

SUMMARY
Many protein systems rely on coupled dynamic networks to allosterically regulate function. However, the broad conformational space sampled by non-coherently dynamic systems has precluded detailed analysis of their communication mechanisms. Here, we have developed a methodology that combines the high sensitivity afforded by nuclear magnetic resonance relaxation techniques and single-site multiple mutations, termed RASSMM, to identify two allosterically coupled dynamic networks within the non-coherently dynamic enzyme cyclophilin A. Using this methodology, we discovered two key hotspot residues, Val6 and Val29, that communicate through these networks, the mutation of which altered active-site dynamics, modulating enzymatic turnover of multiple substrates. Finally, we utilized molecular dynamics simulations to identify the mechanism by which one of these hotspots is coupled to the larger dynamic networks. These studies confirm a link between enzyme dynamics and the catalytic cycle of cyclophilin A and demonstrate how dynamic allostery may be engineered to tune enzyme function.
INTRODUCTION
Conformational fluctuations within proteins are essential for their function, which includes motions on the microsecond to millisecond timescale that are critical for ligand binding and release, in the conformational search for the transition state in enzymatic reactions, and in allosteric communication (Bhabha et al., 2011; Carroll et al., 2012; Cole and Loria, 2002; Henzler-Wildman et al., 2007; Kern et al., 2005; McElheny et al., 2005; Xiao et al., 2014). With recent methodological advances, a growing number of studies have now also recognized that these motions often comprise contributions from non-coherent segments that are nonetheless coupled to form larger dynamic networks (Khirich and Loria, 2015; Kleckner et al., 2012; McDonald et al., 2012; Xiao et al., 2014). A range of approaches has been applied to elucidate the pathways through which allosteric communications propagate (Cole and Loria, 2002; Doucet et al., 2009; Popovych et al., 2006; Selvaratnam et al., 2011; van den Bedem et al., 2013). For example, nuclear magnetic resonance (NMR)-based techniques, such as chemical shift covariance analysis, provide an elegant means to identify coupled networks using multiple ligands together with their induced chemical shift changes. However, the inherently broad conformational space sampled by non-coherently coupled dynamic systems often results in small changes to chemical shifts that may be below the detection limit and has limited our ability to identify allosteric networks in some systems. Moreover, such chemical shift-based methods offer little insight into the potential underlying dynamic basis of allostery. To address these limitations, we have developed a novel but simple methodology that affords the high sensitivity of NMR relaxation techniques and single-site multiple mutations, referred to as RASSMM, which expands upon previous studies that have combined mutagenesis with NMR metrics as a means to monitor allostery (Boyer and Lee, 2008; Du et al., 2015). Even in the absence of large chemical shift changes, we show that this method can monitor dynamic changes distal to an enzyme’s binding site and identify networks of dynamic, allosteric communication that can be exploited to tune enzyme function.
The RASSMM approach is depicted in Figure 1. It first utilizes a sensitive NMR relaxation parameter to monitor the dynamic impacts of active-site perturbations, such as mutagenesis or substrate binding, in order to identify distal dynamic regions (hotspots) that are coupled to active-site dynamics. Subsequently, multiple mutations are made to these hotspots themselves and subjected to the same NMR relaxation experiment in order to identify how these distal regions are allosterically coupled to the active site. Recent advances in NMR relaxation techniques have provided powerful methodologies to probe the conformational landscape of proteins, which allows us to choose the particular relaxation experiment that adequately suits the dynamics of any particular system. Here, we have selected R2 relaxation dispersion (R2-RD), which allows for atomic-resolution, quantitative measurement of low-population states that are dynamically sampled with an exchange rate of ~100–5,000 s−1 (Carr and Purcell, 1954; Meiboom and Gill, 1958), as our NMR relaxation measure for RASSMM to probe dynamic allostery within the enzyme cyclophilin A (CypA).
Figure 1. Schematic of Our Experimental Approach.
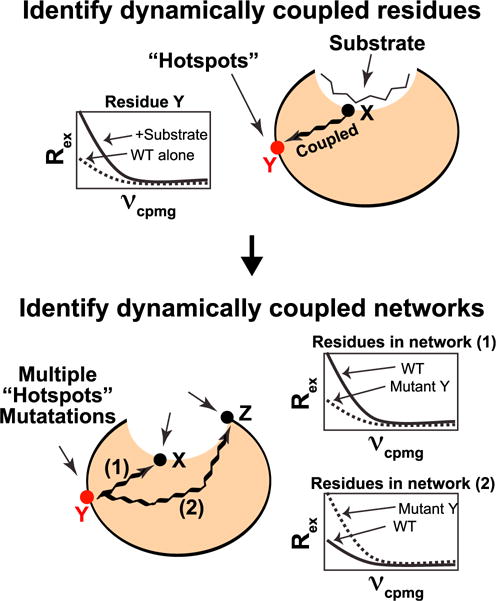
Top, distal hotspot residues are identified by comparing R2-RD dispersions between apo CypA and CypA in the presence of saturating concentrations of a peptide substrate. For residues that are dynamically linked through allosteric networks to the active site, we expect that, upon addition of substrate, the dynamics of distal hotspot residues will be altered, which will manifest in measurable changes to the R2-RD dispersion. Bottom, identifying networks of allosteric communication. We generate multiple single-site mutations to an identified hotspot and monitor global dynamic response to these mutations by R2-RD. We expect that residues along a given allosteric network will exhibit a similar response signature, allowing distinct networks to be identified.
CypA is a peptidyl-prolyl isomerase (PPIase) with a wide range of biologically and pathologically relevant roles (Fischer et al., 1989; Gothel and Marahiel, 1999; Jin et al., 2004; Kim et al., 2005; Lee and Kim, 2010), which we have found represents a compelling example of a dynamically coupled allosteric system. Utilizing deuteration, data collection at multiple field strengths, and temperature variation, we have recently shown that micro- to millisecond dynamics in apo CypA are more prevalent and segmental than originally observed (Eisenmesser et al., 2005; Schlegel et al., 2009). Specifically, R2-RD experiments have revealed that nearly 50% of residues within CypA exhibit micro- to millisecond timescale dynamics and that they comprise rates of motion spanning nearly an order of magnitude. More recently, the segmental nature of dynamics in CypA has also been independently confirmed by variable-temperature X-ray crystallography (Keedy et al., 2015) and we have shown that distinct segmental dynamics persist in solution in the presence of saturating concentrations of substrate (Holliday et al., 2015a). Collectively, these studies reveal that significant global communication occurs between non-coherent regions such that mutagenesis or substrate binding affects motions across the protein at sites reporting on distinct structural transitions. However, it remains to be determined how these localized motions are organized to mediate function and whether they can be exploited to engineer catalytic turnover. Here, we have used the RASSMM approach to identify residues within CypA that are remotely coupled to active-site dynamics, characterized dynamically coupled allosteric networks, determined mechanistically how these networks fine-tune substrate binding/release, utilized this knowledge to modulate CypA turnover, and provided insight into the mechanism of hotspot coupling to these networks via analysis of chemical shift-based molecular dynamics (MD) ensembles.
