

Abstract
Organellar ion channels are essential for cell physiology. Their activities are often regulated by Ca2+ and H+, which are concentrated in many organelles. Here we report a novel structural element critical for Ca2+/pH dual regulation of TRPML1, a Ca2+ release channel crucial for endolysosomal functions. TRPML1 mutations cause mucolipidosis type IV (MLIV), a severe lysosomal storage disorder characterized by neurodegeneration, mental retardation and blindness. We obtained high-resolution crystal structures of a 213-amino acid luminal domain of human TRPML1 that harbors three missense MLIV-causing mutations. This domain forms a tetramer with a highly electronegative central pore formed by a novel luminal pore-loop. Cysteine crosslinking and cryo-EM confirm this structure in the full-length channel. Structure-function studies demonstrate that Ca2+ and H+ interact with the luminal pore to exert physiologically important regulation. The MLIV-causing mutations disrupt the luminal domain structure and cause TRPML1 mislocalization. Our study provides a structural underpinning for TRPML1's regulation, assembly and pathogenesis.
Introduction
Many types of ion channels are present in the membranes of intracellular organelles such as the endoplasmic reticulum (ER), Golgi apparatus, mitochondria, endosomes and lysosomes1-6. These channels help establish and maintain ionic concentration gradients across organellar membranes, and/or serve as release channels for key signaling ions, including Ca2+ and H+. Many organelles have high concentrations of Ca2+ and H+ in their lumen1,3-5,7. These ions, in turn, regulate the activities of numerous organellar ion channels8-12. Elucidating the molecular mechanisms underlying this universal regulation is instrumental in understanding the physiological and pathophysiological functions of organellar ion channels.
TRPML1 is a member of the transient receptor potential mucolipin (TRPML) channel subfamily, first identified as a genetic determinant of mucolipidosis type IV (MLIV)13-15, one of ∼50 lysosomal storage disorders. Children with MLIV often show cognitive, linguistic and motor deficits in their early years of age and are sometimes blind6,16-18. TRPML1 is located primarily in lysosomes where its main role is to conduct Ca2+ from the lysosome lumen to the cytoplasm5-6,19-23. This Ca2+ release step is important for many lysosome-dependent cellular events, including exocytosis5-6,21,24-25, membrane trafficking5-6,25-27, and autophagy5-6,24-25,28-30. TRPML1 activity is markedly regulated by luminal/extracellular Ca2+ and H+6,19-23,31-32. In the presence of divalent ions, TRPML1 currents are greatly potentiated by H+, via unknown mechanisms6,19-23,31-32. The dual regulation by Ca2+ and H+ has clear physiological relevance: TRPML1 exists primarily in lysosomes, and during biogenesis and lysosomal exocytosis, it can also be present in other organelles, including late endosomes and the plasma membrane23-25,33. These distinct subcellular compartments have different pH and Ca2+ concentrations1,5-6,23. The extracellular side of the plasma membrane has 1.8-2 mM Ca2+ and a near-neutral pH of 7.2-7.4, whereas late endosomes have 0.5 mM Ca2+ and an acidic pH of 5.5-6.0. Lysosomes have 0.5-0.6 mM Ca2+ and an even more acidic pH of 4.5-5.0, a condition crucial for the activity of their native hydrolases23,34. Thus, depending on its subcellular location, TRPML1 may be regulated to different extents by Ca2+ and pH to exhibit different activities. The molecular mechanism of this dual regulation is unknown.
The crucial role of TRPML1 in cellular processes is further demonstrated by the existence of >20 mutations in the TRPML1 gene that have been linked to MLIV6,18,35. Of these mutations, some (such as frameshift or nonsense mutations) completely abolish TRPML1 expression, but many (>12) are single amino acid missense mutations6. How these missense mutations alter the structure and function of TRPML1 is largely unclear.
We undertook a structural approach to investigate the molecular mechanism of Ca2+/pH dual regulation of TRPML1 and MLIV pathogenesis, focusing on the linker between the first two transmembrane segments (S1 and S2) of TRPML1, which we refer to as the I-II linker. Like other TRP channel subunits, TRPML1 contains six transmembrane segments (S1-S6, Supplementary Fig. 1a). The I-II linker accounts for more than one third of the channel's length (Supplementary Fig. 1b). In lysosomes and endosomes, this linker faces the lumen (it is therefore also referred to as the ‘luminal linker’); on the plasma membrane, it faces the extracellular solution (Supplementary Fig. 1a). Moreover, it contains three single-amino acid missense mutations that cause MLIV (Supplementary Fig. 1b)6,17-18,36-37. We solved the crystal structures of the luminal linker at three different pH conditions (corresponding to the pH in lysosomes, endosomes and the extracellular milieu) and elucidated its role in the dual regulation of TRPML1 by luminal Ca2+ and pH, TRPML1 channel assembly, and MLIV pathogenesis.
Results
TRPML1 channel activity is regulated by luminal Ca2+ and pH
To obtain human TRPML1 currents, we followed a well-established method by expressing a constitutively active mutant channel named TRPML1VP (tagged with EGFP on its N-terminus) in HEK 293T cells20,38. TRPML1VP contains the V432P mutation, which mimics a spontaneous gain-of-function mutation (A419P) in mouse TRPML3 that causes the varitint-waddler (Va) phenotype39-41. The V432P mutation enables TRPML1 to traffic to the plasma membrane and become spontaneously active20,38. In whole-cell patch-clamp recordings, TRPML1VP produced a strong inward-rectifying current in a nominal divalent ion-free (NDF) extracellular/luminal solution at pH 7.4 (Fig. 1a, b). Addition of Ca2+ to the extracellular/luminal solution inhibited the current in a dose-dependent manner. A dose-response curve yielded an apparent IC50 of 0.27 mM, with a Hill coefficient of 1 (Fig. 1c), suggesting a one-to-one blocking mechanism.
Figure 1.
