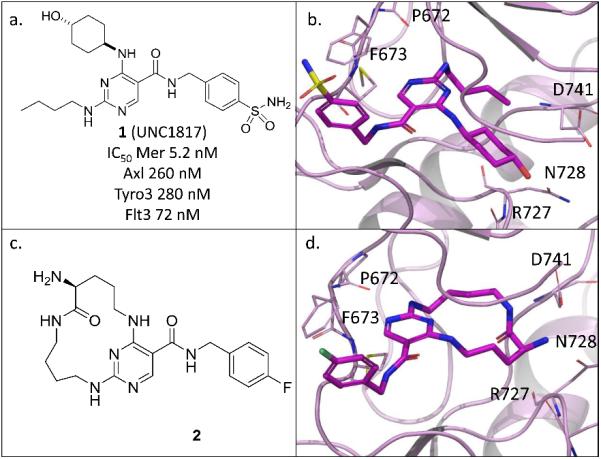
a. Structure of 1; b. X-ray crystal structure of 1 in complex with MerTK (kinase domain) (PDB ID code 4MHA); c. Structure of 2; d. Docking model of macrocyclic pyrimidine 2.
The syntheses of the designed macrocyclic compounds are straight forward. A general synthetic route is shown in Scheme 1 (see Supporting Information for details). Commercially available 2,4-dichloropyrimidine-5-carbonyl chloride reacted with an amine or alcohol to form the amide/ester I. In a one-pot reaction Boc protected amino acids with differing length alkyl chains and various diamines were attached to the pyrimidine core to provide intermediate II. The macrocycle was closed using an intramolecular amide coupling reaction, followed by cleavage of the Boc protecting group to yield the desired macrocycles III. To explore SAR at the R′ position, the starting acid chloride was first converted into an ethyl ester. After the formation of the desired macrocycle, the ethyl ester was hydrolyzed under basic conditions and the resulting free acid was converted to the final amide IV via an amide bond coupling reaction and cleavage of the Boc protecting group.
Scheme 1.
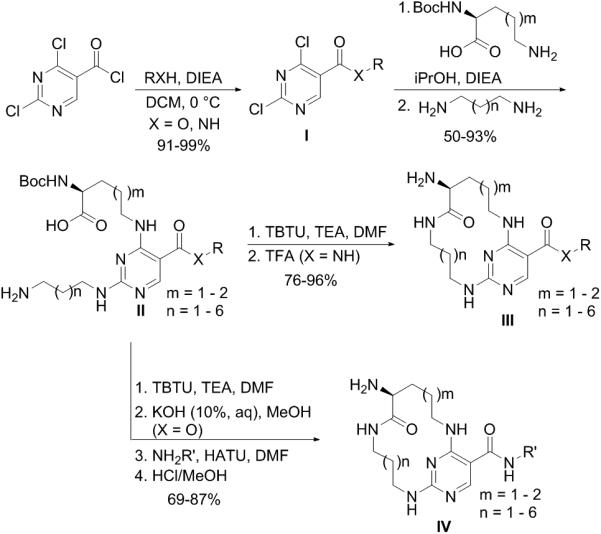
The synthetic route for macrocyclic compounds
Compound 2 was synthesized using the route presented in Scheme 1 and was tested using in-house microfluidic capillary electrophoresis (MCE) assays at the ATP Kms (details see supporting information Table S1).18-20 As shown in Table 1, compound 2 demonstrated exceptional selectivity over Flt3 (110-fold vs 14-fold for compound 1) along with weaker activity against MerTK (12-fold lower IC50 than compound 1). To improve the MerTK activity of compound 2, we initially explored the ring size of the macrocycle since this would simultaneously vary the position of the hydrogen-bond donor, the amino group, and the flexibility of the ring. As shown in Table 1, when m = 1, the inhibitory activity of macrocycles varied depending on the ring size. Compound 2 (n = 2) was 3-fold more potent than compound 3 (n = 1), however, compound 4 (n = 3) was 12-fold less active than compound 2. When n ≥ 4, the potency of the macrocycles was improved as the macrocyclic ring size was increased (compounds 4-7). The trend was also evident when m = 2. Compound 8 (n = 1) was a weak MerTK inhibitor with micromolar potency while compound 11 (UNC2541) (n = 4) was very potent with a low nanomolar IC50 against MerTK. The MerTK inhibitory activities of compound 9 (n = 2) and 10 (n = 3) were intermediate. When the macrocyclic ring became larger (n = 5 or 6), the MerTK activity of the corresponding analogues 12 and 13 was retained, however, the selectivity over Flt3 decreased (73-fold for 11 vs 59-fold for 12 & 19-fold for 13). In general, most of the analogues had excellent selectivity for MerTK over Flt3 and good selectivity over Axl and Tyro3. To evaluate the inhibitory activity of these compounds in a cell-based assay of tyrosine phosphorylated MerTK (pMerTK), we used a 384-well format enzyme-linked immunosorbent assay (ELISA),17 in which HEK293 cells are transfected with a cDNA encoding chimeric protein consisting of the extracellular and transmembrane domains from the epidermal growth factor receptor (EGFR) and the MerTK intracellular kinase domain. Activity is then stimulated with EGF. A chimeric protein was utilized because consistent stimulation of native MerTK kinase with the complex, natural ligand (GAS6 plus PtdSer) was not practical. Compound 11 proved to be the most active in this pMerTK ELISA assay with an EC50 510 nM. The 100-fold IC50 shift from the MCE assay to the pMerTK ELISA assay isn't unexpected due to the ATP concentration difference (5.0 μM in the MCE assay vs low millimolar range in the pMerTK ELISA assay).