RESULTS
An Active-Site Distal Residue Allosterically Linked to Active-Site Dynamics
To determine the role of dynamic allostery in regulating CypA function, we developed an approach to identify active-site distal hotspots in the protein by examining the impact of substrate addition on R2-RD-monitored dynamics at distal sites. We reasoned that if we could identify sites distal to the substrate binding site that were dynamically coupled, we would be able to modify these distal sites in order to modulate active-site dynamics without perturbing the ground-state structure of the substrate interface, permitting examination of the role of long-range dynamically coupled networks in enzymatic function (see experimental schemes, Figure 1). To identify such sites in CypA, we first collected R2-RD data on CypA alone and during isomerization of a peptide substrate (GSFGPDLRAGD, herein referred to as the FGP peptide). The R2-RD experiment consists of a series of refocusing pulses with frequency νcpmg; line broadening due to chemical exchange-induced relaxation (Rex) on the micro- to millisecond timescale is refocused by these pulses with a signature dependent upon the rate of exchange (kex), the minor state population (PB), and the chemical shift difference (Δω) between the states. As shown in Figure 2A, many residues within the binding site exhibit altered Rex values during isomerization relative to apo CypA, as would be expected due to changes in both the local dynamics and the local chemical environment. Because the non-coherent nature of CypA dynamics in both the apo and isomerizing states prevents simultaneous fitting of multiple residues and because FGP-bound CypA dynamics are potentially influenced by both catalysis and the binding/release cycle (Holliday et al., 2015a), we hesitate to directly interpret these Rex changes mechanistically. However, compared with the large number of residues with altered Rex proximal to the substrate, we detect an increase in Rex for only two residues distal to the active site, Val29 and Val6 (Figure 2A). These putative hotspots are thus utilized within this study to map mechanisms of allosteric coupling and to determine their influence on dynamics in regulating CypA function.
Figure 2. Localized Dynamics Are Allosterically Coupled throughout CypA.
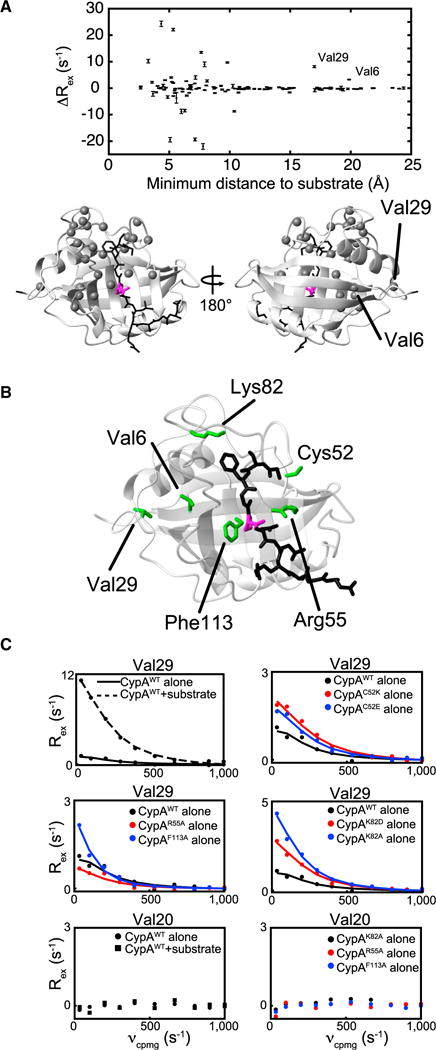
(A) Rex changes measured at 900 MHz upon FGP substrate binding for all residues for which data is available in both the free and bound states, plotted as a function of the minimum distance between a given residue’s amide nitrogen and any heavy atom in FGP in a previously generated MD ensemble (Camilloni et al., 2014). Rex values shown are the fit values to the Carver-Richards equations for the slowest refocusing frequency (33 s−1). Bars represent error propagation of SDs of the residuals of R2-RD data to the Carver-Richards fit calculated for each residue individually. See Figure S2 for Rex values of CypAWT in the apo and bound state on a per-residue basis. A representative CypA structure is shown below with the FGP peptide (black) and the isomerized proline (magenta). Residues exhibiting Rex changes of greater than 2 s−1 upon FGP substrate binding are shown as gray spheres.
(B) CypA structure bound to FGP with mutated residues in green.
(C) R2-RD data (dots) collected at 900 MHz for CypAWT (black, solid lines), CypA mutants (red or blue), and CypAWT in the presence of a saturating concentration (6 mM) of FGP (black, dotted line), as measured for the amide nitrogen of Val29 or Val20. Lines are determined from single residue fitting to the Carver-Richards equation (Carver and Richards, 1972).
Focusing first on Val29, which is >15 Å from the substrate and exhibits the larger Rex change of the two distal hotspots, we observed a dramatic increase in Rex upon addition of the FGP substrate, corresponding to an increased population of a minor conformer (discussed further below) and indicating a link to active-site dynamics. We likewise mutated several residues within the active site which are involved in direct substrate interactions (R55 and F113) or substrate gatekeeping (K82) and have been previously demonstrated to participate in active-site dynamic interactions (Davis et al., 2010; Eisenmesser et al., 2005; Fraser et al., 2009); in addition, we introduced mutations to C52, which interacts with and we predicted would affect the dynamic active-site loop comprising residues 165–175. For each of these mutations, we observed an altered Val29 R2-RD signature (Figures 2B and 2C), further illustrating an allosteric link to the active site in both the isomerizing and apo forms of CypA. As a comparison, Val20, which is representative of the majority of distal residues unaffected by changes in active-site dynamics, exhibits no increase in Rex in response to either substrate addition or active-site mutation. Our identification of Val29 here as a hotspot has recently prompted us to examine this residue by standard MD simulations and functional studies, which revealed that the conformational distributions within the active site are altered upon introducing a single mutation to Val29 (V29L) with a slower rate of catalytic turnover monitored experimentally (Doshi et al., 2016). However, the underlying communication network mediating this allostery remained unclear. The use of R2-RD along with active-site perturbations via either substrate binding or active-site mutations have thus identified residues distal to the CypA active site, which represents the first step in our RASSMM approach (Figure 1).
Single-Site Multiple Mutations to a Hotspot Residue Identifies Pathways of Dynamic Allostery
To determine the pathway(s) by which the Val29 hotspot allosterically communicates with the active site, we utilized the RASSMM strategy of generating multiple mutations to a single hotspot in order to discern the networks through which dynamic allostery is propagated (Figure 1). We expected that residues communicating along a non-coherent network would nonetheless exhibit similar responses to a given mutation.
We mutated Val29 to numerous charged, polar, and hydrophobic residues and found that only three other amino acids that were tolerated at this site, CypAV29A, CypAV29T, and CypAV29L, with all other mutants tested yielding insoluble proteins. CypAV29A, CypAV29T, and CypAV29L, however, are stable, with no global perturbations to the overall structure of the enzyme (Figure S1). As shown in Figures 3 and S2, these mutations alter micro- to millisecond dynamics for nearly every residue that exhibits chemical exchange in the enzyme, consistent with the involvement of Val29 in dynamic allostery within CypA. Furthermore, the pattern of change in Rex falls into two distinct groupings: one for which the alanine mutation has no effect, but CypAV29T and CypAV29L exhibit comparable increases in Rex over CypAWT (Figures 3 and S2, response 1), and a second group for which CypAV29A and CypAV29T exhibit comparable small increases in Rex over CypAWT, while CypAV29L induces a much larger increase over CypAWT (Figure 3 and S2, response 2). Strikingly, the patterns are sharply delineated within the CypA structure, as exemplified by the distinct patterns exhibited by adjacent residues Phe112 and Phe113 within the active site. By mapping residues exhibiting each response pattern onto the CypA structure (Figure 3), two clear groupings emerge that correspond to distinct networks of communication. In contrast to the clearly delineated responses observed by R2-RD, we observe minimal changes in 15N chemical shifts for most of the residues within the two pathways and no clear response pattern among the Val29 mutants (Figure S2), highlighting the improved sensitivity of our approach.
