

Abstract
Rationale
Endothelial progenitor cells (EPCs) respond to SDF-1 through receptors CXCR7 and CXCR4. Whether SDF-1 receptors involves in diabetes induced EPCs dysfunction remains unknown.
Objective
To determine the role of SDF-1 receptors in diabetic EPCs dysfunction.
Methods and Results
CXCR7 expression, but not CXCR4 was reduced in EPCs from db/db mice, which coincided with impaired tube formation. Knockdown of CXCR7 impaired tube formation of EPCs from normal mice, while up-regulation of CXCR7 rescued angiogenic function of EPCs from db/db mice. In normal EPCs treated with oxidized low-density lipoprotein (ox-LDL) or high glucose (HG) also reduced CXCR7 expression, impaired tube formation and increased oxidative stress and apoptosis. The damaging effects of ox-LDL or HG were markedly reduced by SDF-1 pretreatment in EPCs transduced with CXCR7 lentivirus (CXCR7-EPCs) but not in EPCs transduced with control lentivirus (Null-EPCs). Most importantly, CXCR7-EPCs were superior to Null-EPCs for therapy of ischemic limbs in db/db mice. Mechanistic studies demonstrated that ox-LDL or HG inhibited Akt and GSK-3β phosphorylation, nuclear export of Fyn and nuclear localization of Nrf2, blunting Nrf2 downstream target genes HO-1, NQO-1 and catalase, and inducing an increase in EPC oxidative stress. This destructive cascade was blocked by SDF-1 treatment in CXCR7-EPCs. Furthermore, inhibition of PI3K/Akt prevented SDF-1/CXCR7-mediated Nrf2 activation and blocked angiogenic repair. Moreover, Nrf2 knockdown almost completely abolished the protective effects of SDF-1/CXCR7 on EPC function in vitro and in vivo.
Conclusions
Elevated expression of CXCR7 enhances EPC resistance to diabetes-induced oxidative damage and improves therapeutic efficacy of EPCs in treating diabetic limb ischemia. The benefits of CXCR7 are mediated predominantly by an Akt/GSK-3β/Fyn pathway via increased activity of Nrf2.
Keywords: Endothelial progenitor celsl, chemokine receptor CXCR7, angiogenesis, hind limb ischemia, chemoreceptor
Subject Terms: Angiogenesis, Basic Science Research, Cell Signaling/Signal Transduction, Cell Therapy, Mechanisms
Introduction
Diabetes mellitus is a common, chronic, metabolic disease producing great social and economic burden. Microvascular and macrovascular complications associated with type 2 diabetes are leading causes of morbidity and mortality for diabetic patients1. Vascular complications in diabetes are associated with dysregulation of vascular remodeling and vascular growth, decreased responsiveness to ischemic/hypoxic stimuli, impaired or abnormal neovascularization, and a lack of endothelial regeneration2. Thus, there is a great need for therapeutic interventions aimed at accelerating repair of dysfunctional endothelium and restoring blood flow in diabetic patients.
Accumulating evidence suggests that bone-marrow derived circulating endothelial progenitor cells (EPCs) contribute to vascular repair. Following tissue ischemia or endothelial damage, EPCs are mobilized from the bone marrow to the circulation. EPCs then home to sites of vascular injury where they contribute to new blood vessel formation and recovery3. Recently, EPC transplantation has become an experimental therapy for ischemic disease4. Several studies show that EPC transplantation provides some benefit for myocardial infarction5 and limb ischemia6, 7, and transplanted EPCs may compensate for a shortage of endogenous EPCs in diabetic retinopathy8. However, to improve therapeutic efficacy of EPC transplantation it is necessary to improve EPC survival and enhance EPC homing to ischemic tissue.
EPC-induced neovascularization is a highly coordinated, temporally regulated, and complex set of events including mobilization, migration, and homing9, 10. Chemokines play an essential role in each of these steps. Stromal cell–derived factor 1 (SDF-1, aka CXCL12), is a key chemokine in regulating hematopoietic stem cell trafficking between the bone marrow and peripheral circulation, and attracts EPCs to areas of ischemia. For some time, CXCR4 was thought to be the only SDF-1 receptor, and numerous studies demonstrated a role of the SDF-1/CXCR4 axis in mobilizing stem and progenitor cells from bone marrow, homing to target tissue, chemotaxis, adhesion, survival and angiogenesis11, 12. In 2005, a second SDF-1 receptor was identified, CXCR7 that binds SDF-1 with 10-fold higher affinity than CXCR413. EPCs express both CXCR4 and CXCR714, 15 and recently CXCR7 is shown to influence EPCs clinical status, i.e., EPCs of hypertensive patients express low levels of CXCR7 and this contributes to impaired angiogenesis and re-endothelialization16.
In diabetes, the number of EPCs is reduced and EPC function is attenuated17. Diabetes may alter expression and function of SDF-1 receptors in EPCs but this remains untested. Our previous studies demonstrated that CXCR7 has a critical role regulating adhesion and survival of EPCs from rat bone marrow and human cord blood14, 15. Extrapolating from those studies we hypothesized that elevating CXCR7 expression would improve angiogenic function of EPCs in type 2 diabetes. The aims of the present study were to determine the level of SDF-1 receptors in diabetic EPCs and determine if over-expression of CXCR7 improved angiogenic function of EPCs in diabetes and to reveal mechanisms connecting EPC function and CXCR7 expression.
Methods
Animals
FVB wild type (WT) and db/db (FVB background) male mice at age of 10-12 weeks were used in this study. All animal procedures were approved by the Animal Policy and Welfare Committee of Wenzhou Medical University and the Institutional Animal Care and Use Committee of the University of Louisville, which conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (publication No. 85-23, revised 1996).
Isolation and culture of bone marrow-derived EPC
EPCs were isolated from the bone marrow of WT (WT-EPCs) and db/db (db/db-EPCs) mice, and cultured afterward according to our established methods with minor modifications15. Briefly, bone marrow mononuclear cells (MNCs) were isolated from the femurs and tibias of mice by density gradient centrifugation with histopaque-1083 (Sigma-Aldrich, St. Louis, MO). After two washing steps, MNCs were plated on vitronectin-coated culture dishes (Sigma-Aldrich, St. Louis, MO) and maintained in endothelial growth factor-supplemented media (EGM-2 bullet kit, Lonza, Basel, Switzerland) with 10% fetal bovine serum (FBS). Cells were cultured at 37°C with 5% CO2 in a humidified atmosphere. EGM-2 medium was replaced after the first 24 h and every 3 days thereafter. Cell colonies that appeared after 7 days of culture were defined as early EPC, and cell colonies that appeared after 14 days of culture were defined as late EPC as previous reported18.
Characterization of bone marrow-derived MNCs
After 7 or 14 days in endothelial-specific media and the removal of non-adherent MNCs, the remaining cells were characterized by immunofluorescent staining and flow cytometry analysis. For immunofluorescent staining, cells were seeded on vitronectin coated 8 well μ-slide (ibidi, Martinsried, Germany) one day before, and then the cells were incubated with 5μg/mL Acetylated DiI lipoprotein from human plasma (Dil-Ac-LDL, Thermo Fisher Scientific, Waltham, MA) at 37°C for 4 h followed by 3 washes with Dulbecco's phosphate buffered saline (DPBS) and incubated with 10μg/mL fluorescein isothiocyanate labeled ulex europaeus lectin-1 (FITC-UEA-1, Sigma-Aldrich, St. Louis, MO) for 1h at room temperature. After incubation, cells were rinsed with DPBS for 3 times and were visualized via a confocal microscopy. For flow cytometry analysis, cells were incubated with 5% bovine serum albumin (BSA, Sigma) for 15 min for blockade of nonspecific binding, and then stained with anti-mouse CD34-PE, CD31-PE, CD14-PE, CD144-PE, Scal-1-FITC, and c-kit-FITC (BD Biosciences, San Jose, CA), and VEGFR2-APC, CD45-APC (Biolegend, San Diego, CA) at room temperature for 1h, respectively. The same fluorescein labeled isotype IgG served as control to define the negative populations for each stain. Cells were analyzed with a BD FACSAria cell sorter (BD Biosciences, San Jose, CA), and data were analyzed using FloJo software version 8.8.4 (TreeStar, Inc, Ashland, OR).
Lentiviral vector construction, virus production, and infection
To upregulate the CXCR7 expression in EPCs, recombinant lentiviruses encoding CXCR7 (pLVX-CXCR7-EGFP-3FLAG-Puro) was constructed by cloning the CXCR7 gene into the pLVX-EGFP-3FLAG-Puro vector (Shanghai Sunbio Medical Biotechnology, Shanghai, China) via EcoRI and BamHI sites. The CXCR7 cDNA was amplified using the following primers: forward: 5′-CGGAATTCCGCCTCAGAACGATGGATC -3; reverse: 5′-CGGGAGCCAACAAGTAAACCCGTCCCAGA-3′.
Viral supernatants were produced in HEK293T cells after co-transfection of CXCR7 recombinant vector (pLVX-CXCR7-EGFP-3FLAG-Puro) or control vector (pLVX-EGFP-3FLAG-Puro) with the packaging plasmid psPAX2 and the envelope plasmid pMD2. G using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The supernatant was harvested at 72 h post transfection, filtered through Millex-HV 0.45μm PVDF filter (Millipore, Billerica, MA), and stored at −80°C until use.
Early EPCs or late EPCs (passages 3∼4) were infected with the purified lentivirus carrying recombinant CXCR7 (CXCR7-EPCs) or control vector (Null-EPCs) overnight at multiplicity of infection (MOI) of 25 with 2.5 μg/mL polybrene (Santa Cruz, Dallas, TX), and the medium was replaced with fresh growth medium 24 h after infection. After transfection for 72 h, the infection efficiency was determined by flow cytometry analysis of GFP expression. The levels of CXCR7 expression were detected by Western blot and flow cytometry assay.
To knockdown CXCR7 expression in early EPCs from WT mice, specific siRNAs against mouse CXCR7, alone with Silencer Select Negative Control (Thermo Fisher, Waltham, MA) were transfected into EPCs using Lipofectamine 2000 (Thermo Fisher, Waltham, MA). After transfection for 48 h, the expression of CXCR7 was determined by Western blot.