Table 1.
Preliminary SAR of macrocycle ring size
| |||||||
|---|---|---|---|---|---|---|---|
| IC50 (nM)a | EC50 (nM)a | ||||||
| Compound | n | m | MerTK | Axl | Tyro3 | Flt3 | pMerTK ELISA |
| 2 | 2 | 1 | 61 ± 46 | 1600 ± 370 | 1800 ± 970 | 6700 ± 6300 | >1000 |
| 3 | 1 | 1 | 200 ± 140 | 21000 ± 7300 | 8200 ± 7000 | inactive | >1000 |
| 4 | 3 | 1 | 760 ± 330 | 28000 ± 2900 | inactive | inactive | >1000 |
| 5 | 4 | 1 | 230 ± 54 | 7800 ± 2800 | 8500 ± 5300 | 14000 ± 14000 | >1000 |
| 6 | 5 | 1 | 91 ± 28 | 2700 ± 1000 | 3400 ± 1500 | 10000 ± 1800 | >1000 |
| 7 | 6 | 1 | 62 ± 25 | 1100 ± 340 | 2700 ± 2000 | 2100 ± 560 | >1000 |
| 8 | 1 | 2 | 3200 ± 2500 | inactive | 24000 ± 3300 | inactive | >1000 |
| 9 | 2 | 2 | 140 ± 96 | 4000 ± 1400 | 6900 ± 5800 | inactive | >1000 |
| 10 | 3 | 2 | 36 ± 14 | 1700 ± 350 | 1600 ± 910 | 3200 ± 610 | >1000 |
| 11 | 4 | 2 | 4.4 ± 0.5 | 120 ± 60 | 220 ± 82 | 320 ± 11 | 510 ± 300 |
| 12 | 5 | 2 | 4.1 ± 0.2 | 130 ± 16 | 150 ± 4.4 | 240b | ND |
| 13 | 6 | 2 | 6.2 ± 3.3 | 170 ± 13 | 160 ± 110 | 120 ± 36 | 790 ± 110 |
Values are the mean of two or more independent assays ± SD.
Only test once.
With the optimal ring size and linker of macrocycle 11, the SAR at the R′ position was explored using the synthetic route presented in Scheme 1 (Table 2). Introducing a larger and basic dimethyl amine group in the para-position of the benzyl group at the R′ position didn't affect the activity or selectivity of the corresponding analogue 14 in either the MCE or pMerTK ELISA assays. However, 3,5-dimethoxyl benzyl group at the R′ position led to a 6-fold less active analogue 15. A para-substituted phenyl group at the same position (16–18) yielded analogues with dramatically diminished selectivity for MerTK over Flt3 although their MerTK enzymatic activities were similar to 11. No MerTK cellular activity was observed for these derivatives at a concentration of 1.0 μM in the pMerTK ELISA assay. When placing heterocycles such as 3-pyridyl and 4-pyridyl at the R′ position, the resulting analogues 19 and 20 had similar inhibitory enzymatic activity and selectivity for MerTK over Flt3 compared to 11, and again, no cellular activity was observed at a concentration of 1.0 μM. 2-Pyridyl at the same position led to a 5-fold less active analogue 21. However, when 3-pyridyl at the R′ site was further substituted at the para-position with 1-pyrazole, the resulting analogue 22 was 4-fold more active than the parental analogue 19 and exhibited improved Flt3 selectivity (193-fold for 22 vs 152-fold for 19) and cellular activity (IC50 420 nM). Saturated heterocycles such as 4-piperidine (23) and 4-tetrahydropyran (24) were also tolerated at this position. Analogues 23 and 24 were equally potent to 19 in either the MCE or pMerTK ELISA assays. Similarly, further substitution on the nitrogen of 4-piperidine with a benzyl group at the R′ position increased the activity of the corresponding analogue 25, especially in the pMerTK ELISA assay (IC50 250 nM).