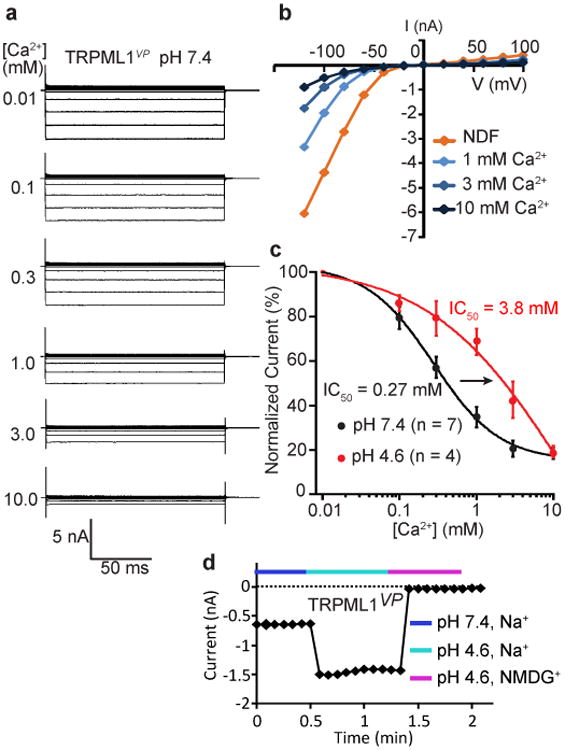
Dual regulation of TRPML1 by Ca2+ and pH. (a) Families of TRPML1vp currents at pH 7.4 and the indicated concentrations of extracellular Ca2+. (b) Current-voltage relationship of the currents in a at the indicated Ca2+ concentrations. NDF: nominal divalent cation free. (c) Dose-response relationships of Ca2+ inhibition of TRPML1vp at pH 7.4 and 4.6, at a potential of -80 mV. Error bars represent SEM. Number of recordings is shown in parentheses. Solid curves are fits to the Hill equation. (d) Time course of TRPML1vp currents at the indicated pH, with 1 mM Ca2+ and either Na+ or NMDG+ as the charge carrier.
Ca2+ inhibition of TRPML1 was strongly dependent on extracellular/luminal pH, becoming much weaker when the pH of the extracellular/luminal solution was lowered from 7.4 to 4.6, the pH of the lysosome lumen. The dose-response curve was shifted to the right at pH 4.6 (Fig. 1c), increasing the apparent IC50 by 14-fold to 3.8 mM. Intriguingly, the shape of the dose-response curve was different and the Hill coefficient was changed from 1 to 0.5 (Fig. 1c), suggesting negative cooperativity between extracellular/luminal Ca2+ and H+. Due to attenuated Ca2+ inhibition at pH 4.6, TRPML1VP currents were augmented when the extracellular/luminal pH was lowered from 7.4 to 4.6 (Fig. 1d).
It should be mentioned that the dual regulation by luminal Ca2+ and pH described above was observed in a gain-of-function mutant rather than the wildtype (WT) channel. Since the V432P mutation enables the channel to open constitutively, any effect of luminal Ca2+ and H+ on gating.
Crystal structure of the luminal linker
Because the TRPML1 luminal linker faces the endolysosomal lumen and constitutes a large part (>1/3) of the channel, we investigated its role in the Ca2+/pH regulation of TRPML1. We first determined the X-ray crystal structure of the luminal linker (G84-S296) of human TRPML1 at a 2.3 Å resolution at pH 6.0 (Fig. 2 and Table 1). The structure shows that the linker forms a tetramer with a four-fold rotational symmetry (Fig. 2a). The structure is ∼90 Å wide (outer diameter) and ∼30 Å tall. At the center of the tetramer exists a highly electronegative pore, named the luminal pore (Fig. 2b). Each protomer has a structural fold that consists of two long α-helices (α1 and α2, with 22 and 17 residues, respectively), two short α-helices (α3 and α4), eight β-strands, and three loops with >15 residues (between α1 and α2, α3 and β1, β4 and β5, respectively) (Fig. 2c). The four α-helices are packed tightly against a 5-stranded β sheet formed by β1, β4, β5, β6 and β7. One side of this β sheet faces the luminal pore, and the other side faces the outside of the tetramer (Fig. 2a, c). Two intrasubunit disulfide bonds are present, one between C166 in β1 and C192 in β4, and another between C253 in β6 and C284 in the short loop between β7 and β8. The structure of E199-K219 (which constitutes part of the loop between β4 and β5) was unresolved, presumably due to its flexibility.
Figure 2.
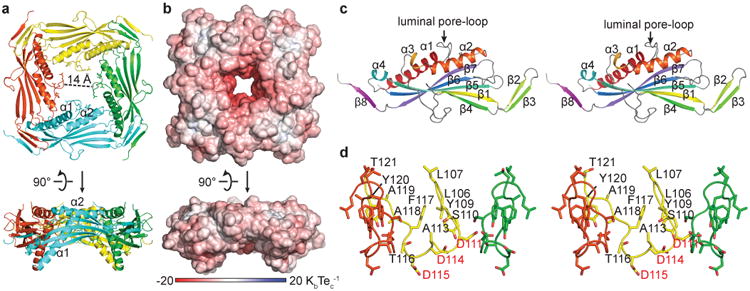
Crystal structure of the TRPML1 I-II linker. (a, b) Ribbon representation a and electrostatic potential surface representation b of the I-II linker structure at pH 6.0. Upper panels, top down views from the extracellular/luminal side of the membrane. Lower panels, side views parallel to the membrane. (c) Stereo top down view of a protomer. (d) Stereo side view of the luminal pore-loop. The front subunit is removed for clarity.
Table 1.
Data collection and refinement statistics.
| TRPML1 I-II linker at pH 6.0 (PDB 5TJA) | TRPML1 I-II linker at pH 6.0, K2Pt(CN)4 derivative | TRPML1 I-II linker at pH 4.5 (PDB 5TJB) | TRPML1 I-II linker at pH 7.5 (PDB 5TJC) | |
|---|---|---|---|---|
| Data collection | ||||
| Space group | I422 | I422 | P4212 | F432 |
| Cell dimensions | ||||
| a, b, c (Å) | 125.3, 125.3, 76.7 | 125.3, 125.3, 76.5 | 94.4, 94.4, 50.7 | 182.8, 182.8, 182.8 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Wavelength | 1.5418 | 1.5418 | 1.5418 | 1.0750 |
| Resolution (Å) | 20.0 - 2.3 (2.38 - 2.30)a | 20.0 - 2.6 (2.69 - 2.60)a | 20.0 - 2.4 (2.49 - 2.40)a | 20.0 - 2.4 (2.49 - 2.40)a |
| Rsym (%) | 9.9 (37.7) | 13.8 (64.4) | 6.8 (43.4) | 5.9 (34.0) |
| I/σ(I) | 20.0 (4.6) | 14.6 (2.8) | 19.0 (2.8) | 22.3 (3.7) |
| CC1/2 | 0.982 (0.944) | 0.961 (0.866) | 0.968 (0.872) | 0.969 (0.882) |
| Completeness (%) | 99.9 (99.6) | 99.9 (99.8) | 99.3 (98.3) | 98.0 (98.6) |
| Redundancy | 7.2 (6.1) | 6.9 (6.4) | 4.7 (3.8) | 4.2 (4.1) |
| Refinement | ||||
| Resolution (Å) | 20 - 2.3 | 20 - 2.4 | 20 - 2.4 | |
| No. reflections | 13298 (1337) | 8777 (927) | 9088 (1033) | |
| Rwork / Rfree | 0.228 / 0.253 | 0.212 / 0.252 | 0.216 / 0.253 | |
| No. atoms | ||||
| Protein | 1518 | 1268 | 1426 | |
| Water | 111 | 104 | 66 | |
| B factors | ||||
| Protein | 33.0 | 42.1 | 50.3 | |
| Water | 37.2 | 45.5 | 47.6 | |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.010 | 0.007 | 0.006 | |
| Bond angles (°) | 1.7 | 1.5 | 1.4 |
Each data set was collected from a single crystal. Values in parentheses are for the highest resolution shell.