To knockdown Nrf2 expression in late EPCs from WT mice, EPCs were infected with the lentiviruses containing shRNA against Nrf2 or nonsense shRNA constructed by Shanghai Sunbio Medical Biotechnology (Shanghai, China). We selected 3 target sequences for Nrf2 and nonsense sequence as follows:
Nrf2-shRNA 1: 5′-CTTGAAGTCTTCAGCATGTTA-3′;
Nrf2-shRNA 2: 5′-GCCTTACTCTCCCAGTGAATA-3′;
Nrf2-shRNA 3: 5′-GACCTCCTTAGACTAAATCC-3′.
Nonsense shRNA: 5′-TTCTCCGAACGTGTCACGTTT-3′.
The transfection was performed following the procedure described above. After transfection for 48 h, the expression of Nrf2 was determined by Western blot. Then CXCR7-EPCs or Null-EPCs were infected with the lentiviruses containing shRNA against Nrf2 with the best knockdown efficiency or nonsense shRNA following the same protocol described above.
Apoptosis assay
EPCs transduced with control lentivirus or lentivirus encoding CXCR7, Nrf2 shRNA or non-target shRNA were seeded on 12-well plates (1× 105 cells/well) and maintained under basal culture conditions or with oxidized low-density lipoprotein (ox-LDL, 50μg/mL, Athens Research & Technology, Inc., Athens, GA), or high glucose (HG, 25 mmol/L, Sigma Chemical Co., St. Louis, MO) along with a same dose of mannitol (Sigma Chemical Co.) as osmotic control for 24 h in the presence or absence of SDF-1 (100 ng/mL). Non-adherent cells were removed by washing with PBS. Subsequently, adherent cells were released with 0.25% trypsin without EDTA. EPCs were collected by centrifugation and stained with APC-conjugated Annexin V Apoptosis Detection Kit with propidium iodide (PI), according to the manufacturer's instructions (Biolegend, San Diego, CA). The apoptotic EPCs were detected by a flow cytometry. Early apoptotic cells were defined as AnnexinV+/PI−.
Angiogenesis assay in vitro
The in vitro angiogenic capability of EPCs was determined by matrigel tube formation assay. Briefly, 48-well plates were coated with growth factor–reduced matrixgel (150μL/per well, BD Biosciences). EPCs transduced with control lentivirus or lentivirus encoding CXCR7, Nrf2 shRNA or non-target shRNA were plated (5 × 104 cells/well) in 200 μL basal culture medium or medium containing ox-LDL or HG in the presence or absence of SDF-1 and incubated at 37°C with 5% CO2 for 12 h to form tubes. Images of tubes in each well were taken using an inverted microscopy (Nikon Eclipse E600, Nikon, Kanagawa, Japan). The tube lengths were calculated by Image J software.
Trans-endothelial migration (TEM) assay
TEM is one of most important abilities for EPCs participating in angiogenesis. We evaluated TEM abilities of EPCs by a trans-well assay according to our previous report15. Human umbilical vein endothelial cells (HUVEC, 1 × 104 cells/well) were cultured in the upper chamber of a 24- trans-well insert (8.0-μm pores; Falcon 353097, BD, Bedford, MA). Before each experiment, monolayer confluency was confirmed by inverted fluorescence microscopy. EPCs transduced with control lentivirus or lentivirus encoding CXCR7, Nrf2 shRNA or non-target shRNA, were cultured in the presence or absence of ox-LDL for 12 h or HG for 24 h. Then EPCs were harvested and re-suspended in basal culture medium (EBM-2, 0.5% BSA). EPC suspension (0.2 mL) was added to the upper chamber of the trans-well, and 0.6 mL EBM-2 medium supplemented with either PBS or SDF-1 (100 ng/mL) was added to the lower chamber of the trans-well. Cells were cultured for 12 h at 37°C, and EPCs traversing from the upper to the lower chamber of the trans-well was quantified by independent investigators blinded to treatment.
Quantitative determination of oxidative stress
To detect the reactive oxygen species (ROS) level of EPCs with ox-LDL or HG treatment, dihydroethidium (DHE; Molecular Probes, Eugene, OR) probe was used to stain EPCs. DHE is cell permeable and able to react with superoxide to form ethidium, which in turn intercalates with DNA and produces nuclear fluorescence. EPCs were seeded on 24-well plates and treated with ox-LDL or HG in presence or absence SDF-1 for 6 h, then incubated with 5 μmol/L DHE in PBS for 30 min at 37°C. Nuclear DHE positive staining indicates superoxide generation in cells. The fluorescence intensity was detected by a microplate reader (SpectraMax M3, Molecular Devices, Sunnyvale, CA) under specific wave length conditions (excitation = 518 nm; fluorescence = 605 nm). Meanwhile, the fluorescence images of random EPCs were captured at 20× magnification (XI 71 Olympus, Tokyo, Japan).
Western blot assay
Western blot was performed as previous study15. For cellular protein extraction, EPCs were rinsed twice with PBS and then suspended in ice-cold RIPA lysis buffer (Santa Cruz Biotechnology, Dallas, TX) and incubated for 15 to 30 min on ice. Nuclear proteins from EPCs were extracted by a nuclear extraction kit (Abcam, Cambridge, MA). Proteins were collected by centrifugation, 12,000g for 15 min at 4°C. The protein concentration was determined using a Bradford protein assay kit (Bio-Rad, Hercules, CA). The total and nuclear proteins were separated on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). The membranes were blocked in tris-buffered saline with 5% non-fat milk and 0.5% BSA for 1h, and then incubated with primary antibody overnight at 4°C, followed by incubation with the secondary antibodies for 1h at room temperature after standard washing procedures. The primary antibodies against Nrf2 (1:1,000), histone H3 (1:10,000), heme oxygenase-1 (HO-1, 1:2,000), NAD(P)H dehydrogenase quinone 1 (NQO-1, 1:1,000), catalase (1:5,000), glyceraldehyde 3-phosphate dehydrogenase (GAPDH, 1:5,000) and β-actin (1:3,000) were purchased from Santa Cruz Biotechnology; Akt and p-Akt (Ser473) (1:2,000), GSK-3β and p-GSK-3β (Ser9) (1:2,000), and Fyn (1:2,000) were purchased from Cell Signaling Technology (Danvers, MA); 3-nitrotyrosine (3-NT, 1:2,000) was purchased from Millipore (Billerica, MA); CXCR4 and CXCR7 (1:2,000) were purchased from Abcam (Cambridge, MA). All horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology. Blots were visualized with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, Waltham, MA) and quantified with Quantity 5.2 software System (Bio-Rad).
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted using Trizol reagent (RNA STAT 60, Tel-Test Inc., Austin, TX) and reversed transcribed using a GoScript™ Reverse Transcription System (Promega, Madison, WI) following the manufacturer's protocol. Primers of Nrf2 (Mm00477784), catalase (Mm00437992_m1), NQO1 (Mm01253561), HO-1 (Mm00516005), and β-actin (Mm00607939) were purchased from Thermo Fisher Scientific (Waltham, MA). qRT-PCR was performed in duplicate with a 20 μL reaction system, which contains 10 μL TaqMan Universal PCR master mix, 6μL H2O, 3μL cDNA, and 1 μL primer, using the ABI 7500 RT-PCR system (Applied Biosystems, Foster City, CA). The comparative cycle time (Ct) method was used to determine fold differences between samples, and the amount of target genes was normalized to β-actin as an endogenous reference (2−ΔΔCt).
Hind limb ischemia (HLI) model and cell therapy
The db/+ founder mice [FVB. BKS(D)-Leprdb/+/ChuaJ, FVB background] were purchased from the Jackson Laboratory (Bar Harbor, ME) and maintained under specific pathogen-free conditions at the University of Louisville Animal Facility (Louisville, Kentucky, USA). The db/db mice were generated by breeding male db/+ to female db/+ mice following Jackson Laboratory's instructions. Male db/db mice were used to develop HLI model as in our previous report19. Briefly, under sufficient anesthesia with isoflurane (1–3% isoflurane in 100% oxygen at a flowrate of 1 L/min), the hind limbs were shaved and the entire right superficial femoral artery and vein (from just below of deep femoral arteries to popliteal artery and vein) were ligated with 6-0 silk sutures, cut, and excised with an electrical coagulator (Fine Science Tools Inc., Foster City, CA). The overlying skin was closed with 4-0 silk sutures.
After surgery, 1 × 106 EPCs, infected with lentivirus vector, lentivirus carrying CXCR7, shRNA against Nrf2 or non-specific shRNA sequence, were infused via tail vein. To evaluate limb perfusion ratio [ischemic limb (right)/normal limb (left)], real-time microcirculation imaging analysis was performed using a Pericam Perfusion Speckle Imager (PSI) based on the laser speckle contrast analysis technology (Perimed Inc., Kings Park, NY) at day 0, 3, 7 14, 21 and 28 post ischemia.
Histological assessment
To examine the homing and incorporation of EPCs in the ischemic muscle, ischemic gastrocnemius muscle and/or soleus muscle were embedded in OCT medium for frozen section. The GFP positive EPCs were counted in randomly selected fields for a total of 20 different fields (×40 magnification) per section and 3 sections per animal. The EPC incorporation was expressed as the percentage of GFP positive capillaries.
The extent of angiogenesis and arteriogenesis at day 28 post ischemic surgery was assessed by measuring capillary density in gastrocnemius muscle and/or soleus muscle using isolectin B4 staining, and arteriole area in adductor muscle. Ischemic gastrocnemius muscle, soleus muscle and/or adductor tissues were fixed with 4% paraformaldehyde and embedded with paraffin. Paraffin sections were cut at 5 μm and stained with Alexa Fluor® 594 conjugated isolectin GS-IB4 (Thermo Scientific, Waltham, MA)or anti-alpha smooth muscle actin (α-SMA) antibody (Abcam) to evaluate the capillary density and arteriole area respectively. The number of capillaries or arteriole area were calculated in randomly selected fields for a total of 20 different fields (×40 magnification) per section and 3 sections per animal. The capillary density was expressed as capillary number per muscle fiber, and the arteriole area was expressed as α-SMA positive arteriole area per fiber and normalized by control.