Table 2.
SAR Study of the R′ position
| ||||||
|---|---|---|---|---|---|---|
| Compound | R′ | IC50 (nM)a | EC50 (nM)a | |||
| MerTK | Axl | Tyro3 | Flt3 | pMerTK ELISA | ||
| 14 |
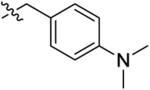
|
6.4 ± 1.3 | 180 ± 98 | 330 ± 110 | 740 ± 210 | 470 ± 79 |
| 15 |
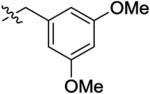
|
25 ± 21 | 500 ± 30 | 920 ± 470 | 5900 ± 470 | >1000 |
| 16 |
|
6.0 ± 2.8 | 100 ± 34 | 340 ± 210 | 49 ± 11 | >1000 |
| 17 |
|
9.5 ± 2.9 | 300 ± 130 | 800 ± 340 | 38 ± 43 | >1000 |
| 18 |
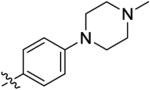
|
4.3 ± 1.1 | 83 ± 54 | 150 ± 27 | 22 ± 3.1 | >1000 |
| 19 |
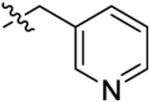
|
6.3 ± 3.3 | 330 ± 270 | 340 ± 230 | 960 ± 410 | >1000 |
| 20 |
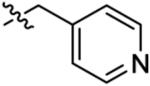
|
4.4 ± 4.1 | 110 ± 41 | 360 ± 230 | 780 ± 110 | >1000 |
| 21 |
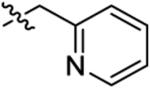
|
22 ± 16 | 540 ± 170 | 680 ± 470 | 890 ± 260 | >1000 |
| 22 |
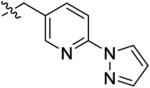
|
1.5 ± 0.4 | 72 ± 31 | 86 ± 2.1 | 290 ± 25 | 420b |
| 23 |
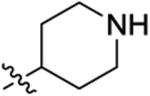
|
6.5 ± 2.0 | 150 ± 66 | 290 ± 220 | 850 ± 280 | >1000 |
| 24 |
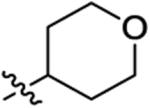
|
14 ± 8.5 | 300 ± 89 | 970 ± 680 | 860 ± 220 | >1000 |
| 25 |
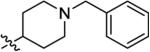
|
2.9 ± 1.9 | 50 ± 22 | 150 ± 200 | 730 ± 260 | 250 ± 28 |
Values are the mean of two or more independent assays ± SD.
Only test once.
The weak cellular activity of these analogues may be due to the polar α-amino amide linker. Therefore, we varied the linker of the macrocycle to explore if any other linker would improve the cellular activity (Table 3). When the primary amine was protected by a Boc group (26) or removed (27), the activity against MerTK was dramatically reduced (over 10-fold in the MCE assay compared to compound 11) due to lack of the key hydrogen bond with either D741 or R727 or N728. On the other hand, a similarly active compound 28 was obtained by substitution of the primary amine to yield a secondary amino group. Unfortunately, the cellular activity of 28 wasn't improved by the reducing the polarity and removing a hydrogen bond donor of the amino group because these changes decreased the MerTK inhibitory activity of 28 (3-fold) as well. In addition, replacement of the amide group in 11 with a less polar ester group such as in 29, preserved activity in either the MerTK MCE or pMerTK ELISA assays. Another attempt to reduce the polarity while retaining the hydrogen bond donor property of the α-amino amide linker was to replace it with a secondary hydroxyl group. Three analogues were prepared (30–32). The ring size and the position of the hydrogen bond donor of analogue 30 was the same as analogue 10. Both analogues had similar MerTK activity, however, the Flt3 selectivity of 30 was diminished from 89-fold (10) to 24-fold (30). The activity and selectivity were worse for analogue 31. Although the ring size and the position of the hydrogen donor of 31 was the same as 12, analogue 31 was 34-fold less active than 12 in the MerTK MCE assay and only had 5-fold selectivity over Flt3. Interestingly, analogue 32 had the same ring size and the same position of its hydrogen bond donor as 3, was 10-fold more active than 3 in the MCE assay. It also had weak activity in the pMerTK ELISA assay (IC50 600 nM). Linkers containing oxygen as the hydrogen bond acceptor such as epoxide (33) or ether (34 & 35), showed much lower MerTK inhibitory activity. More rigid macrocycles containing a phenyl ring resulted in analogues 36 and 37. Analogue 36 had the same-sized macrocycle with its hydrogen bond acceptor, an amino group, at the same position as 22. However, it was 27-fold less active than 22 in the MCE assay. Similarly, analogue 37 was 7-fold weaker than 25 with similar selectivity over Flt3. Overall, the amide linker was optimal in this series.