The TRPML1 I-II linker structure has a number of unique and noteworthy features: (1) The luminal pore is lined by a novel 16-amino acid, reentrant luminal pore-loop (L106-T121) that connects to helices α1 and α2 (Fig. 2a, d). The narrowest region of the luminal pore is 14 Å, wide enough to allow fully hydrated Na+, K+ and Ca2+ ions to pass through. (2) The luminal pore contains twelve Asp residues (D111, D114, and D115 in each linker) (Fig. 2a, d), making this region highly electronegative at neutral pH (Fig. 2b). (3) The first 22 amino acids of the luminal linker (G84-F105) form an α-helix (α1) that may be of strategic importance for allosteric modulation: it connects directly to S1 at the amino end and to the luminal pore-loop at the carboxyl end (Fig. 2a). When the TRPML1 I-II linker was placed on top of the transmembrane segments of the TRPV1 structure42, α1 aligns reasonably well with S1 (Supplementary Fig. 2). Indeed, α1 appears to form a continuous α-helix with S1, thereby anchoring the I-II linker to the rest of the channel. We speculate that in the full-length channel, sequential movements of cytoplasmic and transmembrane regions may propagate to the luminal pore through α1. (4) Another α-helix (α2, R122-A138), whose N-terminus is connected to the luminal pore-loop, forms a flat perimeter on the top of the luminal pore (Fig. 2a).
Confirmation of the luminal linker structure in full-length TRPML1
The structure described above was obtained from an isolated channel domain. Is this tetrameric structure preserved in the full-length channel? We took two approaches to address this question. First, the crystal structure shows that each luminal linker interacts with two neighboring linkers (Fig. 2a), so we performed biochemical experiments to examine crosslinking among them. In this experiment, we used C. elegans instead of human TRPML1 because for unknown reasons the human TRPML1 protein remained tetrameric even in reducing SDS-PAGE (data not shown). Based on the intersubunit interface of the human TRPML1 I-II linker, we engineered three complementary pairs of cysteines in full-length C. elegans TRPML1 that are predicted to form intersubunit disulfide bonds; these include D104C in linker A and T293C in linker B, H111C in linker A and Y136C in linker B, and D158C in linker A and D137C in linker B (Fig. 3a). We also engineered six non-complementary pairs of cysteines in two neighboring linkers that are predicted not to form intersubunit disulfide bonds: D104C and Y136C, D104C and D137C, H111C and T293C, H111C and D137C, D158C and T293C, and D158C and Y136C. The results were as predicted: Under non-reducing conditions (i.e., without DTT), purified full-length TRPML1 proteins containing complementary cysteine mutations formed oligomeric bands in SDS-PAGE at the expense of the monomeric bands (Fig. 3b, upper panel, lanes 2, 6 and 10). However, those proteins containing non-complementary cysteine mutations remained monomeric (Fig. 3b, upper panel, lanes 1, 3, 4, 5, 7 and 8). (The D158C/Y136C protein in lane 9 appeared to have been degraded or precipitated and did not show a band.) Under reducing conditions (i.e., with DTT), all proteins existed as monomers (Fig. 3b, lower panel). That the oligomer bands exist only in the complementary cysteine pairs and only under non-reducing conditions is consistent with disulfide bond formation between the engineered cysteines, even though the molecular weight (MW) of the oligomer bands was larger than that of a predicted tetramer, which could be due to tetramer aggregation.
Figure 3.
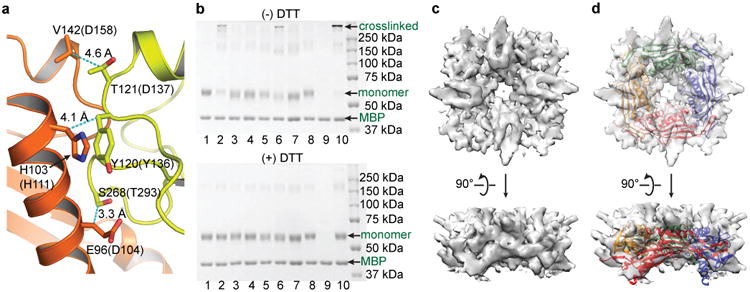
Verification of the I-II linker structure in the full-length protein. (a) Location of three pairs of amino acids in the I-II linker structure, with distances between the Cβ atoms indicated. Residue without or with parentheses correspond to human and C. elegans amino acids, respectively. (b) SDS-PAGE of full-length WT and mutant TRPML1 proteins under non-reducing (upper panel) and reducing (lower panel) conditions. The lanes are: 1 (WT), 2 (D104C/T293C), 3 (D104C/Y136C), 4 (D104C/D137C), 5 (H111C/T293C), 6 (H111C/Y136C), 7 (H111C/D137C), 8 (D158C/T293C), 9 (D158C/Y136C), 10 (D158C/D137C). (c,d) Cryo-EM structure of the I-II linker in full-length TRPML1, without c or with d, superposition of the I-II linker crystal structure. Upper panels, top down views from the extracellular/luminal side of the membrane. Lower panels, side views parallel to the membrane.