Statistical analysis
All data are presented as mean ± SD. Statistical analysis was performed using Origin 7.5 (OriginLab data analysis and graphing software) with one-way or two-way ANOVA, followed by post-hoc multiple comparisons with the Scheffe' test. Statistical significance was considered as p < 0.05.
Results
Characterization of bone marrow-derived EPCs
MNCs isolated from mouse bone marrow were cultured in EGM-2 medium on vitronectin-coated culture dishes. Three days after plating cell colonies appeared with centrally rounded cells and spindly peripheral cells (Online Figure I, A, left panel). By 7 days spindle-shaped adherent early EPCs were observed (Online Figure I, A, middle). By 14-21 days of culture, endothelium-like cells with cobblestone-like morphology late EPCs were nearly confluent (Online Figure I, A, right). Both early and late EPCs are positive for DiI-acLDL and UEA-1 stains (Online Figure I, B, C). Flow cytometry demonstrated that many early EPCs were positive for CD34, CD31, CD14, CD45, Scal-1, c-Kit and VEGFR2, but mostly negative for CD144 (Online Figure I, D with quantitation). The cell surface profile of late EPCs was positive for CD34, CD31, VEGFR2, CD144, c-Kit and Scal-1, but low or negative for both CD14 and CD45 (Online Figure I, E with quantitation). These characteristics were consistent with previous of descriptions EPCs18, 20, 21.
Diabetes attenuates CXCR7 expression and impairs angiogenic function of EPCs
To determine how EPCs were affected by prolonged in vivo diabetes, early EPCs were isolated from bone marrow of WT and db/db mice and assayed for expression of SDF-1 receptors and for tube formation. Total and cell surface expression of CXCR7 were significantly decreased in EPCs from db/db mice compared to those from WT mice, while no significant differences were seen for CXCR4 expression (Figure 1A&B). Tube formation by EPCs from db/db mice was also functionally deficient as indicated by the significantly shorter length of the tubes db/db EPCs produced compared to WT EPCs (Figure 1C).
Figure 1.
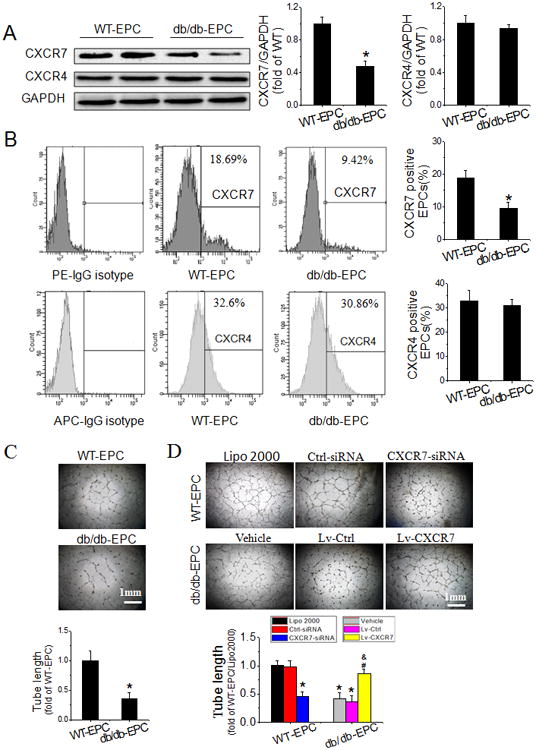
Diabetes attenuates CXCR7 expression and impairs the angiogenic function of EPCs. A-C, Early EPCs were isolated from bone marrow of WT (WT-EPCs) and db/db (db/db-EPCs) mice at 10-12 weeks of age and assayed within 7 days of isolation. Expression of CXCR7 and CXCR4 was detected by Western blot (A) and flow cytometry (B). Angiogenic function of EPCs was evaluated by tube formation assay (C). D, Early WT-EPCs were transfected with specific siRNA against mouse CXCR7 (CXCR7-siRNA) or Silencer Select Negative Control (Ctrl-siRNA), and the transfection reagent (Lipofectamine 2000, Lipo 2000) was used as a blank control; early db/db-EPCs were infected with purified lentivirus carrying recombinant CXCR7 (Lv-CXCR7) or control vector (Lv-Ctrl), and phosphate buffered saline (PBS) was used as vehicle control (Vehicle); Efficiency of CXCR7 knockdown or upregulation was determined by Western blot (shown in Online Figure II) and angiogenic function of EPCs was evaluated by tube formation assay. n=6 mice per group for A, B and C, and three independent experiments were performed for D. Data shown in graphs represents the Means ± SD. *p<0.05, vs WT-EPC for A-C or WT-EPC with Lipo 2000 treatment for D, # P< 0.05, vs db/db-EPCs with vehicle treatment; & P<0.05, vs db/db-EPCs with Lv-Ctrl infection.
To determine if reduced CXCR7 expression in diabetic EPCs was a cause of angiogenic dysfunction, gain and loss of function studies were performed by CXCR7 siRNA knockdown in early EPCs from WT mice, and CXCR7 over-expression with CXCR7 lentivirus in early EPCs from db/db mice. CXCR7 levels were significantly reduced by the siRNA in WT EPCs (Online Figure II) and this was accompanied by impaired tube formation (Figure 1D). Conversely, increasing CXCR7 levels in db/db EPCs with CXCR7 lentivirus (Online Figure II) completely reversed impaired tube formation function of diabetic EPCs (Figure 1D). Most importantly, transplantation of CXCR7 recombinant lentiviral vector transfected db/db EPCs time-dependently improved blood perfusion in HLI in db/db mice (Figure 2A), which was accompanied by promoted angiogenesis mirrored by increased capillary density and exogenous EPCs incorporation mirrored by GFP encoded by the transduced lentivirus vector and isolectin co-localization in both soleus muscle (Figure 2B) and gastrocnemius muscle (Figure 2C) measured 28 days after HLI, but without obvious effects on arteriogenesis mirrored by no apparent changes in α-SMA positive arteriole area in adductor muscle (Figure 2D). These results suggest that diabetic downregulation of CXCR7 plays a causative role in diabetes-induced EPCs angiogenic dysfunction.
Figure 2.
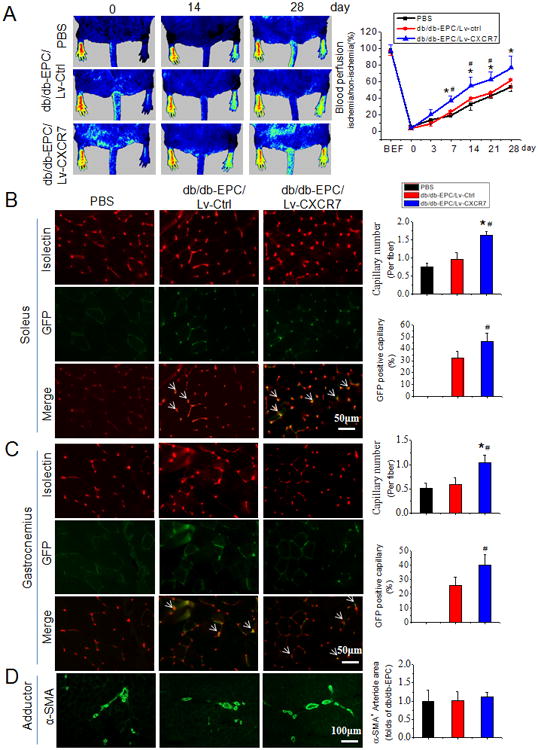
Upregulation of CXCR7 expression rescues angiogenic function of db/db-EPCs in ischemic limb of db/db diabetic mice. A, Time course of blood perfusion shown in images and quantitative analysis after hind limb ischemia (HLI) surgery with or without EPC transplantation. Blood perfusion is the ratio of ischemic to non-ischemic limb perfusion measured by a Pericam Perfusion Speckle Imager (PSI). B & C, Images and quantitation of immunofluorescent isolectin- and/or GFP-positive capillaries in transverse sections of soleus muscle (B) and gastrocnemius muscle (C) tissue from ischemic hind limbs. Capillary density was expressed as isolectin-positive capillaries per muscle fiber. The exogenous EPC incorporation (white arrows) was expressed as the percentage of GFP positive capillaries. D, Representative images and quantitation of alpha smooth muscle actin (α-SMA) positive arteriole in the ischemic adductor muscle tissue. Arteriogenesis was expressed as α-SMA positive arteriole area per fiber normalized by PBS control. Data shown in graphs represents the Means ± SD. n=8 mice per group. * p<0.05, vs PBS; # p<0.05, vs Lv-Ctrl.
Upregulating CXCR7 expression improves survival and angiogenic function of EPCs treated with ox-LDL and HG
In order to overcome the shortage of number limitation of early EPCs and facilitate gene manipulation, late EPCs from WT mice were used for the following mechanistic studies. Late EPCs from WT mice were transduced with CXCR7 recombinant lentiviral vector or control vector. At 72 h after transduction, transfection efficiency was over 70% as measured by GFP expression (Online Figure III, A). CXCR7 expression was dramatically increased in CXCR7-EPCs compared with Null-EPCs and WT-EPCs as determined by flow cytometry (Online Figure III, B) and Western blot assay (Online Figure III, C).
To determine if the effects of diabetes could be reproduced in vitro, late EPCs from WT mice were treated with ox-LDL or HG to mimic conditions of diabetic dyslipidemia or hyperglycemia as previous report22. As found with db/db diabetes, both ox-LDL and HG dose-dependently decreased expression of CXCR7 but not CXCR4, except at a highest dose of HG (Online Figure IV, A, C). Ox-LDL and HG also impaired tube formation by EPCs from WT mice (Online Figure IV, B, D). These results show that both ox-LDL and HG treatment can recapture the effects of diabetes on CXCR7 expression and EPCs dysfunction.