Figure 3. Multiple Mutations of the CypA Val29 Hotspot Monitored via R2-RD Identify Allosterically Coupled Dynamic Networks.
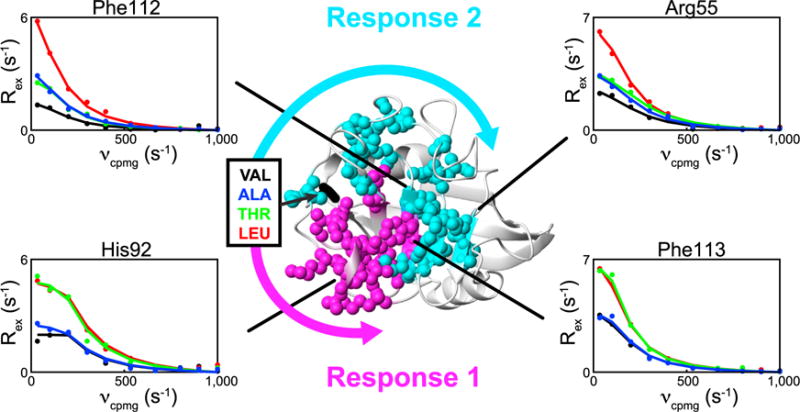
R2-RD data collected at 900 MHz (dots) and individual fits (lines) for CypAWT (black), CypAV29T (green), CypAV29A (blue), and CypAV29L (red). Two response patterns, labeled response 1 and response 2, were identified based on distinct patterns of Rex responses among the three mutants: response 1 residues exhibit near equal and elevated responses in CypAV29T and CypAV29L, with little effect in CypAV29A. Response 2 residues exhibit modest increases in Rex in CypAV29A and CypAV29T, with a much larger increase in CypAV29L. Two representative residues are shown for each of the two response patterns. All residues exhibiting response 1 (magenta spheres) and response 2 (cyan spheres) are plotted onto the CypA structure. Arrows represent putative communication pathways through which mutation to Val29 affects the active site. Only residues for which data were available for CypAWT and all mutants are included. See Figure S1 for the impact of mutation on CypA chemical shifts and Figure S2 for the impact of mutation on Rex values and 15N chemical shifts for all residues within response 1 and response 2.
Changes in Rex, due to mutation or substrate binding, can in principle result from the alteration of any of three different properties of the exchange: Δω, the chemical shift difference between states; kex, the exchange rate constant; and PB, the minor state population occupancies. For residues that may be sampling more than two states, the specific dependence of Rex is more complex, but remains affected by the equivalent parameters. Because of the non-coherent nature of the exchange of CypA, determination of the specific property or properties affected at a given site is not generally possible. However, given the conserved response of Rex across residues within responses 1 and 2 in Figure 3, we can eliminate Δω and kex changes as being the predominant parameter altered across the responses. Specifically, Rex increases with an increase in Δω, and so to observe the response-wide increases in Rex shown in Figure 3, Δω would have to increase for all residues in the response, which is highly unlikely given the overall conservation of the major state (Figure S1). Likewise, Rex is maximal when kex and Δω are equal (Millet et al., 2000), such that across a given non-coherent response, residues exist with kex < Δω and kex > Δω and a response-wide increase or decrease in kex would result in disparate impacts on Rex across a response. Thus, given the uniform response signature across each response in Figure 3, we interpret the Rex increases as being predominantly mediated by increasing minor state occupancy across the responses, with additional relatively minor and residue-specific impacts from alterations to kex or Δω.
Multiple Mutations to a Second Hotspot Identifies Consistent Allosteric Networks
In addition to Val29, Val6 was also identified as the second distal hotspot (Figure 2A) that exhibits increased chemical exchange upon addition of substrate (Figure 4A), thereby providing an additional residue to probe the allosteric connection to the active site within CypA. Unlike Val29, no observed changes appear in the R2-RD profiles of Val6 upon mutation to active-site residues in the apo form, suggesting that the allosteric link to the active site in CypAWT exists only in the presence of the FGP substrate. Nonetheless, we applied our single-site multiple mutation approach in accord with RASSMM to this second hotspot to determine if networks connecting this site to the active site could be identified. As with Val29, only structurally conservative mutations were tolerated for Val6 (CypAV6A, CypAV6I, CypAV6S, CypAV6T, and CypAV6L), and those with significant impacts on isomerization, CypAV6S, CypAV6T, and CypAV6L (see Table 1) were utilized for dynamic studies. Mutations to Val6 do not lead to clearly delineated Rex signatures as are seen for Val29 in Figure 3, preventing us from identifying the specific pathways by which Val6 communicates with the active site. The structural basis for this discrepancy remains unclear, but may result from Val6 mutations impinging upon the active site through multiple interacting pathways that interfere with one another in unpredictable ways. Nonetheless, by identifying those residues for which one or more mutations at Val6 alters Rex-monitored dynamics we converge on a comparable group of residues as are identified in response 2, providing support for the distinct dynamic networks identified in Figure 3.
Figure 4. Multiple Mutations of the Val6 Hotspot Monitored via R2-RD Reveal Its Connection to the Dynamic Networks of CypA.
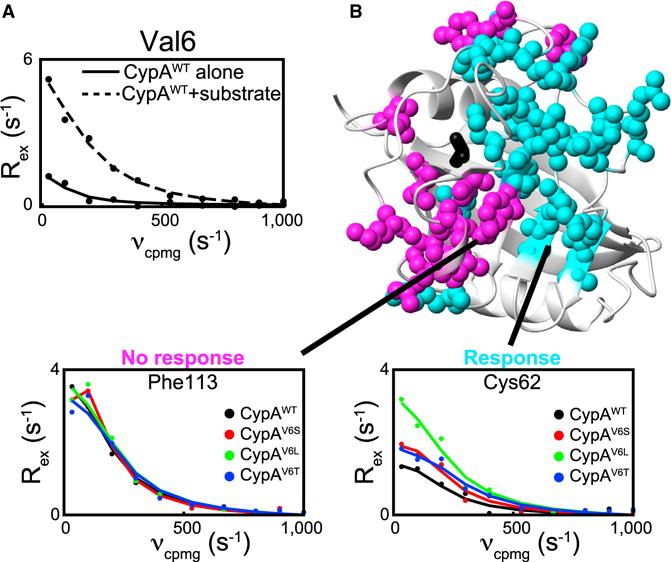
(A) R2-RD data collected for CypAWT alone (solid line) or in the presence of 6 mM FGP (dashed line), measured at the amide nitrogen of Val6.
(B) Representative R2-RD data for CypAWT (black), CypAV6L (green), CypAV6T (blue), and CypAV6S (red), plotted as in Figure 3. Residues for which dynamics are altered (cyan) or not altered (magenta) in the V6 mutants are shown with their side chains as spheres. See Figure S3 for complete Rex values among residues responsive and non-responsive to mutation at Val6. Only residues for which data were available for CypAWT and all mutants are included.
Table 1.