Second, we carried out single-particle electron cryo-microscopy (cryo-EM) analysis of the purified full-length WT TRPML1 protein (Supplementary Fig. 3). The cryo-EM density of the extracellular domain (ED) was clear and robust, but that of the transmembrane domain (TMD) was unclear and heterogeneous (Supplementary Figs. 3c, d). The ED and TMD could be distinguished and recognized based on their dimensions and the gross similarity of the TRPML1 TMD and TRPV1 TMD. We therefore used a masking procedure that allowed us to segregate the ED from the TMD during subsequent cryo-EM analysis (Supplementary Fig. 3c). A cryo-EM density map of the ED was obtained at an overall resolution of 5.28 Å with C4 symmetry imposed (Fig. 3c and Supplementary Fig. 3c). At this resolution, it can be seen that the ED, ∼79% of which is made up of the luminal linker, is a tetramer and has a large pore in the center (Fig. 3c), confirming two of the most salient hallmarks of the crystal structure of the luminal linker. Due to the limited resolution, we did not build an atomic model or identify secondary structures on this density map; however, the crystal structure of the luminal linker can fit into the density map with a correlation coefficient of 0.75, suggesting a reasonably good fit (Fig. 3d). The extra density not covered by the luminal linker crystal structure in the composite may come from the E199-K219 loop (which is unresolved in the crystal structure), other extracellular loops, and glycosylation.
Together, the above biochemical and cryo-EM studies suggest that the TRPML1 luminal linker has the same structure in the full-length channel as in the isolated domain: it forms a tetramer and contains a large central pore. This conclusion is further supported by structure-guided mutagenesis studies described directly below.
The luminal linker tetramer is crucial for full-length TRPML1 assembly
The crystal structure of the luminal linker shows that it forms a tight tetramer through extensive intersubunit interactions. These interactions include hydrogen bonding, ion pairs, hydrophobic interactions, and an antiparallel, domain-swapped 3-stranded β-sheet formed between two neighboring subunits (Fig. 4a and Supplementary Fig. 1b). The interface between two adjacent linkers has a buried surface area of 1430 Å2. To examine whether the tetrameric assembly of the luminal linker is important for the tetrameric assembly of full-length TRPML1, we mutated L144 and R146, two residues that are engaged in intersubunit interactions (Fig. 4a), to lysine and serine, respectively, in both the isolated luminal linker and full-length human TRPML1. We then examined the oligomeric state of wild-type (WT) and mutant domain proteins or full-length proteins by native gel electrophoresis. In a non-denaturing gel, the isolated luminal linker containing the L144K/R146S mutation (Fig. 4b, upper panel, lane 2) migrated at a much lower molecular weight than did the WT linker (Fig. 4b, upper panel, lane 1), consistent with them being monomer and tetramer, respectively, and indicating that the double mutation disrupted the tetrameric assembly of the isolated linker. In a blue native gel, full-length WT TRPML1 showed a distinct band, which presumably corresponds to tetramer (Fig. 4b, lower panel, lane 1). This band was absent with the mutant full-length TRPML1, which migrated as aggregated oligomers (Fig. 4b, lower panel, lane 2), suggesting that the L144K/R146S mutation disrupted the tetrameric assembly of full-length TRPML1.
Figure 4.
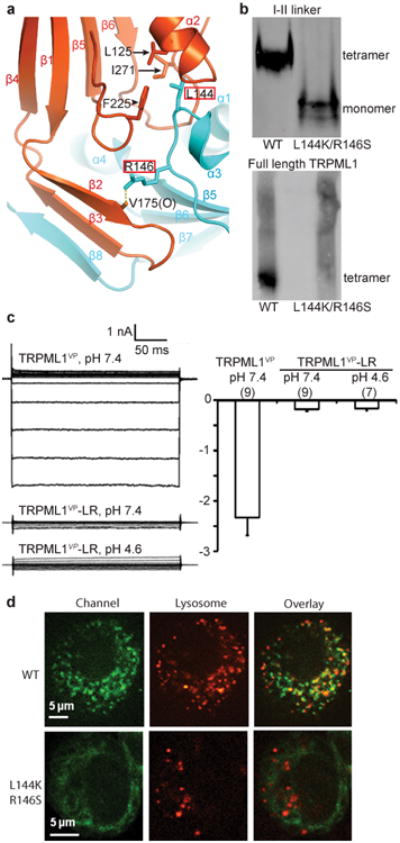
I-II linker intersubunit interactions contribute to TRPML1 assembly. (a) The I-II linker intersubunit interface. L144 and R146 are boxed in red, and their interacting partners are indicated. (b) Native gel electrophoresis of WT and mutant I-II linker proteins (upper panel) or full-length proteins (lower panel). (c) Left, families of currents of the indicated channels at pH 7.4 or pH 4.6. Right, average current amplitude at -80 mV of the indicated channels at pH 7.4 or pH 4.6. Number of recordings is shown in parentheses. Error bars represent SEM. (d) Confocal images of HeLa cells expressing the indicated GFP-tagged channels. Red indicates LysoTracker-labeled lysosomes.
Consistent with the above biochemical observations, a mutant TRPML1VP channel containing the L144K/R146S mutation (named TRPML1VP-LR) produced little whole-cell currents when expressed in HEK 293T cells, in a NDF solution at either pH 7.4 or 4.6 (Fig. 4c). We then examined the subcellular localization of TRPML1-LR, which contains the L144K/R146S mutation, by using confocal fluorescence microscopy. The wild-type and mutant channels tagged with EGFP were expressed in HeLa cells, which were labeled with the lysosomal marker LysoTracker to identify lysosomes. WT TRPML1 had a punctuate distribution and localized to lysosomes, as previously observed43-46; TRPML1-LR, however, had a diffuse distribution throughout the cell and did not localize to lysosomes (Fig. 4d). Altogether, these results indicate that proper formation of the luminal linker tetramer is critical for the proper assembly and proper subcellular organelle targeting of TRPML1.
MLIV-causing mutations in the luminal linker disrupt TRPML1 assembly and localization
Three MLIV-causing single amino acid missense mutations (L106P, C166F, and T232P) are present in the TRPML1 luminal linker (Fig. 5a). These mutations produce milder MLIV symptoms than do truncation/frame-shift mutations18,47-48, which generally result in a complete loss of the TRPML1 protein. The pathogenic mechanisms of these missense mutations at the channel level are unknown. The crystal structure of the luminal linker provides a structural context to investigate how these mutations affect TRPML1 structure, assembly, localization and activity.
Figure 5.