To test the effects of CXCR7 manipulation on EPC function in vitro, EPC dysfunction was induced by 24 h exposure late EPCs from WT mice to ox-LDL at 50μg/mL or HG at 25 mmol/L, chosen on the basis of the dose-response study in Online Figure IV. Apoptosis, measured with Annexin V/PI staining (Figure 3A and Online Figure V, A) was greatly increased in Null-EPCs by both ox-LDL and HG. This was slightly but not significantly reduced by SDF-1 pretreatment. In CXCR7-EPCs both ox-LDL and HG also induced apoptosis but it was significantly less than in Null-EPCs. Most striking was the almost complete protection against apoptosis in CXCR7-EPCs by SDF-1 pretreatment.
Figure 3.
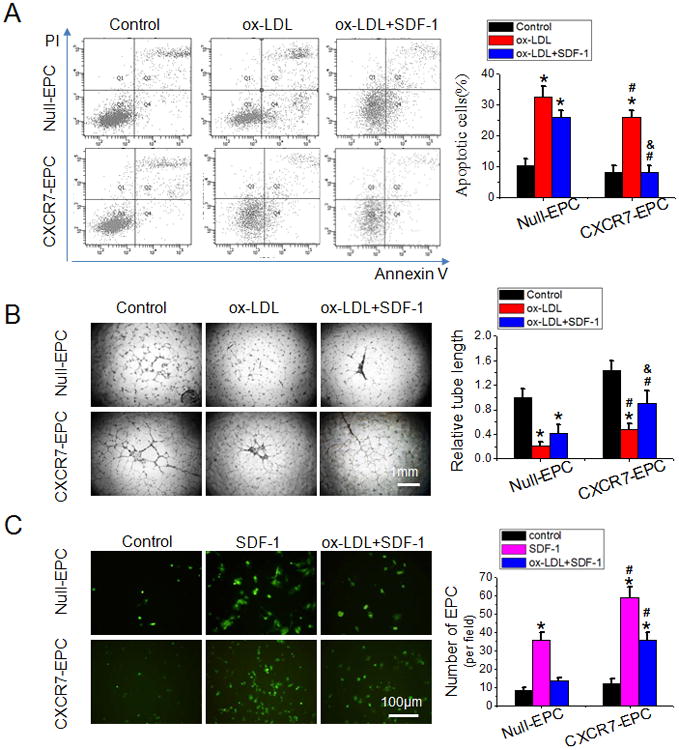
Upregulating CXCR7 expression protects EPCs from ox-LDL-induced apoptosis and angiogenic dysfunction. A, The apoptosis of EPCs was analyzed by flow cytometry analysis using Annexin V/propidium iodide (PI) staining after exposure to ox-LDL (50μg/mL, 24 h). Apoptotic cells were defined as Annexin V+/PI- (Quadrant 4). B, The effects of CXCR7 upregulation on the angiogenic function of EPCs under ox-LDL treatment condition were determined by tube formation assay. Tube length was normalized to the control Null-EPC group. C, The trans-endothelial migration of EPCs was analyzed by trans-well assay. Images are representatives of 3 independent experiments. Data shown in graphs represents the Mean ± SD. * P<0.05, vs respective control in Null-EPCs or CXCR7-EPCs; # P< 0.05, vs Null-EPCs with the same treatment; & P<0.05, vs CXCR7-EPCs with ox-LDL treatment. CXCR7-EPCs or Null-EPCs: EPCs from WT mice were transduced with CXCR7 recombinant lentiviral vector or control vector.
Tube formation assays were used as one measure of in vitro angiogenic function (Figure 3B and Online Figure V, B). Both Ox-LDL and HG significantly impaired the tube formation abilities in Null-EPCs. As with apoptosis, the negative effect on tube formation was slightly but not significantly reduced by SDF-1 pretreatment in Null-EPCs. In CXCR7-EPCs ox-LDL and HG had less of an inhibitory effect on tube formation and SDF-1 pretreatment provided much more effective protection against ox-LDL and HG inhibition than it did in Null-EPCs.
TEM is essential for EPC homing to the sites of blood vessel repair and formation and was assayed by trans-well migration assay (Figure 3C and Online Figure V, C). The assay showed no significant difference in basal TEM between Null-EPCs and CXCR7-EPC. When EPCs were treated with SDF-1, TEM of CXCR7-EPCs was much higher than that of Null-EPCs. Ox-LDL and HG pretreatment almost completely abolished SDF-1-stimulated increase of TEM in Null-EPCs, but in CXCR7-EPCs the ability of SDF-1 to increase TEM was significantly preserved.
Upregulating CXCR7 expression promotes EPCs-mediated angiogenesis and blood perfusion in HLI in db/db type 2 diabetes
To determine if elevated CXCR7 improves late EPC-mediated neovascularization in type 2 diabetes in vivo, we transplanted CXCR7-EPCs or Null-EPCs into db/db mice after HLI surgery. Both Null-EPCs and CXCR7-EPCs transplantation time-dependently increased blood perfusion compared to mice without transplantation. Notably, CXCR7-EPCs were significantly more effective than Null-EPCs at all-time points from day 7 to 28 (Online Figure VI, A). The benefit of CXCR7-EPCs was also apparent in increased capillary density and exogenous EPCs incorporation measured 28 days after HLI (Online Figure VI, B). These findings indicate that CXCR7 overexpression also promotes late EPCs homing and incorporating in endothelium in the ischemic tissue.
Upregulating CXCR7 expression attenuates ox-LDL-and HG-induced oxidative stress in EPCs
Oxidative stress is a major factor responsible for the dysfunction of diabetic EPCs23, 24. Here we show that ROS levels and oxidative damage produced by ox-LDL and HG is significantly reduced by elevated expression of CXCR7. Both ox-LDL and HG induced superoxide production in Null-EPCs, as measured by DHE staining and superoxide was slightly reduced by SDF-1 pretreatment (Figure 4A and Online Figure VII, A). CXCR7 upregulation significantly attenuated ox-LDL-and HG-induced superoxide production in CXCR7-EPCs, and almost completely blocked superoxide production after SDF-1 pretreatment (Figure 4A and Online Figure VII, A). Using 3-NT as an oxidative damage marker we obtained similar results (Figure 4B and Online Figure VII, B): CXCR7 upregulation reduced 3-NT levels that were elevated by ox-LDL and HG exposure and CXCR7 completely blocked elevated 3-NT in the presence of SDF-1.
Figure 4.

Upregulating CXCR7 expression attenuates the superoxide level and oxidative damage in EPCs induced by ox-LDL. A, Fluorescent Images and quantitation of superoxide levels in EPCs treated with or without ox-LDL (50 μg/mL) in the presence or absence of SDF-1 (100 ng/mL) for 6 h. Superoxide was determined with the fluorescent indicator DHE, and the fluorescent intensity of DHE was measured by a fluorescent microplate reader. B, Levels of the oxidative damage marker 3-nitroryrosine (3-NT) in EPCs treated with or without ox-LDL (50μg/ml) in the presence or absence of SDF-1 (100 ng/mL) for 12 h was detected by Western blot. Three independent experiments were performed. Data shown in graphs represents the Mean ± SD. * P<0.05, vs respective control in Null-EPCs or CXCR7-EPCs; # P< 0.05, vs Null-EPCs with the same treatment; & P<0.05, vs CXCR7-EPCs with ox-LDL treatment.
Upregulating CXCR7 expression activates Nrf2 via Akt/GSK-3β/Fyn pathway in EPCs
Nrf2 plays a critical role in the cellular response to oxidative stress25, 26. Because increased CXCR7 expression decreased both ox-LDL-and HG-induced oxidative stress, we tested whether CXCR7 upregulation activate Nrf2 and its downstream target genes. There was no significant change in total Nrf2 protein and/or Nrf2 mRNA expression under any treatment condition in either Null-EPCs or CXCR7-EPCs (Figure 5A&B and Online Figure VIII, A). However when treated with ox-LDL or HG nuclear Nrf2 levels significantly declined in Null-EPCs but not in CXCR7-EPCs. In fact, SDF-1 pretreatment significantly increased nuclear Nrf2 levels in CXCR7-EPCs compared with Null-EPCs (Figure 5A and Online Figure VIII, A) which was also supported by studies of Nrf2 nuclear immunofluorescent localization in Null-EPCs and CXCR7-EPCs (Figure 5C). These results indicate that CXCR7 upregulation increased Nrf2 levels in the nucleus but did not increase total Nrf2 expression. Most importantly, we found that protein and/or mRNA expression of antioxidant Nrf2 target genes HO-1, NQO1 and catalase were significantly higher in CXCR7-EPCs compared with Null-EPCs in the presence of ox-LDL or HG, especially with SDF-1 pretreatment (Figure 5A&B and Online Figure VIII, A). More importantly, CXCR7 upregulation mediated Nrf2 nuclear translocation and downstream gene activation were also confirmed in early EPCs from db/db mice (Online Figure VIII, B). These changes in Nrf2 target gene expression are fully consistent with the changes found in Nrf2 nuclear localization produced by CXCR7 upregulation (Figure 5A&C and Online Figure VIII, A, B) and suggest that SDF-1/CXCR7 activates Nrf2 transcriptional function.
Figure 5.