Impact of Active-Site Distal Mutations on FGP Binding Affinity and Isomerization Rates
| Enzyme | KD-app (μM)a | kiso(s−1)b |
|---|---|---|
| WT | 76 ± 3c | 10.4 ± 0.3c |
| V29A | 57 ± 2 | 8.9 ± 0.2 |
| V29T | 73 ± 3 | 12.3 ± 1.1 |
| V29L | 38 ± 2c | 6.9 ± 0.3 |
| V6A | 82 ± 5 | 10.1 ± 0.2 |
| V6I | 86 ± 4 | 10.2 ± 0.1 |
| V6S | 65 ± 3 | 8.9 ± 0.2 |
| V6T | 48 ± 2 | 9.1 ± 0.1 |
| V6L | 69 ± 3 | 9.4 ± 0.1 |
| V6TV29L | 37 ± 3 | 6.1 ± 0.2 |
See Table S1 for select ITC binding data and Figure S4 for representative titration spectra, fitted titration data, raw ITC data, and fitted ITC data. WT, wild-type.
See Figure S3 and Table S1 for equivalent binding and catalytic data with the hemagglutinin peptide.
Errors in KD-app are in fits to a single experiment.
Errors in kiso are SDs of two or more independent experiments.
Published previously by Doshi et al. (2016) and Holliday et al. (2015b).
In addition, through combined mutation of Val6 and Val29, we found that the influences of the two mutated hotspots interfere both constructively and destructively, such that the Val6 mutation can complement or short-circuit the influence of Val29 in different regions of the active site (Figure S3). These findings highlight the potential to engineer specific dynamic motions within an enzyme active site by modulating allosterically, coupled dynamic networks at distant sites with functional consequences, as described below.
Tuning CypA Catalytic Function through Dynamic Networks
The distinct advantage of altering active-site dynamics through distal hotspots is that the impact of dynamics on function can be assessed without directly disturbing the enzyme/substrate interface. Specifically, we monitored the functional effects of perturbing active-site dynamics by assaying CypAWT and each of the distal hotspot mutants for both binding and isomerization activity toward multiple substrates.
Binding was assayed via titration of the FGP substrate into 15N-labeled CypA and mutants, which provides an apparent dissociation constant (KD-app) that includes contributions from binding to both the trans and cis isoforms of the substrate (Holliday et al., 2015b). Single mutation of CypA hotspots induced small, but significant, changes in binding affinity for FGP, inducing up to a 50% change in KD-app compared with CypAWT (Table 1). The apparent binding affinity of CypAWT and Val29 mutants toward FGP were additionally measured via isothermal titration calorimetry (ITC). When utilizing ITC, we systematically measured binding affinities (KD-app-ITC) of about 2-fold weaker than by NMR titration, likely due the two-species nature of the substrate and the different physical parameters being measured (i.e., the heat given off by the bulk combination of cis and trans isoform binding as measured by ITC does not directly correspond to the fraction of bound CypA as measured by NMR). Nonetheless, KD-app-ITC values indicate a similar relative change in affinity for the substrate as when measured by NMR titration (Table S1 and Figure S4).
CypA-mediated isomerization was monitored by ZZ exchange as described previously (Farrow et al., 1994; Holliday et al., 2015b). In brief, ZZ exchange spectra were collected for 15N-labeled FGP upon addition of catalytic concentrations of unlabeled enzyme. This experiment provides an observed isomerization rate, kiso, where kiso = kcis⟶trans + ktrans⟶cis. Single mutation of CypA hotspots caused a 35% decrease to 20% increase in isomerization (Figure 5A and Table 1), demonstrating a role for active-site dynamics in modulating CypA catalysis. Notably, the double mutants CypAV6TV29L and CypAV6TV29T exhibit combinatorial effects on isomerization rates, highlighting the promise of rationally engineering enzyme function through combinations of mutants that alter active-site dynamics through allosteric communication.
Figure 5. Engineering CypA Function through Shifting Conformational Sampling in the Active Site.
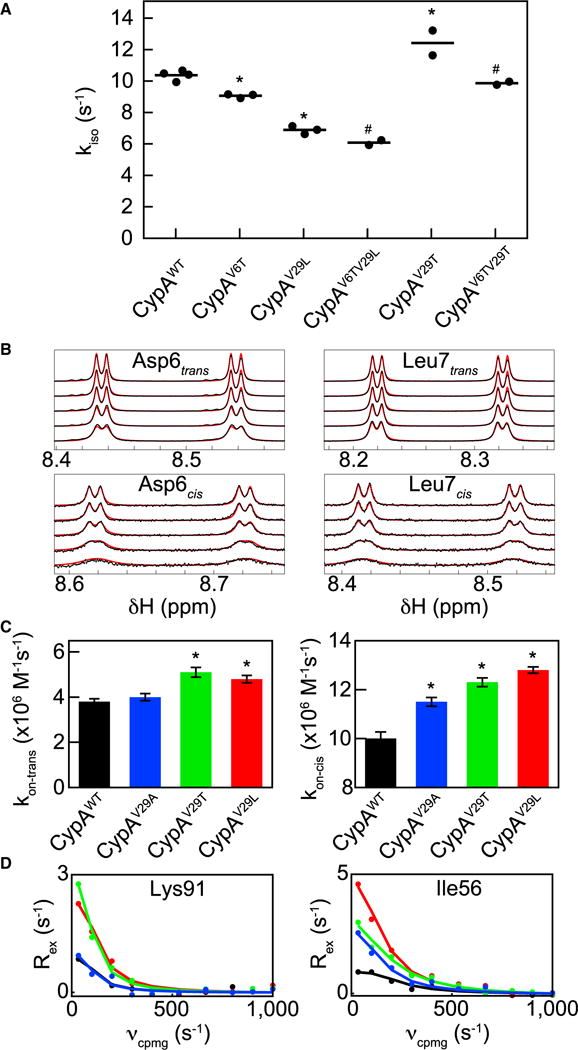
(A) Isomerization rate, kiso, monitored for CypAWT; single mutants CypAV6T, CypAV29L, and CypAV29T; and double mutants CypAV6TV29L and CypAV6TV29T. Dots represent independent experiments with lines at the mean value. By two-sided t test, *p < 0.05 compared with CypAWT, #p < 0.05 compared with CypAV29L (CypAV6TV29L), or CypAV6T (CypAV6TV29T). kiso values for CypAWT and CypAV29L have been published previously (Doshi et al., 2016; Holliday et al., 2015b). See Figure S3 for the dynamic impacts of CypAV6TV29L.
(B) Representative lineshape data that are slices extracted from high-resolution 15N-HSQC spectra (black dots) and fits (red lines) for the cis and trans isoforms of residues Asp6 and Leu7 of 15N-FGP with CypAV29L. For clarity, data are only plotted for 1 mM 15N-FGP and 5, 10, 20, 50, and 100 μM unlabeled CypAV29L (top to bottom for each isoform of each residue). Peak-splitting results in four peaks per residue per isoform as described in Experimental Procedures. For visualization purposes, cis peaks intensities are magnified 5-fold compared with trans peaks. See Figure S6 for full lineshape fitting results.
(C) On rates for the cis and trans peptide isoforms as determined by lineshape analysis for CypAWT and Val29 mutants. Error bars represent SEs, by two-sided t test, *p < 0.05 compared with CypAWT.
(D) Examples of residues exhibiting response 1 (left) and response 2 (right) to Val29 mutants, plotted as in Figure 3. See Figure S4 for representative ZZ exchange spectra and fitted data and Figure S7 for isomer specific root-mean-square deviation comparisons from MD ensembles.