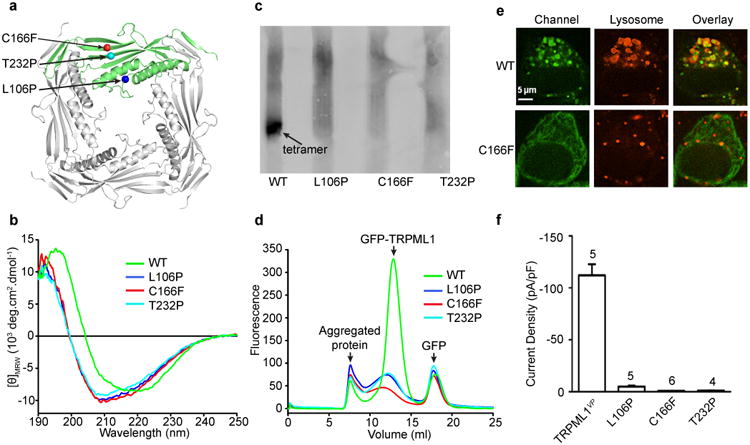
Effect of MLIV-causing mutations. (a) Location of three MLIV-causing missense mutations in the I-II linker structure. (b) Circular dichroism spectra of WT and mutant I-II linker proteins. (c) Native gel electrophoresis of WT and mutant full-length proteins. (d) Fluorescence-detection size-exclusion chromatography profiles of the indicated GFP-tagged proteins. (e) Confocal images of HeLa cells expressing the indicated GFP-tagged channels. Red indicates LysoTracker-labeled lysosomes. The scale bar represents 5 μm. (f) Average current density at -80 mV of the indicated channels at pH 7.4. Number of recordings is indicated in above the bars. Error bars represent SEM.
In the structure, L106 is at the junction of the anchoring α-helix (α1) and the luminal pore-loop, C166 forms a disulfide bond with C192, and T232 is located on β5 of a core β sheet (Fig. 5a and Supplementary Figs. 1b and 4a). The luminal linker protein containing any one of the three pathogenic mutations could still be expressed but with low yields. Circular dichroism measurements of the purified proteins revealed marked changes in the secondary structure of all three mutant linkers (Fig. 5b). In a blue native gel, the full-length WT protein migrated as a distinct putative tetramer band (Fig. 5c, lane 1), whereas the full-length mutant proteins migrated as aggregated oligomers (Fig. 5c, lanes 2, 3 and 4). Size-exclusion chromatography further confirmed increased aggregation of the mutant proteins (Fig. 5d). These results indicate that all three mutations disrupt the structure of the luminal pore and the tetrameric assembly of the full-length channel. These detrimental effects are not unexpected since two of the mutations (L106P and T232P) introduce a proline and one of the mutations (C166F) disrupts an intrasubunit disulfide bond and replaces a small amino acid with a larger hydrophobic one.
Consistent with the biochemical characterizations, confocal imaging showed that the mutant channels expressed in HeLa cells were diffusely distributed in the cells and did not localized to lysosomes, where the WT channels were mostly found (Fig. 5e and Supplementary Fig. 4b). As expected, because of abnormal assembly, the mutant channels produced little or no currents in HEK 293T cells when the mutations were introduced in TRPML1VP (Fig. 5f).
The luminal pore is critical for Ca2+ and pH regulation of TRPML1
The existence of a luminal pore is a distinct feature of TRPML1. The manual docking model shown in Supplementary Figure 2 suggests that the luminal pore is a part of the ion conduction pathway. To test this hypothesis, we simultaneously mutated three amino acids with relatively small side chains (S110, G112 and A113) in the luminal pore-loop to cysteine in TRPML1VP, and examined the effect of MTSET (a sulfhydryl-specific modifying reagent with a head group of 5.8 Å wide) on ion conduction in the mutant channel (named TRPML1VP-3C). The whole-cell current of TRPML1VP did not change significantly after extracellular application of 5 mM MTSET, but the current of TRPML1VP-3C was quickly and irreversibly inhibited (Supplementary Fig. 5), supporting the hypothesis above. Consistent with the structural revelation of a wide luminal pore, the current reduction after MTSET modification was only partial (Supplementary Figs. 5b-d). Further supporting the notion of a wide luminal pore and the specificity of thiol modification, the currents of TRPML1VP channels carrying a S110C/G112C or G112C/A113C double mutation were not inhibited by 5 mM extracellular MTSET (data not shown).
The presence of 12 Asp residues in close proximity within the luminal pore (Figs. 2a, d) raises a strong possibility that this pore region plays a role in Ca2+ and pH regulation. We tested this by simultaneously mutating all 12 Asp residues (D111, D114, and D115 in each subunit) to glutamine in TRPML1VP, generating a mutant channel named TRPML1VP-3DQ. These mutations greatly reduced Ca2+ inhibition at pH 7.4, increasing the apparent IC50 by 20-fold, from 0.27 mM to 5.5 mM while keeping the Hill coefficient at 1 (Fig. 6a). As shown in Figure 1c, decreasing the pH from 7.4 to 4.6 attenuated Ca2+ inhibition of TRPML1VP. The Asp→Gln mutations further decreased Ca2+ inhibition at pH 4.6, increasing the apparent IC50 from 3.8 mM to >10 mM (Fig. 6b). These results indicate that the luminal pore Asp residues are important for Ca2+ and pH regulation of TRPML1.
Figure 6.
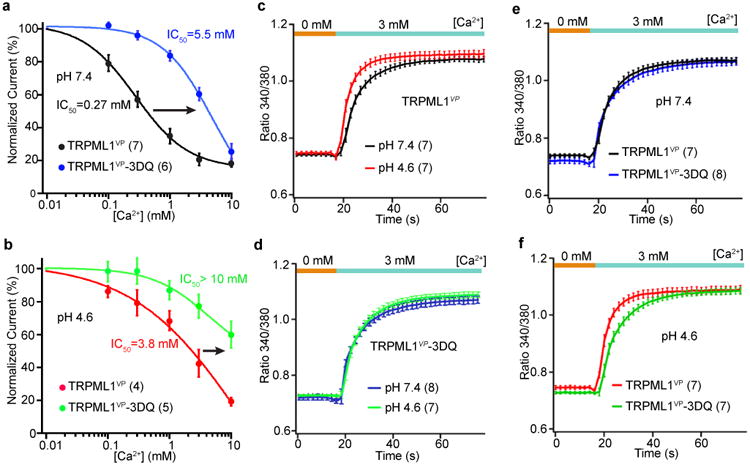
Effect of luminal pore aspartate mutations. (a, b) Dose-response relationships of Ca2+ inhibition of the indicated channels at pH 7.4, a, and 4.6, b, at a potential of -80 mV. Error bars represent SEM. Number of recordings is shown in parentheses. Solid curves are fits to the Hill equation. (c-f) Time course of intracellular Ca2+ increase at pH 7.4 or pH 4.6, following a step increase of extracellular Ca2+ from 0 to 3 mM in cells expressing the indicated channels. Error bars represent SEM. Number of recordings is shown in parentheses.