Table 3.
Exploration of different macrocycle linkers
| Compound | Structure | IC50 (nM)a | EC50 (nM)a | |||
|---|---|---|---|---|---|---|
| Mer | Axl | Tyro3 | Flt3 | pMerTK ELISA | ||
| 26 |
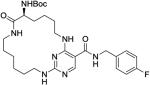
|
54 ± 2.7 | 1300 ± 190 | 2200 ± 350 | inactive | >1000 |
| 27b |
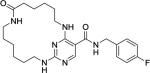
|
59 | 670 | 2300 | 4500 | 750 |
| 28 |
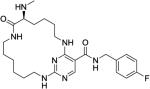
|
11 ± 2.7 | 210 ± 92 | 850 ± 350 | 840 ± 350 | 410 ± 140 |
| 29 |
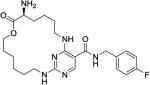
|
3.9 ± 0.1 | 220 ± 11 | 190 ± 49 | 65 ± 13 | 430 ± 42 |
| 30b |
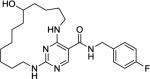
|
68 | 1600 | 1500 | 1600 | >1000 |
| 31b |
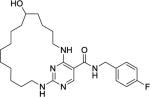
|
140 | 1300 | 2500 | 630 | >1000 |
| 32 |
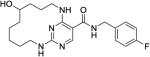
|
21b | 920b | 1100b | 900b | 600 ± 76 |
| 33 |
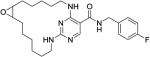
|
480 ± 150 | 4400 ± 1100 | 9200 ± 4100 | 3100 ± 1700 | ND |
| 34 |
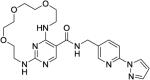
|
770 ± 600 | 20170±17020 | 17350 ± 15500 | 20080 ± 17190 | ND |
| 35 |
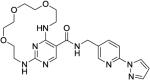
|
14000 ± 10000 | inactive | inactive | inactive | ND |
| 36 |
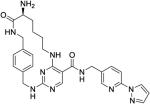
|
40 ± 13 | 300 ± 150 | 670 ± 470 | 9700 ± 6500 | >1000 |
| 37 |
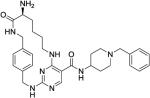
|
21 ± 7.0 | 590 ± 340 | 1200 ± 1500 | 15140 ± 21010 | >1000 |
Values are the mean of two or more independent assays ± SD.
Only test once.
To confirm our initial design strategy and gain better insight into ligand-protein interactions, we solved the X-ray structure of MerTK in complex with 11 at a resolution of 2.23 Å. Overall, the structure corroborated the preliminary docking hypothesis (Figure 1). However, there was one notable difference. In the crystal structure (Figure 2), both the carbonyl and primary amine groups of the macrocycle pointed outward with respect to the residues R727, N728 and D741, while we had hypothesized that the main interaction in this region would occur between only the primary amine and the above three residues. Our docking hypothesis was based on previous non-macrocyclic amino-pyridines, such as 1, which all needed either a hydroxyl or a primary amino group to gain potency through interactions with R727, N728 or D741.
Figure 2.

a. Structure of 11; b&C. X-ray crystal structure of 11 in complex with Mer (kinase domain) (PDB ID code 5K0X).