To determine the degree to which the observed functional effects are substrate specific, we identified another peptide substrate with little sequence similarity to the FGP peptide, the hemagglutinin (HA) tag sequence YPTDVPDYA, originally identified in influenza HA (Green et al., 1982). Although previous work has shown that host CypA acts as a restriction factor for influenza infection through interactions with the viral M1 protein (Liu et al., 2009), our discovery here, which has detailed one of the highest CypA affinity interactions for a substrate, reveals another potential role of CypA in influenza infection that may warrant future studies (Figure S5). Since Pro2 of the HA peptide is not isomerized by CypA, we generated a P2A mutant (HAP2A) which exhibits comparable affinity with CypA as the standard HA peptide, yet simplifies the spectrum sufficiently to allow collection of Pro6 isomerization data (Figure S5). As shown in Table S2, KD-app and kiso are affected in a generally similar manner by CypA hotspot mutations for the HAP2A substrate as for the FGP substrate, indicating that the CypA mutations influence enzyme function generally, and not in a manner limited to a single substrate. However, some variation does exist between the functional effects on each substrate, most notably in the catalytic activity of CypAV29T, which increases kiso for the FGP substrate, but decreases it for the HAP2A substrate, and in the minimal impact of the V6T mutations on the binding affinity toward the HAP2A substrate. This variability indicates that, while altering conformational sampling does influence enzymatic function in CypA toward multiple substrates, the specific outcomes are somewhat substrate specific, likely due to differences in the substrate/active-site interactions within each enzyme/substrate complex. While previous correlative measures have hinted at a role for micro- to millisecond dynamics in the catalytic cycle of CypA (Eisenmesser et al., 2005; Fraser et al., 2009; Holliday et al., 2015a), here we have shown a direct connection between the dynamic networks of CypA identified via our RASSMM approach and catalysis of multiple substrates.
Distal Hotspot Mutants Impact Conformational Selection in CypA
Although the RASSMM approach allowed us to identify dynamic networks within CypA and alter CypA catalytic function via mutations to key hotspots, the underlying mechanism modulated by these mutations remained unknown. The minimal catalytic cycle of PPIases consists of six microscopic rate constants defining the on and off rates for each isomer, as well as the two rate constants defining the on-enzyme interconversion (Figure S6). Lineshape analysis has provided a powerful means to elucidate these rate constants, which are not readily accessible by classic enzymatic assays (De et al., 2012; Greenwood et al., 2011; Holliday et al., 2015a; Kern et al., 1995). Thus, to determine the specific impact that perturbations to the dynamic networks of CypA have on the catalytic cycle, we collected high-resolution 15N-heteronuclear single quantum coherence (HSQC) spectra of 15N-FGP in solution with varying concentrations of CypAWT or mutants (representative example shown in Figure 5B).
An examination of the microscopic rate constants that define the full catalytic cycle mediated by CypA reveals that on-enzyme isomerization rates remain relatively unaffected when the hotspots are mutated (Figure S6). In contrast, as shown in Figure 5C, lineshape analysis demonstrates that both CypAV29T and CypAV29L exhibit an increase in the trans peptide on rate and that CypAV29A, CypAV29T, and CypAV29L exhibit an increase in cis peptide on rate.
These increases in on rates monitored through lineshape analysis qualitatively parallel the observed increases in Rex, which predominantly report on the sampled minor conformations, as described previously (Figures 3, 5D, and S2). Residues within the dynamic network defined by response 1 mirror the changes for kon-trans and residues within the dynamic network of response 2 mirror the changes for kon-cis. Consistent with this finding, in chemical shift-restrained MD simulations of the CypA:FGP complex (Camilloni et al., 2014), both protein and peptide root-mean-square deviations indicate isomer-specific, localized constraints in mobility. Specifically, the N terminus of the peptide, which interacts predominantly with residues in response 2, is more constrained for the cis peptide, while the C terminus, which interacts predominantly with residues in response 1, is more constrained for the trans peptide (Figure S7). Mutations that differentially affect conformational sampling with responses 1 and 2 would thus be expected to differentially affect binding to the cis and trans substrate isoforms.
These findings suggest that mutation of the CypA hotspots has altered catalytic function via shifting the inherent conformational sampling of the CypA active site. Lineshape analysis indicates that the V29L mutation additionally imparts a decrease in the cis peptide off rate alone both alone and in the context of the V6T mutation (Figure S6), implicating conformational fluctuations as potentially relevant to substrate release in CypA as well as in substrate binding. Collectively, these data reveal that modulations imparted by mutations to active-site distal hotspots within CypA serve to fine-tune active-site dynamics to regulate substrate binding and release.
Chemical-Shift-Restrained MD Ensembles Identify Coupling to CypA Dynamic Networks
Given the surprisingly distinct networks identified via conservative mutation to Val29 (Figure 3), we sought an atomistic view to further understand how Val29 is coupled to each of these networks. We have previously demonstrated that chemical shifts can be utilized as replica-averaged structural restraints that can accurately recapitulate the free-energy landscape of a dynamic protein (Camilloni et al., 2012) and we have previously applied this approach to CypA as a means to probe the catalytic mechanism (Camilloni et al., 2014). Moreover, this ensemble method also accurately recapitulates observed flexibility in CypA, as shown in Figure S7. Thus, here we utilized chemical-shift restrained MD ensembles to determine the interactions through which Val29 is linked to the dynamic networks of CypA.
First, we determined whether the chemical-shift-restrained MD simulations qualitatively corroborated the existence of chemical exchange measured by Val29 (Figure 2A). Specifically, we back-calculated the chemical shifts of the Val29 amide nitrogen in free CypA by applying the SPARTA+ (Shen and Bax, 2010) method of empirical chemical shift prediction to every structure within the ensemble. As shown in Figure 6A, we were clearly able to identify two distinct chemical environments experienced by the Val29 amide that correspond to rotation of its side chain about the χ1 angle.
Figure 6. Mapping Dynamic Interactions through Chemical-Shift Restrained MD Ensembles.
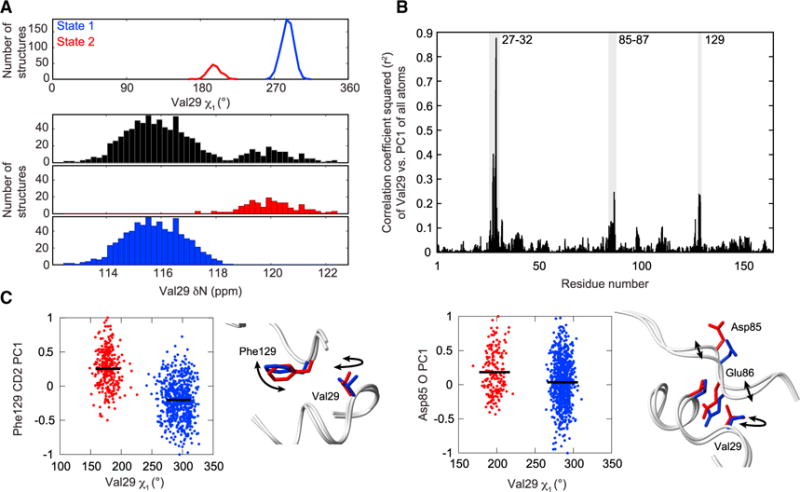
(A) Top, Val29 samples two rotameric states. Bottom, the predicted chemical shift of the Val29 amide nitrogen over all structures (black), structures corresponding to Val29 χ1 state 1 (blue), and structures corresponding to Val29 χ1 state 2 (red).
(B) Maximum squared correlation coefficient for Val29 χ1, Cγ1 PC1, or Cγ2 PC1 compared with the PC1 values for all other heavy atoms in CypA. Highlighted peaks (gray) correspond to residues that are partially coupled to Val29.