The high electronegativity of the luminal pore is also expected to enhance Ca2+ conduction by attracting and concentrating Ca2+. However, the effect on Ca2+ conduction is likely complex and pH-dependent. On the one hand, the luminal pore aspartates attract and concentrate Ca2+ and hence increase Ca2+ conduction; on the other hand, they bind Ca2+ to block the luminal pore and thereby decrease Ca2+ conduction. Thus, the end result on Ca2+ conduction is a balance of these two opposite effects, depending on the protonation state of the luminal pore aspartates.
To examine the importance of the luminal pore on Ca2+ conduction, we measured (by using Ca2+ imaging) Ca2+ influx into HEK 293T cells expressing TRPML1VP upon switching from an extracellular solution without Ca2+ to one with 3 mM Ca2+. This concentration was chosen to achieve a balance between a strong enough Ca2+ signal and physiological relevance. Intracellular Ca2+ increased gradually with time upon switching from 0 to 3 mM Ca2+ (Fig. 6c). The slow rise time was not due to a slow solution exchange, which was complete in 100-300 ms; it might be due to a slow accumulation of Ca2+ inside the cells. Regardless, the rise time was significantly faster at pH 4.6 than at pH 7.4, presumably reflecting a faster Ca2+ conduction at pH 4.6. This outcome is likely mainly due to a weaker inhibition of the channel by Ca2+, even though the Ca2+-concentrating effect is also attenuated at this acidic pH.
We next examined the effect of the Asp→Gln mutations on Ca2+ conduction. These mutations are expected to produce two opposite effects: (i) less Ca2+ inhibition and hence increased Ca2+ conduction, and (ii) less Ca2+-concentrating effect and hence decreased Ca2+ conduction. The initial rise time of the Ca2+ increase was similar at pH 4.6 and 7.4 for TRPML1VP-3DQ, consistent with the expected effect of the Asp→Gln substitutions (Fig. 6d). At pH 7.4, the time course of Ca2+ increase of TRPML1VP-3DQ was similar to that TRPML1VP (Fig. 6e), which was strongly inhibited by Ca2+ at this pH. However, at pH 4.6, the Asp→Gln mutations significantly slowed the time course of Ca2+ increase (Fig. 6f). This result can probably be attributed to a much reduced Ca2+-concentrating effect in the mutant channel. Altogether, these results suggest that the luminal pore Asp residues play a role in Ca2+ conduction through TRPML1. It is noticeable, however, that the TRPML1VP-3DQ channel currents still exhibited strong inward rectification, indicating that this property is not controlled by the luminal pore Asp residues (Supplementary Fig. 6).
The luminal pore structure is not significantly altered by pH
There are two possibilities of how acidic pH attenuates Ca2+ inhibition of TRPML1. First, low pH changes the structure of the luminal pore, either globally or locally. Second, low pH simply protonates the luminal pore aspartates without causing significant structural changes. To distinguish between these possibilities, we obtained crystal structures of the TRPML1 luminal linker at pH 4.5, mimicking its native physiological state, and 7.5, mimicking the pH at the plasma membrane. Both structures were determined at a 2.4 Å resolution (Fig. 7a and Table 1). Since residues E199-K219 were unresolved in the pH 6.0 structure, we removed R200-E213 in the constructs used in the pH 4.5 and pH 7.5 structures. The different structures show some differences in the periphery; e.g., at pH4.5, the first 9 amino acids of α1 became unstructured and several β strands (β2, β3 and β8′) could not be resolved. These differences might be caused by different crystal packing in different pH conditions. Remarkably, the structures are almost identical in most regions, including the luminal pore-loop (Fig. 7a, b). Even the side-chain orientation of the luminal pore aspartates is virtually the same (Fig. 7b), although the crystals at different pH are in completely different crystal forms (Table 1). These structures indicate that the I-II linker exists in a highly stable state. Thus, the attenuated Ca2+ inhibition at the acidic pH (Fig. 1) is likely due to protonation of the luminal pore aspartates rather than large conformational changes in the luminal pore.
Figure 7.
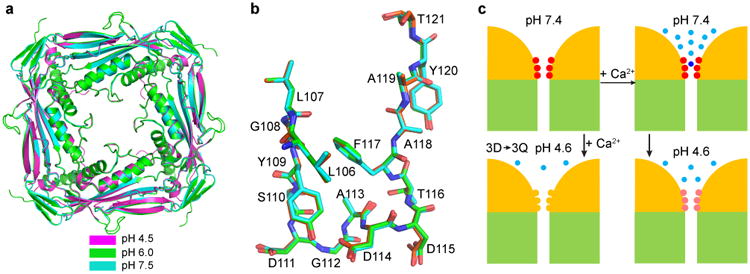
Structures of the TRPML1 I-II linker at different pH and model of Ca2+/pH regulation. (a) Superposition of the I-II linker crystal structure obtained at pH 4.5, 6.0 and 7.5, viewed from the extracellular/luminal side of the membrane. (b) Side view of the superimposed luminal pore-loop structures obtained at pH 4.5, 6.0, and 7.5. (c) Model of Ca2+/pH dual regulation of TRPML1. Two of the four channels are schematized, with yellow representing the ED and green the TMD. The luminal pore aspartates are illustrated in red or pink, depending on luminal pH, and in yellow when mutated to glutamine. Light blue dots represent free Ca2+ ions, and the dark blue dot represents a bound Ca2+ ion. See text for details.
Discussion
The long luminal/extracellular linker between the first two transmembrane segments is a unique but shared feature of the mucolipin (TRPML) and polycystin (TRPP) subfamilies of TRP channels. Our crystal structure of the TRPML1 I-II linker is the first high-resolution structure of a TRPML channel. Since the I-II linkers of TRPML2 and TRPML3 share high amino acid sequence homology with TRPML1 (Supplementary Fig. 1b), they likely have the same or similar structures. Recently, a 3.0 Å-resolution cryo-EM structure of a human TRPP2 construct containing the ED and TMD was obtained 49. Comparisons of the structures of the TRPML1 and TRPP2 I-II linkers show that despite divergent primary sequences (Supplementary Fig. 7a), the two linkers have a very similar overall structural fold (Supplementary Fig. 7b) and form tightly bound tetramers (Supplementary Fig. 7c, d). Given its unique presence in both TRPP and TRPML channels, we suggest that the I-II linker domain be named “the polycystin-mucolipin domain” instead of “the polycystin domain’, as proposed recently49. On the other hand, structural comparisons also reveal that the highly electronegative luminal pore-loop is a unique hallmark of TRPML1 (Supplementary Fig. 7e). In TRPML1 this loop extends downward toward the ion selectivity filter, whereas in TRPP2 it bends upward toward the luminal/extracellular entryway.