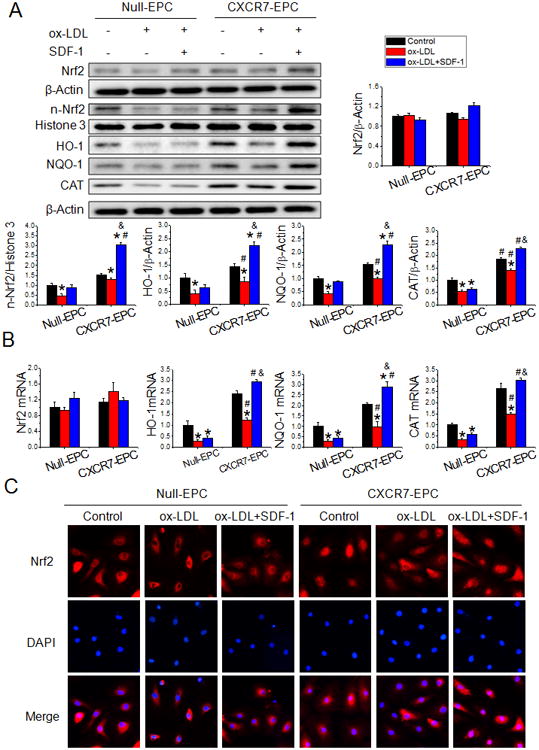
Upregulating CXCR7 activates nuclear Nrf2 signaling in EPCs. Null-EPCs or CXCR7-EPCs were exposed to ox-LDL (0 or 50μg/ml) in the presence or absence of SDF-1 (100 ng/mL) for 12 h. A, Protein levels of Nrf2 and its downstream target genes HO-1, NQO-1 and CAT and nuclear expression of Nrf2 (n-Nrf2) were detected by Western blot. B, The mRNA expression of Nrf2 and its downstream target genes HO-1, NQO-1 and CAT were determined by real-time PCR. C, Nrf2 nuclear translocation was determined in fixed cells by immunofluorescent staining. Three independent experiments were performed. Data shown in graphs represents the Means ± SD. * P<0.05, vs respective control in Null-EPCs or CXCR7-EPCs; # P< 0.05, vs Null-EPC with the same treatment; & P<0.05, vs CXCR7-EPC with ox-LDL treatment. NQO-1: NAD(P)H dehydrogenase quinone 1; HO-1: Heme oxygenase-1; CAT: catalase.
SDF-1/CXCR7 is thought to promote CD34+ hematopoietic stem/progenitor cell cycling and survival via Akt signaling27 and SDF-1/CXCR7 also regulates cell migration28 by Akt signaling. Furthermore, the function of Nrf2 can be regulated by the Akt/GSK-3β/Fyn pathway in fibroblasts and PC12 cells by controlling Fyn-mediated export and degradation of nuclear Nrf229, 30. We found that exposure of Null-EPCs and CXCR7-EPCs to ox-LDL or HG significantly decreased the phosphorylation of Akt and GSK-3β and increased the level of Fyn in the nucleus (Figure 6A and Online Figure IX, A). In CXCR7-EPCs, SDF-1 pretreatment prevented ox-LDL- and HG-induced decreases in Akt and GSK-3β phosphorylation, and nuclear accumulation of Fyn (Figure 6A and Online Figure IX, A). Furthermore, CXCR7 upregulation mediated preservation Akt/GSK-3β/Fyn pathway was also confirmed in early EPCs from db/db mice (Online Figure IX, B). More importantly, the ability of SDF-1 and CXCR7 to reverse the effects of ox-LDL and HG on Akt, GSK-3β and Fyn were blocked by the specific pharmacological inhibitor of PI3K/Akt, Wortmannin (Figure 6A and Online Figure IX, A). To determine if the PI3K/Akt pathway was critical to all of the protective effects observed in CXCR7-EPCs treated with SDF-1, Wortmannin was tested against parameters. Wortmannin blockade completely abolished SDF-1/CXCR7 axis-mediated Nrf2 nuclear localization and upregulation of its downstream target genes at both protein and/or mRNA levels (Figure 6B&C and Online Figure IX, C), resulting in a complete abolishment of SDF-1/CXCR7 axis-preserved cell survival (Figure 6D and Online Figure IX, D), tube formation (Figure 6E and Online Figure IX, E) and TEM ability (Figure 6F and Online Figure IX, F) in CXCR7-EPCs. These results suggest that upregulation of SDF-1/CXCR7 axis preserves EPCs function via regulating Akt/GSK-3β/Fyn pathway to activate Nrf2 function.
Figure 6.


Upregulating CXCR7 activates Nrf2 via Akt/GSK-3β/Fyn pathway. Null-EPCs or CXCR7-EPCs were pretreated with or without PI3K inhibitor wortmannin for 30 min, and then exposed to ox-LDL (50μg/ml) for 12 h in the presence or absence of SDF-1 (100 ng/mL). A, The phosphorylation of Akt and GSK-3β, and the nuclear translocation of Fyn (n-Fyn) were evaluated by Western blot. B, C, The expression of nuclear Nrf2 (n-Nrf2) and its downstream target genes (HO-1, NQO-1 and CAT) was evaluated by Western blot (B) and real-time PCR (C). The results were normalized to the control group of Null-EPCs. D, The apoptosis of EPCs was analyzed by flow cytometry using Annexin V/PI staining. E, The angiogenic function of EPCs was determined by tube formation assay, the tube length was normalized to the control group of Null-EPCs. F, The trans-endothelial migration abilities of EPCs was analyzed by trans-well assay. Three independent experiments were performed for each study. Data shown in graphs represents the Means ± SD. * P<0.05 vs respective control in Null-EPCs or CXCR7-EPCs; # P< 0.05 vs Null-EPC with the same treatment; & P<0.05 vs CXCR7-EPC with ox-LDL treatment in presence of SDF-1.
Knockdown of Nrf2 impairs the protective effects of CXCR7 on EPCs
To confirm the pivotal role of Nrf2 in SDF-1/CXCR7 protection we performed knockdown of Nrf2 gene expression with Nrf2-shRNA (Online Figure X, A). The vector sh-Nrf2 2 had the best knockdown efficiency among the 3 Nrf2-shRNA (Online Figure X, A) vectors tested and this vector was used to knockdown Nrf2 in subsequent studies: Nonsense shRNA did not alter Nrf2 expression and was used as control vector. Knockdown of Nrf2 expression blocked most of the protective effects we had seen with SDF-1 and CXCR7 (Figures 2-6 and Figures S5-9):In SDF-1 treated CXCR7-EPCs Nrf2-shRNA eliminated the effect of elevated CXCR7 on nuclear accumulation of Nrf2 (Online Figure X, B, C), induction of antioxidant genes (Online Figure X, B, C), superoxide production (Online Figure X, D, E), apoptosis (Figure 7A and Online Figure XI, A) and tube formation (Figure 7B and Online Figure XI, B) under ox-LDL or HG treatment conditions. The one assay not effected by Nrf2 knockdown in SDF-1 treated CXCR7-EPCs was TEM ability (Figure 7C and Online Figure XI, C), implying that Nrf2 is not important for TEM of EPCs. Knockdown of Nrf2 expression did not affect increased phosphorylation of Akt and GSK-3β or reduced nuclear accumulation of Fyn in SDF-1 treated CXCR7-EPCs (Online Figure XII, A, B). This result indicates that SDF-1/CXCR7 effects on Nrf2 are downstream of Akt/GSK/Fyn. Nrf-2 knockdown tested in vivo by transplantation into db/db mice effectively eliminated the ability of elevated CXCR7 expression in CXCR7-EPCs to augment of EPC-mediated ischemic blood perfusion (Figure 8A) and angiogenesis (Figure 8B).
Figure 7.

Knockdown of Nrf2 attenuates the protective effects of CXCR7 upregulation on EPCs. CXCR7-EPCs or Null-EPCs were transfected with lentivirus vector encoding Nrf2 shRNA (CXCR7/sh-Nrf2-EPCs) or control shRNA (CXCR7/sh-Ctrl-EPCs or Null/sh-Ctrl-EPCs). Following shRNA transfection the protective effects of SDF-1/CXCR7 on EPC survival, angiogenesis and trans-endothelial migration were evaluated as described in Figure 6. A, Apoptosis was analyzed by flow cytometry. B, Angiogenic function was determined by tube formation assay. C, Trans-endothelial migration was analyzed by trans-well assay. Three independent experiments were performed for each study. Data shown in graphs represents the Means ± SD. * P<0.05 vs respective control in Null/sh-Ctrl-EPCs, CXCR7/sh-Ctrl-EPCs or CXCR7/sh-Nrf2-EPCs; # P< 0.05 vs Null/sh-Ctrl-EPC with the same treatment; & P<0.05 vs CXCR7/sh-Ctrl-EPC with the same treatment.
Figure 8.