(C) The Val29 χ1 angle is plotted against the PC1 value of representative atoms within the two coupled clusters. Means are shown as solid lines (black), with structures grouped and colored based on k-means clustering of the Val29 χ1 angle. Representative structures from each cluster are shown next to each correlation graph and colored based on the cluster in which they fall. See Figure S7 for a comparison of experimental and MD simulated global dynamics.
Second, we identified how this rotation of Val29 is coupled to conformational changes in adjacent residues that link Val29 to the larger dynamic networks. To identify correlations between the spatial localization of different residues within CypA over the ensemble, we mapped the 3×N position matrix (i.e., x, y, and z positions for all N ensemble structures) of each atom to a single normalized spatial variable (1×N vector) using principle component analysis (described further in Supplemental Experimental Procedures). This single spatial variable, the first principle component (PC1), compresses the data along the axis of maximum spatial variance and thus provides a single variable that can then be straightforwardly compared between any two atoms by linear least-squares analysis. PC1 values can likewise be directly compared with dihedral angles, allowing identification of correlations between side-chain rotameric states and atomic localizations between residues. We applied this analysis to Val29 and identified direct conformational coupling of Val29 to other residues in the protein (Figure 6B). Two distinct clusters, distant in sequence yet proximal in tertiary structure, comprising residue 129 and residues 85–87, exhibit elevated r2 values when calculated against Val29, indicating a direct conformation coupling between each cluster and Val29 (Figure 6C). As shown in Figure 6C, both Phe129 and Asp85 sample a continuum of states in the MD simulations, suggestive of conformational coupling between micro- to millisecond timescale motions of Val29 and faster motions than those monitored by R2-RD at Phe129 and Asp85; correspondingly, we observe minimal exchange-induced relaxation in CypAWT or mutants directly at these sites.
Phe129 is located between Val29 and those residues identified in response 1, and the 85–87 loop is located between Val29 and those residues identified in response 2, prompting us to hypothesize that Phe129 and the 85–87 loop link Val29 to the networks corresponding to responses 1 and 2, respectively. To test this hypothesis, we generated a mutation to Asp85 (CypAD85N), which dramatically affects many residues within response 2, while minimally affecting residues within response 1 (Figures 7 and S2).
Figure 7. Mutational Impact on a Single Dynamic Network.
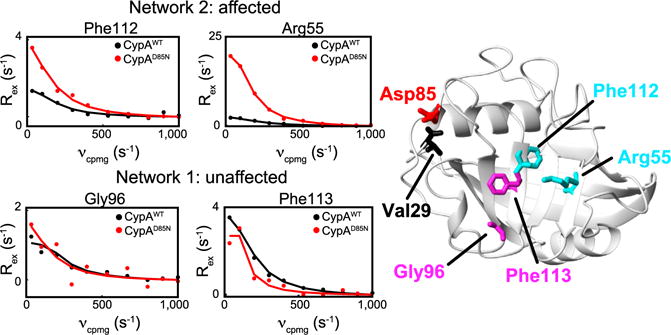
R2-RD data (dots) along with individual fits (lines) are shown collected at 900 MHz for CypAWT (black) and CypAD85N (red). Top, representative residues along network 2 (Figure 3, response 2), and, bottom, residues along network 1 (Figure 3, response 1). CypA structure is shown with relevant residues highlighted. See Figure S2 for Rex data for CypAD85N for all residues within response 1 and response 2.
DISCUSSION
Despite the inherently complex, multidimensional nature of conformational dynamics in propagating allosteric communication and regulating enzyme function, a comprehensive understanding of these processes is necessary to rationally target distal sites pharmacologically or to engineer enzyme dynamics to modulate function. Segmental dynamic systems, such as CypA, provide particular challenges compared with globally coherent motions, namely that a full description of the dynamic landscape of these systems must include mechanisms of intercommunication between these distinct segmental processes. Here, we have developed a straightforward methodology of utilizing NMR relaxation, specifically R2-RD, with perturbations to the active site to identify hotspots within an enzyme and then utilized this information to create single-site multiple mutations that have revealed the dynamically coupled networks. This approach could readily be extended to other timescales for which changes in any dynamic parameter can be measured in response to hotspot mutations. This includes a relatively broad range of timescales, such as fast microsecond motions monitored by R1ρ dispersions (Loria et al., 2008), micro- to millisecond motions monitored here using R2-RD, or slower millisecond motions that can be monitored by chemical exchange saturation transfer experiments (Vallurupalli et al., 2012).
The RASSMM approach has allowed us to alter the conformational sampling of the CypA active site independent of changes to the ground-state substrate binding site in order to modulate catalysis. We contrast this approach with the findings of Fraser et al. (2009), in which the CypAS99T mutant was utilized to invert the population occupancy of many substrate-interacting residues, including Phe113, inducing a severe reduction in substrate isomerization. While the studies outlined by Fraser et al. (2009) were instrumental in identifying the conformational states sampled within the active site and in demonstrating the role of side-chain rotations in mediating dynamics in CypA, this particular mutation dramatically alters the ground-state active-site structure, precluding deconvolution of steric and dynamic effects in modulating substrate interactions.
We have used our approach to additionally identify multiple dynamic pathways that underlie allosteric communication in CypA (Figure 3). Notably, this inter-residue communication occurs between residues mobile on different timescales (Figure 2C). Furthermore, analysis of chemical-shift-restrained MD ensembles indicates communication between residues that occupy two distinct states (Figure 6C, Val29) and those that occupy a continuum of states (Figure 6C, Phe129 and Asp85), demonstrating a representative mechanism to propagate allosteric information between non-coherent residues. Given the complex responses due to crosstalk among multiple interacting pathways (Figure S3), communication across timescales, and the challenges in monitoring dynamics in the substrate-bound state that we have recently reported (Holliday et al., 2015a), the RASSMM approach nonetheless allows identification of thermodynamically coupled networks in a non-coherent system.
The functional changes associated with the mutations characterized in this study (Figure 5, Tables 1 and S1) demonstrate a link between conformational sampling within the CypA active site and progression through the enzymatic cycle, thereby defining a mechanism by which dynamic allostery modulates substrate binding/release. Although significant hurdles remain before precise predictions of macromolecular dynamics and the impact of a given mutation match those now achieved for structural predictions (Das and Baker, 2008), our studies indicate that enzyme dynamics act to fine-tune the catalytic function of CypA. Such findings have powerful implications for understanding the role of dynamics in enzyme function, and particularly for future enzyme engineering efforts.
EXPERIMENTAL PROCEDURES
NMR Relaxation Experiments
15N-transverse relaxation optimized spectroscopy R2-RD experiments were collected on a Varian 900 MHz spectrometer with a cryogenically cooled probe and on a Varian 600 MHz spectrometer using a constant relaxation time of 60 ms and νcpmg of 33.3, 100, 200, 300, 400, 533.3, 666.7, 800, 900, and 1,000 s−1. All R2-RD data were collected on 2H15N-labeled protein samples. Data were collected at 10°C using 1 mM protein for apo samples or 0.5 mM protein with 6 mM peptide for substrate-bound samples. Data were least-squares fit to the Carver-Richards equation (Carver and Richards, 1972) using data collected at both 900 and 600 MHz as described previously (Schlegel et al., 2009). Inherent in the application of the Carver-Richards equation is the assumption that the data are reporting on two-state conformational exchange. Given the localized nature of dynamics within CypA, we were unable to simultaneously fit groups of residues, preventing accurate determination of the exchange parameters available in a more coherent system. Given these limitations, the Carver-Richards equations have been applied only as a means to provide a fit line for the reader to more readily identify changes in Rex, with interpretation limited to qualitative comparison of Rex values. All NMR data were processed using NMRPipe (Delaglio et al., 1995) and analyzed using CCPNmr Analysis (Vranken et al., 2005).