Our studies show that the novel luminal pore of TRPML1 has an important physiological function. Because of TRPML1's crucial role in numerous cellular processes, especially lysosome-dependent events, the subcellular localization and activity of TRPML1 must be tightly regulated. Although TRPML1 localizes mainly to lysosomes, it can be inserted into the plasma membrane under certain circumstances, such as during lysosomal exocytosis23-25,33. Cells appear to employ multiple regulatory mechanisms to ensure proper levels of TRPML1 activity in different subcellular compartments – high activity in lysosomes and low activity in the plasma membrane. One mechanism is subcellular compartment-dependent regulation by phosphoinositides. Thus, TRPML1 is activated by PI(3,5)P222, which is enriched on the lysosome membrane23,50. On the other hand, TRPML1 is inhibited by PI(4,5)P233, which is abundant on the plasma membrane. The Ca2+/pH dual regulation represents another subcellular compartmentalized regulatory mechanism. The acidic environment of the lysosome ensures high TRPML1 conductance in lysosomes by diminishing TRPML1 inhibition by luminal Ca2+. In contrast, the neutral extracellular pH ensures low TRPML1 conductance on the cell surface by enhancing TRPML1 inhibition by extracellular Ca2+.
It should be mentioned that the dual regulation by luminal Ca2+ and pH we described was observed in a gain-of-function mutant rather than the wildtype (WT) channel. Since the V432P mutation enables the channel to open constitutively, any allosteric effect of luminal Ca2+ and H+ on channel gating might not have been revealed. Our studies suggest, however, that the Ca2+/pH dual regulation is due to Ca2+ block of the luminal pore and modulation of this block by protonation. This dual regulation is largely conferred by the luminal pore Asp residues, which either form a low-affinity Ca2+ binding site or attract extracellular/luminal Ca2+ through electrostatic interactions to bind a site in the transmembrane pore. Based on our structural and functional studies, we propose a molecular model for the dual regulation of TRPML1 by extracellular/luminal Ca2+ and pH. At pH 7.4, virtually all luminal pore aspartates carry negative charges (Fig. 7c, upper left). These negative charges attract and bind extracellular/luminal Ca2+, thereby blocking Ca2+ as well as monovalent cation conduction (Fig. 7c, upper right). Lowering the pH to 4.6, which is close to the pKa of the aspartate side chain, results in protonation of the aspartates, reducing the net amount of negative charges and hence attenuating Ca2+ block (Fig. 7c, lower right). There is residual Ca2+ block at pH 4.6, because some of the aspartates are unprotonated and can attract and bind Ca2+. The negative cooperativity observed at pH 4.6 between H+ and Ca2+ (Fig. 1c) is in good agreement with the protonation hypothesis. Mutating the aspartates to glutamines removes all the negative charges in the luminal pore and markedly further reduce Ca2+ block (Fig. 7c, lower left).
A docking model (Supplementary Fig. 2) suggests that the TRPML1 I-II linker constitutes virtually all of the exposed surface of TRPML1 in the endolysosomal lumen and thus may contain all the interaction sites between TRPML1 and the lipids and proteins in the lumen. It is notable that the structure of E199-K219 in the TRPML1 I-II linker was unresolved (Fig. 2 and Supplementary Fig. 1b), probably due to its flexibility. This 21-amino acid loop may protrude into the endolysosomal lumen, beyond an otherwise rigid structure, and is thus a candidate interaction site with endolysosomal lumen molecules. Finally, the architecture and strong electronegativity of the luminal pore suggests that it may be a suitable site for structure-based design of TRPML1-specific blockers.
Accession codes
3D cryo-EM density maps of full-length C. elegans TRPML1 or the I-II linker, without and with low-pass filter and amplitude modification, have been deposited in the Electron Microscopy Data Bank under the accession number EMD-6669 and EMD-6670, respectively. The coordinates of the atomic model of the TRPML1 I-II linker at pH 4.5, 6.0 and 7.5 have been deposited in the Protein Data Bank under the accession number 5TJB, 5TJA and 5TJC, respectively.
Data availability
Source data for Figures 1, 3, 4, 5 and 6 and for Supplementary Figures 5 and 6 are provided with the paper. Other data supporting the findings of this study are available from the corresponding author upon reasonable request.
Online Methods
Mutagenesis and cloning
The human TRPML1 clone (accession number: BC005149) was purchased from OpenBiosystems. The C. elegans TRPML1 (CUP-5) was cloned from a cDNA library. Site-directed mutagenesis was carried out by overlapping PCR or Quick Change Site-Directed Mutagenesis Kit (TransGen Biotech) to produce all the mutations. The DNA sequences of all constructs were verified by DNA sequencing. Depending on the experiment, WT and mutant constructs were cloned into different expression vectors, as detailed below in the respective sections.
Expression and purification of the TRPML1 I-II linker protein
A DNA fragment encoding the I-II linker (residues 84-296) of human TRPML1 was obtained by PCR and cloned into a modified pET26b(+) vector. A maltose-binding protein (MBP) tag was added to the N-terminus of the I-II linker, and a DNA fragment encoding the recognition site of thrombin was inserted between the MBP tag and I-II linker. The resulted construct thus encodes a fusion protein that consists of an N-terminal MBP tag followed by the thrombin recognition site, the I-II linker, and a C-terminal 6×Histidine tag.
Rosetta-gami™ 2 (DE3) cells were used to express the I-II linker to allow proper disulfide bond formation. Transformed cells were selected and grown at 37 °C in 10 liters of Luria-Bertani (LB) medium containing 50 ug/ml kanamycin and 34 ug/ml chloramphenicol in an incubator shaker at 250 rpm. When the optical density at 600 nm reached 1.0, the culture was cooled down to 22 °C and 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) was added. The bacterial cells were incubated for another 12 hours in the shaker at 22 °C before they were harvested by centrifugation at 3000 g for 15 min. The cells were resuspended in a solution containing 50 mM sodium phosphate pH 8.0, 300 mM NaCl, 2.5% (w/w) glycerol (solution A). Five mM imidazole, 0.5 mg/ml lysozyme, 25 ug/ml DNase and 2 mM phenylmethylsulfonyl fluoride (PMSF) were added, and the cells were disrupted by sonication. Insoluble cell debris was removed by centrifugation at 17,000 g for 30 min. The supernatant was incubated with Ni-NTA His-Bind resin (Novagen) with gentle agitation at 4 °C for 1 hour. The beads were spun down at 800 × g for 1 min and transferred to a gravity-flow chromatography column. After the resin was washed with 10 volumes of solution A containing 5 mM imidazole, the bound protein was eluted with 500 mM imidazole in solution A. The eluted protein was incubated with amylose resin (NEB) at 4 °C for 2.5 hours. The resin was collected by centrifugation at 800 g for 1 min, transferred to a column, and washed with solution A. The MBP-tagged protein was eluted by 20 mM maltose in solution A. Thrombin (Sigma-Aldrich) was added at 4 units per milligram protein to cut off the MBP tag. After incubation at 16 °C for overnight, the protein was concentrated and purified on a Superdex 200 column (GE Healthcare) to remove MBP. The gel filtration solution contained 10 mM HPEPS and 150 mM NaCl, pH 7.5. The peak fractions corresponding to the tetrameric I-II linker protein were collected and concentrated to a concentration of 4 mg/ml for crystallization.