In addition, we used a panel of 30 kinases which emphasized tyrosine kinases along with selected serine/threonine kinases to assess the selectivity of promising compounds over other kinase families.8-9 Analogue 11 was evaluated in this panel at a concentration 100 fold above its Mer IC50 (Figure 3) (detailed in supplemental materials). Similar to its open-chained analogue (substituted pyrimidines),8 five tyrosine kinases were inhibited greater than 50% in the presence of 430 nM 11. However, three additional serine/threonine kinases were inhibited greater than 50% and were not inhibited by 1 under the same conditions.
Figure 3.
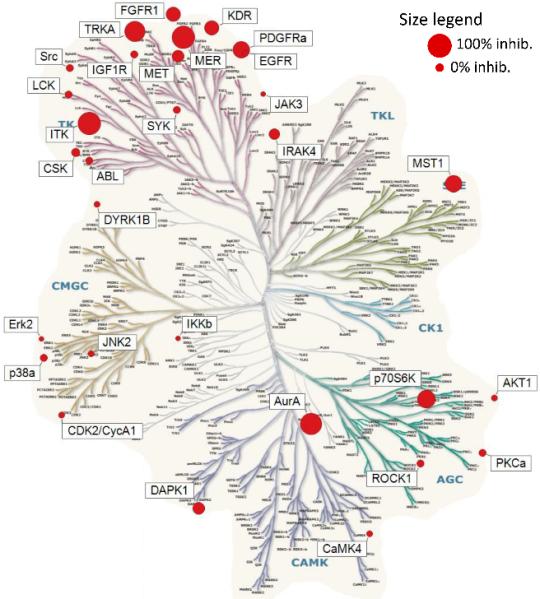
Kinase Tree
In summary, novel and potent macrocyclic pyrimidines have been discovered as MerTK kinase inhibitors via a structure-based drug design approach. The binding mode of these macrocycles was confirmed by an X-ray structure of MerTK protein complexed with analogue 11. A high-throughput ELISA assay was also employed to evaluate the inhibitory activity of the compounds in a cell-based assay of tyrosine phosphorylated MerTK. These macrocycles proved to be MerTK-selective within the TAM family and over Flt3. However, the selectivity over other kinase families and the cellular activity of these macrocycles need to be further improved to yield a lead compound worthy of further progression in this series.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Brenda Temple for her help in depositing the X-ray crystallographic structure to the Protein Data Bank (PDB).
Funding Sources
This work was supported by the University Cancer Research Fund and Federal Funds from the National Cancer institute, National Institute of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
ABBREVIATIONS
- TAM
Tyro3-Axl-Mer
- AML
acute myeloid leukemia
- MCE
microfluidic capillary electrophoresis
- IC50
half maximal inhibitory concentration
- pMerTK
phosphorylated Mer tyrosine kinase
- ELISA
enzyme-linked immunosorbent assay
- EC50
half maximal effective concentration
Footnotes
Author Contributions
This manuscript was written through contributions of all authors. / All authors have given approval to the final version of the manuscript. /
Supporting Information
Experimental details and characterization of all compounds and biological methods. The Supporting Information is available, in the online version, at
REFERENES
- 1.Giordanetto F, Kihlberg J. J Med Chem. 2014;57(2):278–295. doi: 10.1021/jm400887j. [DOI] [PubMed] [Google Scholar]
- 2.Rosenquist Å, Samuelsson B, Johansson P-O, Cummings MD, Lenz O, Raboisson P, Simmen K, Vendeville S, de Kock H, Nilsson M. J Med Chem. 2014;57:1673. doi: 10.1021/jm401507s. [DOI] [PubMed] [Google Scholar]
- 3.De Rosa M, Unge J, Motwani HV, Rosenquist Å, Vrang L, Wallberg H, Larhed M. J Med Chem. 2014;57:6444. doi: 10.1021/jm500434q. [DOI] [PubMed] [Google Scholar]
- 4.Heinis C. Nat Chem Biol. 2014;10:696–698. doi: 10.1038/nchembio.1605. [DOI] [PubMed] [Google Scholar]
- 5.Breslin HJ, Lane BM, Ott GR, Ghose AK, Angeles TS, Albom MS, Cheng M, Wan W, Haltiwanger RC, Wells-Knecht KJ, Dorsey BD. J Med Chem. 2012;55(1):449–464. doi: 10.1021/jm201333e. [DOI] [PubMed] [Google Scholar]
- 6.William AD, Lee ACH, Goh KC, Blanchard S, Poulsen A, Teo EL, Nagaraj H, Lee CP, Wang H, William s M, Sun ET, Hu C, Jayaraman R, Pasha MK, Ethirajulu K, Wood JM, Dymock BW. J Med Chem. 2012;55(1):169–196. doi: 10.1021/jm201112g. [DOI] [PubMed] [Google Scholar]