Measuring Binding Affinity
Binding affinity was measured via collection of 15N-HSQC spectra of 15N-CypA upon serial addition of unlabeled FGP, as described previously (Holliday et al., 2015b). Spectra were collected on a Varian 900 MHz spectrometer upon addition of 0, 0.1, 0.2, 0.5, 1, and 2 mM unlabeled peptide to 0.5 mM 15N-CypA. For all non-overlapping residues, data were first fit individually to the steady-state equilibrium binding equation; data were then fit simultaneously for affected residues exhibiting fast exchange (identified by an individual fit with r2 > 0.99, between 46 and 50 residues per titration experiment). 15N and 1H chemical shifts were treated independently in this analysis. Binding affinity was measured for FGP, HA, and HAP2A via ITC. HA and HAP2A ITC data were collected using 0.5 mM protein and titrating in up to a 1:2 molar ratio of each peptide on a MicroCal iTC200 and processed using MicroCal ITC-ORIGIN. FGP ITC data were collected with 0.5 mM protein and titration up to a 1:5 molar ratio on a TA Nano ITC and processed with NanoAnalyze. All data were collected at 10°C.
Measuring Isomerization
Isomerization was measured for FGP via ZZ exchange using 1 mM 15N-labeled peptide with 20 μM protein and fit as described previously (Holliday et al., 2015b). Data were collected with mixing times of 0.036, 0.072, 0.144, 0.24, 0.3, 0.36, 0.54, 0.72, 0.9, 1.08, and 1.2 s. The intensity of the cis peaks, trans peaks, and cross peaks of peptide residues Asp6 and Leu7 (one cross peak of Asp6 is overlapped with another residue and so was excluded) were simultaneously fit the equation described by Farrow et al. (1994) to determine the effective isomerization rate kiso, where kiso = kcis⟶trans + ktrans⟶cis. At least two independent experiments, comprising data collected on different samples generated from different protein purifications, were performed for each mutant to ensure reproducibility. For HAP2A, data were collected on samples with 1 mM 15N-labeled HAP2A and 50 μM protein, and fits were performed simultaneously using residues Tyr3, Val5, and Tyr8. All data were collected at 10 C.
Additional Experimental Procedures are detailed in Supplemental Experimental Procedures, including detailed descriptions of protein purification, line-shape analysis, and MD simulations.
Supplementary Material
Highlights.
The RASSMM approach allows identification and modulation of allosteric networks
Two distinct networks of dynamic allostery are identified within cyclophilin A
Active-site conformational sampling modulates enzymatic function in cyclophilin A
Non-coherent interactions mediate communication within larger dynamics networks
Acknowledgments
NMR experiments were collected at The Rocky Mountain 900 Facility and the High Magnetic Field Laboratory (NHMFL), which is supported by cooperative agreement between the National Science Foundation and the State of Florida. M.H. was supported by the Earleen and Victor Bolie Scholarship Fund and NIH application numbers 5T32GM008730-13 and 1F31CA183206. E.Z.E. is supported by NIH application number 1RO1GM107262.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.str.2016.12.003.
AUTHOR CONTRIBUTIONS
M.J.H. designed and performed all experiments unless otherwise noted, with guidance from E.Z.E. and M.V. C.C. performed all MD simulations. G.S.A. provided assistance in the design and execution of NMR experiments. M.J.H. and E.Z.E. wrote the manuscript with input from C.C. and M.V.
References
- Bhabha G, Lee J, Ekiert DC, Gam J, Wilson IA, Dyson HJ, Benkovic SJ, Wright PE. A dynamic knockout reveals that conformational fluctuations influence the chemical step of enzyme catalysis. Science. 2011;332:234–238. doi: 10.1126/science.1198542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer JA, Lee AL. Monitoring aromatic picosecond to nanosecond dynamics in proteins via 13C relaxation: expanding perturbation mapping of the rigidifying core mutation, V54A, in eglin c. Biochemistry. 2008;47:4876–4886. doi: 10.1021/bi702330t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilloni C, Robustelli P, De Simone A, Cavalli A, Vendruscolo M. Characterization of the conformational equilibrium between the two major substates of RNase A using NMR chemical shifts. J Am Chem Soc. 2012;134:3968–3971. doi: 10.1021/ja210951z. [DOI] [PubMed] [Google Scholar]
- Camilloni C, Sahakyan AB, Holliday MJ, Isern NG, Zhang F, Eisenmesser EZ, Vendruscolo M. Cyclophilin A catalyzes proline isomerization by an electrostatic handle mechanism. Proc Natl Acad Sci USA. 2014;111:10203–10208. doi: 10.1073/pnas.1404220111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr HY, Purcell EM. Effects of diffusion on free precession in nuclear magnetic resonance experiments. Phys Rev. 1954;94:630–683. [Google Scholar]
- Carroll MJ, Mauldin RV, Gromova AV, Singleton SF, Collins EJ, Lee AL. Evidence for dynamics in proteins as a mechanism for ligand dissociation. Nat Chem Biol. 2012;8:246–252. doi: 10.1038/nchembio.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver JP, Richards RE. A general two-site solution for the chemical exchange produced dependence of T2 upon the Carr-Purcell pulse separation. J Magn Reson. 1972;6:89–105. [Google Scholar]
- Cole R, Loria JP. Evidence for flexibility in the function of ribonuclease A. Biochemistry. 2002;41:6072–6081. doi: 10.1021/bi025655m. [DOI] [PubMed] [Google Scholar]
- Das R, Baker D. Macromolecular modeling with rosetta. Annu Rev Biochem. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- Davis TL, Walker JR, Campagna-Slater V, Finerty PJ, Paramanathan R, Bernstein G, MacKenzie F, Tempel W, Ouyang H, Lee WH, et al. Structural and biochemical characterization of the human cyclophilin family of peptidyl-prolyl isomerases. PLoS Biol. 2010;8:e1000439. doi: 10.1371/journal.pbio.1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S, Greenwood AI, Rogals MJ, Kovrigin EL, Lu KP, Nicholson LK. Complete thermodynamic and kinetic characterization of the isomer-specific interaction between Pin1-WW domain and the amyloid precursor protein cytoplasmic tail phosphorylated at Thr668. Biochemistry. 2012;51:8583–8596. doi: 10.1021/bi3008214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Doshi U, Holliday MJ, Eisenmesser EZ, Hamelberg D. Dynamical network of residue-residue contacts reveals coupled allosteric effects in recognition, catalysis, and mutation. Proc Natl Acad Sci USA. 2016;113:4735–4740. doi: 10.1073/pnas.1523573113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet N, Watt ED, Loria JP. The flexibility of a distant loop modulates active site motion and product release in ribonuclease A. Biochemistry. 2009;48:7160–7168. doi: 10.1021/bi900830g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du JF, Li W, Li L, Wen GB, Lin YW, Tan X. Regulating the coordination state of a heme protein by a designed distal hydrogen-bonding network. ChemistryOpen. 2015;4:97–101. doi: 10.1002/open.201402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- Farrow NA, Zhang O, Forman-Kay JD, Kay LE. A heteronuclear correlation experiment for simultaneous determination of 15N longitudinal decay and chemical exchange rates of systems in slow equilibrium. J Biomol NMR. 1994;4:727–734. doi: 10.1007/BF00404280. [DOI] [PubMed] [Google Scholar]