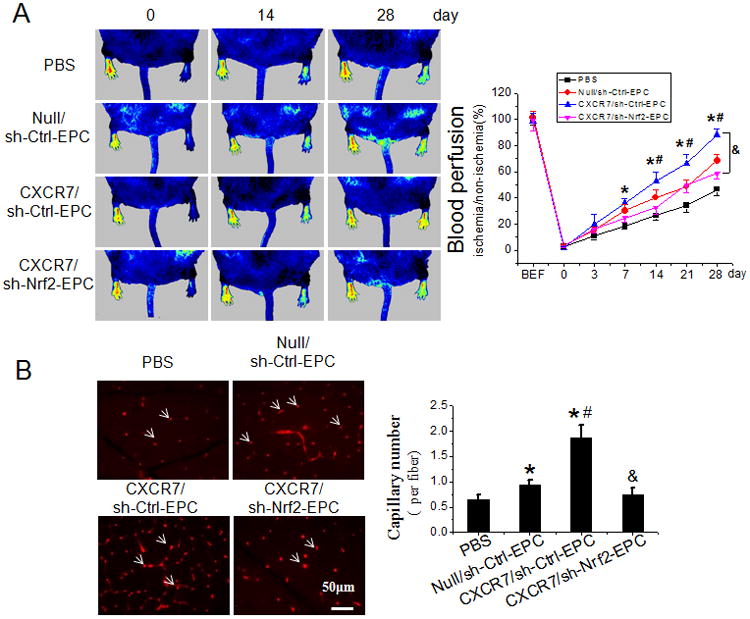
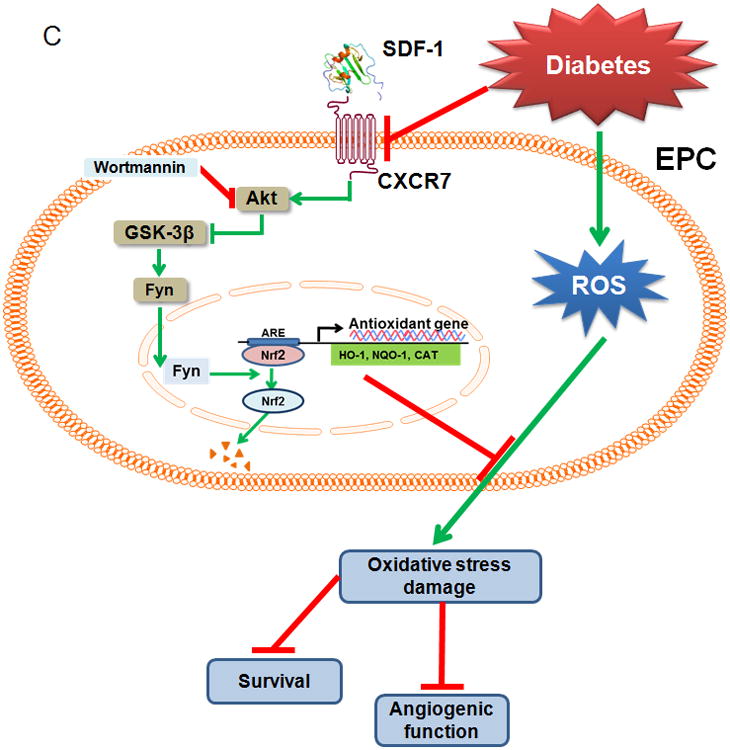
Knockdown of Nrf2 attenuates the beneficial effects of CXCR7 upregulation on EPC mediated angiogenesis in ischemic limb of db/db diabetic mice. The effects of Nrf2 knockdown on the beneficial effects of CXCR7 upregulation on EPC mediated angiogenesis and blood perfusion were evaluated in the HLI model in db/db diabetic mice as described in Figure 2 and Online Figure VI. A, Time course of blood perfusion after HLI surgery with or without EPCs transplantation shown in images and quantitative data analysis. Blood perfusion is the ratio of ischemic to non-ischemic limb perfusion measured by PSI. B, Immunofluorescent staining and quantitation of isolectin-positive capillaries (white arrows) in transverse sections of gastrocnemius muscle tissue from ischemic hind limbs 28 days after HLI surgery. Capillary density was expressed as isolectin-positive capillaries per muscle fiber. n=8 mice per group. Data shown in graphs represents the Means ± SD. * P<0.05 vs PBS, # P< 0.05 vs Null/sh-Ctrl-EPC group; & P<0.05 vs CXCR7/sh-Ctrl-EPC group. C, Schematic illustration of the protective effects of SDF-1/CXCR7 on EPCs under diabetic conditions. Diabetes decreases expression of CXCR7 in EPCs and induces oxidative stress, which impairs the survival and angiogenic function of EPCs. Under diabetic conditions upregulation of SDF-1/CXCR7 signaling improves EPC survival and function predominantly by Nrf2 activation mediated by increasing phosphorylation of Akt and GSK-3β and inhibiting Fyn-mediated export and degradation of nuclear Nrf2.
Discussion
Patients with type 1 or type 2 diabetes have decreased number and impaired angiogenic function of EPCs, which plays a causative role in the development and progression of virtually all diabetic complications31. Despite its critical importance, the mechanism for impaired EPC function in diabetes remains largely unknown. The present study provides three new lines of evidence implicating CXCR7 initiated pathways in diabetic EPC dysfunction: The first novel finding is the strong relationship between CXCR7 levels and diabetic EPC dysfunction: CXCR7 levels are decreased in diabetic EPCs, diabetic dysfunction is mimicked by CXCR7 knockdown and EPC dysfunction is reversed by upregulation of CXCR7 levels. The second innovative finding is that the benefit of increased CXCR7 expression requires activation of Nrf2 antioxidant signaling. Our third novel finding is that the Akt/GSK-3β/Fyn pathway mediates CXCR7 induced Nrf2 activation in diabetic EPCs.
Emerging evidence indicates that CXCR7 is essential for regulation of multiple cellular functions32. Our previous studies demonstrated that CXCR7 plays a critical role in the regulation of adhesion and survival of EPCs from both rat bone marrow and human cord blood14, 15. In the present study we demonstrate that diabetes significantly decreased EPC cellular and cell surface levels of CXCR7 but produces no similar change in CXCR4 levels (Figure 1A&B). We addressed what factors in diabetes might cause down-regulation of CXCR7 and dysfunction of EPCs. To test if the decrease of CXCR7 expression in db/db EPCs could be caused by the dysmetabolic environment of diabetes, we exposed normal EPCs to elevated ox-LDL or HG to mimic dysmetabolic diabetic conditions, as previous reported22. The results show that ox-LDL and HG both dose-dependently decrease CXCR7 but not CXCR4 levels (Online Figure IV, A, C) and indicate that CXCR7 is a strong candidate for impairment of EPC function under dysmetabolic diabetic conditions. However, the exact molecular mechanism by which the diabetic stressors HG and ox-LDL, as well as non-diabetic stressors such as hypertension16, down-regulate CXCR7 levels in EPCs remain unknown.
The most important, new finding of this study is that overexpression of CXCR7 rescued function of both diabetic EPCs and normal EPCs impaired by exposure to diabetic conditions in vitro or in vivo. Upregulation of CXCR7 levels restored angiogenic function of type 2 diabetic, db/db EPCs (Figure 1D), and protected normal EPCs treated with ox-LDL or HG from excess apoptosis and angiogenic dysfunction (Figure 3A&B and Online Figure V, A, B). Moreover, upregulation of CXCR7 expression in normal or db/db EPCs promoted homing of EPCs to ischemic tissue, increased capillary density and improved blood flow perfusion when EPCs were transplanted into a type 2 diabetic HLI model (Online Figure VI and Figure 2). It is well documented that elevated ox-LDL and hyperglycemic conditions can promote endothelial and smooth muscle dysfunction in established arterial and arteriolar vessels during diabetes33, 34. Thus, in addition to a profound impact on capillary density and neovascularization in diabetic animals reported here, a potential contribution of changes in EPC CXCR7 expression to basal myogenic tone development and vasodilatory capacity in small diameter arterioles and its impact on blood flow in diabetes warrants further investigation. Nonetheless, considering that CXCR7 overexpression reverses diabetic EPC dysfunction and improves EPC survival these results strongly support the concept that the decline in CXCR7 levels is a significant, causative factor in development of diabetic EPC dysfunction, and further, indicate that CXCR7 is a new therapeutic target for improving the ischemia-reparative capacity of EPCs in patients with diabetes. Intriguingly, EPCs from hypertensive patients have also been shown to have reduced CXCR7 levels, which coincides with impaired function16, and like diabetic EPCs, their function can be rescued by overexpression of CXCR716. The analogous findings for CXCR7 in diabetic and hypertensive EPCs emphasizes the importance of CXCR7 in maintaining adequate EPC function.
The second innovative finding of this study is that the benefit of increased CXCR7 expression requires activation of Nrf2 antioxidant signaling. Accumulating evidence demonstrates that oxidative stress in type 2 diabetes contributes to endothelial cell and EPC dysfunction23, 24, 35. Intracellular ROS level are elevated in EPCs from diabetic mice or EPCs cultured under HG or high lipid conditions24, 35, 36. Methods that reduced intracellular ROS levels restore function of diabetic EPCs37, 38. In the present study, we found that the ROS level of EPCs cultured with ox-LDL or HG were increased. Upregulating CXCR7 expression, especially when combined with SDF-1 pretreatment, almost completely inhibited the increase in ROS level of EPCs exposed to ox-LDL or HG (Figure 4 and Online Figure VII). This indicates that upregulation of the SDF-1/CXCR7 axis improves EPC antioxidant capacity.
We had previous seen that Nrf2 is a critical redox sensor and one of the master regulators of the antioxidant response39. Nrf2 binds to regulatory antioxidant response elements and activates transcription of many antioxidant genes, such as HO-1, and NQO-1 that counteract ROS40. In this study, we demonstrate for the first time that upregulating CXCR7 markedly increased nuclear accumulation of Nrf2 and increases the expression of downstream antioxidant target genes (HO-1, NQO-1 and catalase, Figure 5 and Online Figure VIII). These results indicate that upregulating CXCR7 improves EPC antioxidant capacity by activating Nrf2 transcriptional function. The essential role Nrf2 plays in endothelial function has been widely appreciated. Florzyk et al reported that lack of Nrf2 attenuated survival, proliferation and angiogenic function of endothelial cells in vitro and in vivo; while angiogenic factors increased the nuclear localization of Nrf2 and the expression of its target genes HO-1and NQO-1, which stimulated tube formation of endothelial cells41. Furthermore, knockdown of Nrf2 inhibits angiogenesis of rat cardiac microvascular endothelial cells under hypoxic conditions42. Consistent with these findings in endothelial cells, our studies in EPCs revealed that lentivirus-mediated, shRNA knockdown of Nrf2 almost completely abrogated the ability of elevated CXCR7 levels to augment EPC survival and tube formation in vitro (Figure 7 and Online Figure XI), and to enhance ischemic angiogenesis and blood perfusion in vivo (Figure 8). These findings prove that Nrf2 plays an essential role in the ability of CXCR7 upregulation to augment EPC function.
Our third important, new finding is that the Akt/GSK-3β/Fyn pathway mediates the activation of Nrf2 produced by elevated CXCR7 in diabetic EPCs. The mechanism of Nrf2 activation has been widely investigated43, 44. In PC12 cells, it was found that puerarin triggers Akt activation, which in turn inhibits GSK-3β activation resulting in more Nrf2 nuclear translocation. Moreover, it was demonstrated that CXCR7 activates PI3K/Akt independently of CXCR427, 28. In this study, diabetes, ox-LDL and HG all reduced Akt phosphorylation in EPCs. Using in vitro studies, we showed that decreased Akt phosphorylation produced by ox-LDL or HG exposure could be prevented by SDF-1 if CXCR7 expression was elevated in EPCs (Figure 6A and Online Figure IX, A, B), showing that augmentation of the SDF-1/CXCR7 axis activates Akt in EPCs. However, the direct linkage between the SDF-1/CXCR7 axis and phosphorylation of Akt in EPCs remains a key unanswered question.