- 7.William AD, Lee ACH, Poulsen A, Goh KC, Madan B, Hart S, Tan E, Wang H, Nagaraj H, Chen D, Lee CP, Sun ET, Jayaraman R, Pasha MK, Ethirajulu K, Wood JM, Dymock BW. J Med Chem. 2012;55(6):2623–2640. doi: 10.1021/jm201454n. [DOI] [PubMed] [Google Scholar]
- 8.Graham DK, Dawson TL, Mullaney DL, Snodgrass HR, Earp HS. Cell Growth Differ. 1994;5(6):647–657. [PubMed] [Google Scholar]
- 9.Graham DK, DeRyckere D, Davies KD, Earp H. Cancer. 2014;14(12):769–785. doi: 10.1038/nrc3847. [DOI] [PubMed] [Google Scholar]
- 10.Liu J, Yang C, Simpson C, DeRyckere D, Van DA, Miley MJ, Kireev D, Norris-Drouin J, Sather S, Hunter D, Korboukh VK, Patel HS, Janzen WP, Machius M, Johnson GL, Earp HS, Graham DK, Frye SV, Wang X. ACS Med. Chem. Lett. 2012;3:129–134. doi: 10.1021/ml200239k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J, Zhang W, Stashko MA, Deryckere D, Cummings CT, Hunter D, Yang C, Jayakody CN, Cheng N, Simpson C, Norris-Drouin J, Sather S, Kireev D, Janzen WP, Earp HS, Graham DK, Frye SV, Wang X. Eur J Med Chem. 2013;65:83–93. doi: 10.1016/j.ejmech.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang W, McIver AL, Stashko MA, Deryckere D, Branchford BR, Hunter D, Kireev D, Miley MJ, Norris-Drouin J, Stewart WM, Lee M, Sather S, Zhou Y, Di Paola JA, Machius M, Janzen WP, Earp HS, Graham DK, Frye SV, Wang X. J Med Chem. 2013;56(23):9693–9700. doi: 10.1021/jm4013888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang W, Zhang D, Stashko MA, DeRyckere D, Hunter D, Kireev D, Miley MJ, Cummings C, Lee M, Norris-Drouin J, Stewart WM, Sather S, Zhou Y, Kirkpatrick G, Machius M, Janzen WP, Earp HS, Graham DK, Frye SV, Wang X. J Med Chem. 2013;56(23):9683–9692. doi: 10.1021/jm401387j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang W, DeRyckere D, Hunter D, Liu J, Stashko MA, Minson KA, Cummings CT, Lee M, Glaros TG, Newton DL, Sather S, Zhang D, Kireev D, Janzen WP, Earp HS, Graham DK, Frye SV, Wang X. J Med Chem. 2014;57(16):7031–7041. doi: 10.1021/jm500749d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor SJ, Dagger SA, Thien CB, Wikstrom ME, Langdon WY. Blood. 2012;120(19):4049–4057. doi: 10.1182/blood-2012-06-436675. [DOI] [PubMed] [Google Scholar]
- 16.Knapper S, Burnett AK, Littlewood T, Kell WJ, Agrawal S, Chopra R, Clark R, Levis MJ, Small D. Blood. 2006;108(10):3262–3270. doi: 10.1182/blood-2006-04-015560. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, Liu J, Zhang W, Stashko MA, Nichols J, Miley MJ, Norris-Drouin J, Chen Z, Machius M, DeRyckere D, Wood E, Graham DK, Earp HS, Kireev D, Frye SV. ACS Med Chem Lett. 2016;7(12):1044–1049. doi: 10.1021/acsmedchemlett.6b00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pommereau A, Pap E, Kannt A. J Biomol Screen. 2004;9(5):409–416. doi: 10.1177/1087057104264175. [DOI] [PubMed] [Google Scholar]
- 19.Dunne J, Reardon H, Trinh V, Li E, Farinas J. Assay Drug Dev Technol. 2004;2(2):121–129. doi: 10.1089/154065804323056468. [DOI] [PubMed] [Google Scholar]
- 20.Bernasconi P, Chen M, Galasinski S, Popa-Burke I, Bobasheva A, Coudurier L, Birkos S, Hallam R, Janzen WP. J Biomol Screen. 2007;12(7):972–982. doi: 10.1177/1087057107306759. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.