- Fischer G, Wittmann-Liebold B, Lang K, Kiefhaber T, Schmid FX. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature. 1989;337:476–478. doi: 10.1038/337476a0. [DOI] [PubMed] [Google Scholar]
- Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, Alber T. Hidden alternative structures of proline isomerase essential for catalysis. Nature. 2009;462:669–673. doi: 10.1038/nature08615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gothel SF, Marahiel MA. Peptidyl-prolyl cis-trans isomerases, a superfamily of ubiquitous folding catalysts. Cell Mol Life Sci. 1999;55:423–436. doi: 10.1007/s000180050299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green N, Alexander H, Olson A, Alexander S, Shinnick TM, Sutcliffe JG, Lerner RA. Immunogenic structure of the influenza virus hemagglutinin. Cell. 1982;28:477–487. doi: 10.1016/0092-8674(82)90202-1. [DOI] [PubMed] [Google Scholar]
- Greenwood AI, Rogals MJ, De S, Lu KP, Kovrigin EL, Nicholson LK. Complete determination of the Pin1 catalytic domain thermodynamic cycle by NMR lineshape analysis. J Biomol NMR. 2011;51:21–34. doi: 10.1007/s10858-011-9538-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler-Wildman KA, Thai V, Lei M, Ott M, Wolf-Watz M, Fenn T, Pozharski E, Wilson MA, Petsko GA, Karplus M, et al. Intrinsic motions along an enzymatic reaction trajectory. Nature. 2007;450:838–844. doi: 10.1038/nature06410. [DOI] [PubMed] [Google Scholar]
- Holliday MJ, Armstrong GS, Eisenmesser EZ. Determination of the full catalytic cycle among multiple cyclophilin family members and limitations on the application of CPMG-RD in reversible catalytic systems. Biochemistry. 2015a;54:5815–5827. doi: 10.1021/acs.biochem.5b00746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday MJ, Camilloni C, Armstrong GS, Isern NG, Zhang F, Vendruscolo M, Eisenmesser EZ. Structure and dynamics of GeoCyp: a thermophilic cyclophilin with a novel substrate binding mechanism that functions efficiently at low temperatures. Biochemistry. 2015b;54:3207–3217. doi: 10.1021/acs.biochem.5b00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin ZG, Lungu AO, Xie L, Wang M, Wong C, Berk BC. Cyclophilin A is a proinflammatory cytokine that activates endothelial cells. Arterioscler Thromb Vasc Biol. 2004;24:1186–1191. doi: 10.1161/01.ATV.0000130664.51010.28. [DOI] [PubMed] [Google Scholar]
- Keedy DA, Kenner LR, Warkentin M, Woldeyes RA, Hopkins JB, Thompson MC, Brewster AS, Van Benschoten AH, Baxter EL, Uervirojnangkoorn M, et al. Mapping the conformational landscape of a dynamic enzyme by multitemperature and XFEL crystallography. Elife. 2015;4 doi: 10.7554/eLife.07574. http://dx.doi.org/10.7554/eLife.07574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern D, Kern G, Scherer G, Fischer G, Drakenberg T. Kinetic analysis of cyclophilin-catalyzed prolyl cis/trans isomerization by dynamic NMR spectroscopy. Biochemistry. 1995;34:13594–13602. doi: 10.1021/bi00041a039. [DOI] [PubMed] [Google Scholar]
- Kern D, Eisenmesser EZ, Wolf-Watz M. Enzyme dynamics during catalysis measured by NMR spectroscopy. Methods Enzymol. 2005;394:507–524. doi: 10.1016/S0076-6879(05)94021-4. [DOI] [PubMed] [Google Scholar]
- Khirich G, Loria JP. Complexity of protein energy landscapes studied by solution NMR relaxation dispersion experiments. J Phys Chem B. 2015;119:3743–3754. doi: 10.1021/acs.jpcb.5b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Kim WJ, Jeon ST, Koh EM, Cha HS, Ahn KS, Lee WH. Cyclophilin A may contribute to the inflammatory processes in rheumatoid arthritis through induction of matrix degrading enzymes and inflammatory cytokines from macrophages. Clin Immunol. 2005;116:217–224. doi: 10.1016/j.clim.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Kleckner IR, Gollnick P, Foster MP. Mechanisms of allosteric gene regulation by NMR quantification of microsecond-millisecond protein dynamics. J Mol Biol. 2012;415:372–381. doi: 10.1016/j.jmb.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kim SS. An overview of cyclophilins in human cancers. J Int Med Res. 2010;38:1561–1574. doi: 10.1177/147323001003800501. [DOI] [PubMed] [Google Scholar]
- Liu X, Sun L, Yu M, Wang Z, Xu C, Xue Q, Zhang K, Ye X, Kitamura Y, Liu W. Cyclophilin A interacts with influenza A virus M1 protein and impairs the early stage of the viral replication. Cell Microbiol. 2009;11:730–741. doi: 10.1111/j.1462-5822.2009.01286.x. [DOI] [PubMed] [Google Scholar]
- Loria JP, Berlow RB, Watt ED. Characterization of enzyme motions by solution NMR relaxation dispersion. Acc Chem Res. 2008;41:214–221. doi: 10.1021/ar700132n. [DOI] [PubMed] [Google Scholar]
- McDonald LR, Boyer JA, Lee AL. Segmental motions, not a two-state concerted switch, underlie allostery in CheY. Structure. 2012;20:1363–1373. doi: 10.1016/j.str.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElheny D, Schnell JR, Lansing JC, Dyson HJ, Wright PE. Defining the role of active-site loop fluctuations in dihydrofolate reductase catalysis. Proc Natl Acad Sci USA. 2005;102:5032–5037. doi: 10.1073/pnas.0500699102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiboom G, Gill D. Modified spin-echo method for measuring nuclear relaxation times. Rev Sci Instrum. 1958;29:688–691. [Google Scholar]
- Millet O, Loria JP, Kroenke CD, Pons M, Palmer AG. The static magnetic field dependence of chemical exchange line broadening defines the NMR chemical shift time scale. J Am Chem Soc. 2000;122:2867–2877. [Google Scholar]
- Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nat Struct Mol Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel J, Armstrong GS, Redzic JS, Zhang F, Eisenmesser EZ. Characterizing and controlling the inherent dynamics of cyclophilin-A. Protein Sci. 2009;18:811–824. doi: 10.1002/pro.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaratnam R, Chowdhury S, VanSchouwen B, Melacini G. Mapping allostery through the covariance analysis of NMR chemical shifts. Proc Natl Acad Sci USA. 2011;108:6133–6138. doi: 10.1073/pnas.1017311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Bax A. SPARTA+: a modest improvement in empirical NMR chemical shift prediction by means of an artificial neural network. J Biomol NMR. 2010;48:13–22. doi: 10.1007/s10858-010-9433-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallurupalli P, Bouvignies G, Kay LE. Studying “invisible” excited protein states in slow exchange with a major state conformation. J Am Chem Soc. 2012;134:8148–8161. doi: 10.1021/ja3001419. [DOI] [PubMed] [Google Scholar]
- van den Bedem H, Bhabha G, Yang K, Wright PE, Fraser JS. Automated identification of functional dynamic contact networks from X-ray crystallography. Nat Methods. 2013;10:896–902. doi: 10.1038/nmeth.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Lee T, Latham MP, Warner LR, Tanimoto A, Pardi A, Ahn NG. Phosphorylation releases constraints to domain motion in ERK2. Proc Natl Acad Sci USA. 2014;111:2506–2511. doi: 10.1073/pnas.1318899111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.