Several recent studies showing that the Akt/GSK-3β/Fyn pathway plays a pivotal role in regulation of Nrf2 nuclear export and degradation25, 29, 30, 45, 46. Based on those studies and Akt phosphorylation results, we hypothesized that SDF-1/CXCR7 signaling activates Nrf2 via the Akt/GSK-3β/Fyn pathway. Consistent with this hypothesis we found that stimulating the SDF-1/CXCR7 axis not only increased phosphorylation of Akt but also inhibited GSK-3β activity which decreased nuclear accumulation of Fyn (Figure 6A and Online Figure IX, A, B). It is known that Fyn can phosphorylate Nrf2 tyrosines, promoting Nrf2 nuclear export and degradation45, 47. Importantly, we found that a PI3K/Akt specific inhibitor (wortmannin) almost completely abolished SDF-1/CXCR7 activation of Nrf2 and expression of its downstream genes (Figure 6B&C and Online Figure IX, C). This was accompanied by almost complete attenuation of the protective effects of SDF-1/CXCR7 activation on EPC survival and angiogenic function under ox-LDL and HG treatment conditions (Figure 6D&E and Online Figure IX, D, E).
In conclusion, upregulation of signaling of the SDF-1/CXCR7 axis improves EPC survival and function under diabetic conditions. This is predominantly due to increased Nrf2 activation, mediated in EPCs by increasing Akt and GSK-3β phosphorylation and inhibiting Fyn-mediated Nrf2 nuclear export and degradation. By preserving Nrf2 nuclear localization Nrf2 function is enhanced and antioxidant gene expression increased, protecting EPCs from diabetes, ox-LDL and HG-induced oxidative damage as illustrated in Figure 8C.
Supplementary Material
Novelty and Significance.
What Is Known?
Diabetes associated dysregulation of endothelial regeneration may attribute to the dysfunction of endothelial progenitor cells (EPCs).
CXC chemokine receptor 7 (CXCR7) plays a critical role in EPC angiogenic function. Diabetes may alter expression and function of CXCR7 in EPCs but this remains untested.
What New Information Does This Article Contribute?
CXCR7 is necessary for diabetic EPCs survival, resistance to oxidative stress and angiogenic capacity.
The benefits of CXCR7 in EPCs under diabetic conditions require activation of nuclear factor (erythroid-derived 2)-like 2 (Nrf2) antioxidant signaling.
The protein kinase B/glycogen synthase kinase 3β/tyrosine kinase Fyn (Akt/GSK-3β/Fyn) pathway mediates CXCR7 induced Nrf2 activation in EPCs under diabetic conditions.
EPCs respond to stromal cell-derived factor 1 through receptors CXCR7 and CXCR4. The reduced CXCR7 but not CXCR4 expression leads to impaired angiogenic function of EPCs under diabetic conditions, whereas elevating CXCR7 expression enhances EPCs resistance to diabetes-induced oxidative damage and improves therapeutic efficacy of EPCs. The benefits of CXCR7 are mediated by an Akt/GSK-3β/Fyn pathway via Nrf2 activation. Our study demonstrates for the first time that reduced CXCR7 expression contributes to diabetes-induced EPCs dysfunction and provides a novel therapeutic target for enhancing endothelial repair capacity in diabetes.
Acknowledgments
We thank Dr. Lu Cai, Department of Pediatrics, University of Louisville, for sharing reagents and equipment, and critical reading the manuscript.
Sources of Funding: This study was supported in part by a Junior Faculty Award (1-13-JF-53) from American Diabetes Association, an Innovative Grant (1-INO-2014-122-A-N) from Juvenile Diabetes Research Foundation, NSFC projects (91639111, 81573435, 81200917, 81200239, 81370917), a Key New Drug Development Grant (2012ZX09103-301-016), Scientific Research Fund of Health and Family Planning Commission of Sichuan Province (130297), a Visiting Scholarship from the China Scholarship Council, and NIH GM103492.
Nonstandard Abbreviations and Acronyms
- EPCs
endothelial progenitor cells
- SDF-1
stromal cell-derived factor 1, also named CXCL12
- CXCR4
C-X-C chemokine receptor type 4
- CXCR7
C-X-C chemokine receptor type 7
- ox-LDL
oxidized low-density lipoprotein
- CXCR7-EPCs
EPCs transduced with CXCR7 lentivirus
- Null-EPCs
EPCs transduced with control lentivirus
- PI3K/Akt
phosphatidylinositol 3-kinase/Protein kinase B
- GSK-3β
glycogen synthase kinase 3β
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- HO-1
heme oxygenase 1
- NQO-1
NAD(P)H dehydrogenase (quinone 1)
- MNCs
marrow mononuclear cells
- MOI
multiplicity of infection
- 3-NT
3-nitrotyrosine
- TEM
trans-endothelial migration
- HUVEC
human umbilical vein endothelial cells
- HLI
hind limb ischemia
- ROS
reactive oxygen species
- DHE
dihydroethidium
- PSI
perfusion speckle imager
Footnotes
Disclosures: None.
References
- 1.Muniyappa R, Sowers JR. Glycogen synthase kinase-3beta and cathepsin b in diabetic endothelial progenitor cell dysfunction: An old player finds a new partner. Diabetes. 2014;63:1194–1197. doi: 10.2337/db14-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Georgescu A. Vascular dysfunction in diabetes: The endothelial progenitor cells as new therapeutic strategy. World J Diabetes. 2011;2:92–97. doi: 10.4239/wjd.v2.i6.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williamson K, Stringer SE, Alexander MY. Endothelial progenitor cells enter the aging arena. Front Physiol. 2012;3:30. doi: 10.3389/fphys.2012.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar AH, Caplice NM. Clinical potential of adult vascular progenitor cells. Arterioscler Thromb Vasc Biol. 2010;30:1080–1087. doi: 10.1161/ATVBAHA.109.198895. [DOI] [PubMed] [Google Scholar]
- 5.Taljaard M, Ward MR, Kutryk MJ, Courtman DW, Camack NJ, Goodman SG, Parker TG, Dick AJ, Galipeau J, Stewart DJ. Rationale and design of enhanced angiogenic cell therapy in acute myocardial infarction (enact-ami): The first randomized placebo-controlled trial of enhanced progenitor cell therapy for acute myocardial infarction. Am Heart J. 2010;159:354–360. doi: 10.1016/j.ahj.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 6.Aicher A, Heeschen C, Sasaki K, Urbich C, Zeiher AM, Dimmeler S. Low-energy shock wave for enhancing recruitment of endothelial progenitor cells: A new modality to increase efficacy of cell therapy in chronic hind limb ischemia. Circulation. 2006;114:2823–2830. doi: 10.1161/CIRCULATIONAHA.106.628623. [DOI] [PubMed] [Google Scholar]
- 7.Grochot-Przeczek A, Kotlinowski J, Kozakowska M, Starowicz K, Jagodzinska J, Stachurska A, Volger OL, Bukowska-Strakova K, Florczyk U, Tertil M, Jazwa A, Szade K, Stepniewski J, Loboda A, Horrevoets AJ, Dulak J, Jozkowicz A. Heme oxygenase-1 is required for angiogenic function of bone marrow-derived progenitor cells: Role in therapeutic revascularization. Antioxid Redox Signal. 2014;20:1677–1692. doi: 10.1089/ars.2013.5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhatwadekar AD, Shaw LC, Grant MB. Promise of endothelial progenitor cell for treatment of diabetic retinopathy. Expert Rev Endocrinol Metab. 2010;5:29–37. doi: 10.1586/eem.09.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silvestre JS, Smadja DM, Levy BI. Postischemic revascularization: From cellular and molecular mechanisms to clinical applications. Physiol Rev. 2013;93:1743–1802. doi: 10.1152/physrev.00006.2013. [DOI] [PubMed] [Google Scholar]
- 10.Dimmeler S. Regulation of bone marrow-derived vascular progenitor cell mobilization and maintenance. Arterioscler Thromb Vasc Biol. 2010;30:1088–1093. doi: 10.1161/ATVBAHA.109.191668. [DOI] [PubMed] [Google Scholar]
- 11.Rosenkranz K, Kumbruch S, Lebermann K, Marschner K, Jensen A, Dermietzel R, Meier C. The chemokine sdf-1/cxcl12 contributes to the ‘homing’ of umbilical cord blood cells to a hypoxic-ischemic lesion in the rat brain. J Neurosci Res. 2010;88:1223–1233. doi: 10.1002/jnr.22292. [DOI] [PubMed] [Google Scholar]
- 12.Shen L, Gao Y, Qian J, Sun A, Ge J. A novel mechanism for endothelial progenitor cells homing: The sdf-1/cxcr4-rac pathway may regulate endothelial progenitor cells homing through cellular polarization. Med Hypotheses. 2011;76:256–258. doi: 10.1016/j.mehy.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 13.Balabanian K, Lagane B, Infantino S, Chow KY, Harriague J, Moepps B, Arenzana-Seisdedos F, Thelen M, Bachelerie F. The chemokine sdf-1/cxcl12 binds to and signals through the orphan receptor rdc1 in t lymphocytes. J Biol Chem. 2005;280:35760–35766. doi: 10.1074/jbc.M508234200. [DOI] [PubMed] [Google Scholar]
- 14.Yan X, Cai S, Xiong X, Sun W, Dai X, Chen S, Ye Q, Song Z, Jiang Q, Xu Z. Chemokine receptor cxcr7 mediates human endothelial progenitor cells survival, angiogenesis, but not proliferation. J Cell Biochem. 2012;113:1437–1446. doi: 10.1002/jcb.24015. [DOI] [PubMed] [Google Scholar]
- 15.Dai X, Tan Y, Cai S, Xiong X, Wang L, Ye Q, Yan X, Ma K, Cai L. The role of cxcr7 on the adhesion, proliferation and angiogenesis of endothelial progenitor cells. J Cell Mol Med. 2011;15:1299–1309. doi: 10.1111/j.1582-4934.2011.01301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang XY, Su C, Cao Z, Xu SY, Xia WH, Xie WL, Chen L, Yu BB, Zhang B, Wang Y, Tao J. Cxcr7 upregulation is required for early endothelial progenitor cell-mediated endothelial repair in patients with hypertension. Hypertension. 2014;63:383–389. doi: 10.1161/HYPERTENSIONAHA.113.02273. [DOI] [PubMed] [Google Scholar]
- 17.Georgescu A, Alexandru N, Constantinescu A, Titorencu I, Popov D. The promise of epc-based therapies on vascular dysfunction in diabetes. Eur J Pharmacol. 2011;669:1–6. doi: 10.1016/j.ejphar.2011.07.035. [DOI] [PubMed] [Google Scholar]
- 18.Fadini GP, Losordo D, Dimmeler S. Critical reevaluation of endothelial progenitor cell phenotypes for therapeutic and diagnostic use. Circ Res. 2012;110:624–637. doi: 10.1161/CIRCRESAHA.111.243386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan Y, Li Y, Xiao J, Shao H, Ding C, Arteel GE, Webster KA, Yan J, Yu H, Cai L, Li X. A novel cxcr4 antagonist derived from human sdf-1beta enhances angiogenesis in ischaemic mice. Cardiovasc Res. 2009;82:513–521. doi: 10.1093/cvr/cvp044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Urbich C, Heeschen C, Aicher A, Dernbach E, Zeiher AM, Dimmeler S. Relevance of monocytic features for neovascularization capacity of circulating endothelial progenitor cells. Circulation. 2003;108:2511–2516. doi: 10.1161/01.CIR.0000096483.29777.50. [DOI] [PubMed] [Google Scholar]
- 21.Kalka C, Masuda H, Takahashi T, Kalka-Moll WM, Silver M, Kearney M, Li T, Isner JM, Asahara T. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc Natl Acad Sci U S A. 2000;97:3422–3427. doi: 10.1073/pnas.070046397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamed S, Brenner B, Abassi Z, Aharon A, Daoud D, Roguin A. Hyperglycemia and oxidized-ldl exert a deleterious effect on endothelial progenitor cell migration in type 2 diabetes mellitus. Thromb Res. 2010;126:166–174. doi: 10.1016/j.thromres.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 23.Dernbach E, Urbich C, Brandes RP, Hofmann WK, Zeiher AM, Dimmeler S. Antioxidative stress-associated genes in circulating progenitor cells: Evidence for enhanced resistance against oxidative stress. Blood. 2004;104:3591–3597. doi: 10.1182/blood-2003-12-4103. [DOI] [PubMed] [Google Scholar]
- 24.Sorrentino SA, Bahlmann FH, Besler C, Muller M, Schulz S, Kirchhoff N, Doerries C, Horvath T, Limbourg A, Limbourg F, Fliser D, Haller H, Drexler H, Landmesser U. Oxidant stress impairs in vivo reendothelialization capacity of endothelial progenitor cells from patients with type 2 diabetes mellitus: Restoration by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Circulation. 2007;116:163–173. doi: 10.1161/CIRCULATIONAHA.106.684381. [DOI] [PubMed] [Google Scholar]
- 25.Cheng Y, Zhang J, Guo W, Li F, Sun W, Chen J, Zhang C, Lu X, Tan Y, Feng W, Fu Y, Liu GC, Xu Z, Cai L. Up-regulation of nrf2 is involved in fgf21-mediated fenofibrate protection against type 1 diabetic nephropathy. Free Radic Biol Med. 2016;93:94–109. doi: 10.1016/j.freeradbiomed.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jyrkkanen HK, Kansanen E, Inkala M, Kivela AM, Hurttila H, Heinonen SE, Goldsteins G, Jauhiainen S, Tiainen S, Makkonen H, Oskolkova O, Afonyushkin T, Koistinaho J, Yamamoto M, Bochkov VN, Yla-Herttuala S, Levonen AL. Nrf2 regulates antioxidant gene expression evoked by oxidized phospholipids in endothelial cells and murine arteries in vivo. Circ Res. 2008;103:e1–9. doi: 10.1161/CIRCRESAHA.108.176883. [DOI] [PubMed] [Google Scholar]
- 27.Torossian F, Anginot A, Chabanon A, Clay D, Guerton B, Desterke C, Boutin L, Marullo S, Scott MG, Lataillade JJ, Le Bousse-Kerdiles MC. Cxcr7 participates in cxcl12-induced cd34+ cell cycling through beta-arrestin-dependent akt activation. Blood. 2014;123:191–202. doi: 10.1182/blood-2013-05-500496. [DOI] [PubMed] [Google Scholar]
- 28.Chen D, Xia Y, Zuo K, Wang Y, Zhang S, Kuang D, Duan Y, Zhao X, Wang G. Crosstalk between sdf-1/cxcr4 and sdf-1/cxcr7 in cardiac stem cell migration. Sci Rep. 2015;5:16813. doi: 10.1038/srep16813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bitar MS. The gsk-3beta/fyn/nrf2 pathway in fibroblasts and wounds of type 2 diabetes: On the road to an evidence-based therapy of non-healing wounds. Adipocyte. 2012;1:161–163. doi: 10.4161/adip.20235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li C, Pan Z, Xu T, Zhang C, Wu Q, Niu Y. Puerarin induces the upregulation of glutathione levels and nuclear translocation of nrf2 through pi3k/akt/gsk-3beta signaling events in pc12 cells exposed to lead. Neurotoxicol Teratol. 2014;46:1–9. doi: 10.1016/j.ntt.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 31.Fadini GP, Sartore S, Agostini C, Avogaro A. Significance of endothelial progenitor cells in subjects with diabetes. Diabetes Care. 2007;30:1305–1313. doi: 10.2337/dc06-2305. [DOI] [PubMed] [Google Scholar]
- 32.Puchert M, Engele J. The peculiarities of the sdf-1/cxcl12 system: In some cells, cxcr4 and cxcr7 sing solos, in others, they sing duets. Cell Tissue Res. 2014;355:239–253. doi: 10.1007/s00441-013-1747-y. [DOI] [PubMed] [Google Scholar]
- 33.Kizub IV, Klymenko KI, Soloviev AI. Protein kinase c in enhanced vascular tone in diabetes mellitus. Int J Cardiol. 2014;174:230–242. doi: 10.1016/j.ijcard.2014.04.117. [DOI] [PubMed] [Google Scholar]
- 34.Galle J, Hansen-Hagge T, Wanner C, Seibold S. Impact of oxidized low density lipoprotein on vascular cells. Atherosclerosis. 2006;185:219–226. doi: 10.1016/j.atherosclerosis.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 35.Ohshima M, Li TS, Kubo M, Qin SL, Hamano K. Antioxidant therapy attenuates diabetes-related impairment of bone marrow stem cells. Circ J. 2009;73:162–166. doi: 10.1253/circj.cj-08-0123. [DOI] [PubMed] [Google Scholar]
- 36.Callaghan MJ, Ceradini DJ, Gurtner GC. Hyperglycemia-induced reactive oxygen species and impaired endothelial progenitor cell function. Antioxid Redox Signal. 2005;7:1476–1482. doi: 10.1089/ars.2005.7.1476. [DOI] [PubMed] [Google Scholar]
- 37.Di Stefano V, Cencioni C, Zaccagnini G, Magenta A, Capogrossi MC, Martelli F. P66shca modulates oxidative stress and survival of endothelial progenitor cells in response to high glucose. Cardiovasc Res. 2009;82:421–429. doi: 10.1093/cvr/cvp082. [DOI] [PubMed] [Google Scholar]
- 38.Chang J, Li Y, Huang Y, Lam KS, Hoo RL, Wong WT, Cheng KK, Wang Y, Vanhoutte PM, Xu A. Adiponectin prevents diabetic premature senescence of endothelial progenitor cells and promotes endothelial repair by suppressing the p38 map kinase/p16ink4a signaling pathway. Diabetes. 2010;59:2949–2959. doi: 10.2337/db10-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tan Y, Ichikawa T, Li JQ, Si QS, Yang HT, Chen XB, Goldblatt CS, Meyer CJ, Li XK, Cai L, Cui TX. Diabetic downregulation of nrf2 activity via erk contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes. 2011;60:625–633. doi: 10.2337/db10-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murakami S, Motohashi H. Roles of nrf2 in cell proliferation and differentiation. Free Radic Biol Med. 2015;88:168–178. doi: 10.1016/j.freeradbiomed.2015.06.030. [DOI] [PubMed] [Google Scholar]
- 41.Florczyk U, Jazwa A, Maleszewska M, Mendel M, Szade K, Kozakowska M, Grochot-Przeczek A, Viscardi M, Czauderna S, Bukowska-Strakova K, Kotlinowski J, Jozkowicz A, Loboda A, Dulak J. Nrf2 regulates angiogenesis: Effect on endothelial cells, bone marrow-derived proangiogenic cells and hind limb ischemia. Antioxid Redox Signal. 2014;20:1693–1708. doi: 10.1089/ars.2013.5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuang L, Feng J, He G, Jing T. Knockdown of nrf2 inhibits the angiogenesis of rat cardiac micro-vascular endothelial cells under hypoxic conditions. Int J Biol Sci. 2013;9:656–665. doi: 10.7150/ijbs.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Um HC, Jang JH, Kim DH, Lee C, Surh YJ. Nitric oxide activates nrf2 through s-nitrosylation of keap1 in pc12 cells. Nitric Oxide. 2011;25:161–168. doi: 10.1016/j.niox.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 44.Bryan HK, Olayanju A, Goldring CE, Park BK. The nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem Pharmacol. 2013;85:705–717. doi: 10.1016/j.bcp.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 45.Kaspar JW, Jaiswal AK. Tyrosine phosphorylation controls nuclear export of fyn, allowing nrf2 activation of cytoprotective gene expression. FASEB J. 2011;25:1076–1087. doi: 10.1096/fj.10-171553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jain AK, Jaiswal AK. Gsk-3beta acts upstream of fyn kinase in regulation of nuclear export and degradation of nf-e2 related factor 2. J Biol Chem. 2007;282:16502–16510. doi: 10.1074/jbc.M611336200. [DOI] [PubMed] [Google Scholar]
- 47.Jain AK, Jaiswal AK. Phosphorylation of tyrosine 568 controls nuclear export of nrf2. J Biol Chem. 2006;281:12132–12142. doi: 10.1074/jbc.M511198200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.