Crystallization, data collection, structure determination and refinement
Crystallization of the TRPML1 I-II linker was carried out by using the hanging drop vapor diffusion method at 16 °C (for pH 6.0) or 20°C (for pH 4.5 and 7.5). For the pH 6.0 crystal, protein solution was mixed with reservoir solution at a 1:1 ratio. The reservoir solution contained 1.38 M sodium phosphate monobasic monohydrate and 0.42 M potassium phosphate dibasic, pH 6.0, and 5% pentaerythritol ethoxylate (3/4 EO/OH) (Hampton Research). Best crystals grew from drops formed by 4 mg/ml protein. For the pH 4.5 crystal, the protein concentration was 4 mg/ml and the reservoir solution contained 200 mM magnesium sulfate, 5.3% PEG 3350, and 100 mM acetate pH 4.5. Crystals at pH 7.5 were obtained by macroseeding in a reservoir solution of 100 mM magnesium sulfate, 4% PEG 3350, 100 mM HEPES at pH 7.5, with a protein concentration of 4 mg/ml. Heavy atom derivative crystals were prepared by soaking crystals in a solution containing 1.53 M sodium phosphate monobasic monohydrate and 0.47 M potassium phosphate dibasic, pH 6.0 and 1 mM K2Pt(CN)4 for 24 hours.
Single crystals were flash-cooled in liquid nitrogen with Paratone-N (Hampton Research) (for pH 4.5 and 6.0 crystals) or 30% glycerol as a cryoprotectant (for pH 7.5 crystals). X-ray diffraction data for native and derivative crystals (at pH 4.5 and 6.0) were collected at 100 K on a RAXIS-IV detector using Cu K radiation (λ = 1.5418 Å) from a Rigaku RuH3R X-ray generator. X-ray diffraction data at pH 7.5 were collected at the National Synchrotron Light Source (NSLS) beamline X29 at Brookhaven National Laboratory.
The diffraction images were processed and scaled with the HKL package51. Structure of the I-II linker at pH 6.0 was solved by the SIRAS (single isomorphous replacement with anomalous scattering) method using native and platinum derivative data sets. Two platinum sites were found by Shake and Bake52, and were fed to SOLVE53 for calculating initial phases. Density modification was done using RESOLVE53 to improve phase accuracy. The quality of the density map from RESOLVE was good enough to locate most residues. The presence of two intrasubunit disulfide bonds helped the tracing. Initial model was built by using COOT54. Refinement by using CNS55 and manual rebuilding were done iteratively. Structures of the I-II linker at pH 4.5 and 7.5 were solved by molecular replacement with the program PHASER56, using the structure at pH6 as the search model. Cycles of refinement by using CNS and manual model correction using COOT were carried out. Crystallographic statistics are summarized in Table 1.
Expression and purification of the TRPML1 I-II linker protein for circular dichroism
TRPML1 I-II linkers were expressed by viral infection in Hi5 insect cells (Expression Systems, not tested for mycoplasma). The linkers were cloned in the pFastBac1 vector, which was modified by the insertion of a honeybee melittin secretion signal peptide coding region followed by BamHI/XhoI restriction sites and a 6 × His tag coding region. DNA fragments encoding the WT or mutant I-II linkers were inserted into the BamHI/XhoI sites of the modified vector.
Recombinant baculovirus was generated by using the Bac-to-Bac method (Invitrogen). The virus was amplified in Sf9 cells (Invitrogen, not tested for mycoplasma) and was used to infect Hi5 cells in ESF920 medium (Expression Systems). The expressed protein containing a 6×His tag on its C-terminus was secreted into the extracellular medium where the honeybee melittin secretion signal peptide was cleaved. Forty eight hours after transfection, cells were removed by centrifugation and the medium containing the desired protein was collected. The medium was concentrated, dialyzed against solution A containing 5 mM imidazole, and incubated with TALON metal affinity resin (Clontech) at 4 °C for one hour. The resin was collected and washed with solution A containing 10 mM imidazole. The His-tagged protein was eluted with 300 mM imidazole in solution A. The protein was concentrated and changed to a solution containing 10 mM sodium phosphate, 150 mM NaCl, pH 7.0 by dialysis for circular dichroism (CD) analysis.
Circular dichroism spectroscopy
CD was performed using a J-815 CD spectrometer (Jasco, inc.). All proteins were adjusted to a concentration of 1 mg/ml and loaded into a quartz cuvette with a path length of 0.01 cm for measurement. A buffer containing no protein was used for subtraction of the baseline signal. Measurements were performed from 185 to 250 nm with a 0.1 nm interval, 1 nm bandwidth, and scanning speed of 50 nm/min. For each protein, three samples were measured, and three accumulations were measured for each sample and averaged. Mean residue ellipticity (MRE) was calculated from raw CD signal, θ, using the equation [θ]MRE = θ/(10 × Cr × l) where Cr is the protein concentration (M × residue number) and l is the cuvette path length (cm).
Crosslinking
The DNA fragment encoding C. elegans full-length TRPML1 was cloned into a modified pFastBac1 vector. An MBP tag was added before the N-terminus of TRPML1, and a 6 × His tag was added before the N-terminus of MBP. A linker region containing a TEV protease recognition sequence (NNNNNNENLYFQGGGGS) was inserted between the MBP tag and TRPML1. Cysteine substitutions were subsequently introduced into this background construct.
The baculovirus of TRPML1 was generated in Sf9 cells using the standard Bac-to-Bac method and used to infect Hi5 insect cells. Forty-eight hours after infection, cells were harvested by centrifugation at 4 °C, and suspended in a buffer containing 50 mM HEPES-NaOH, pH 7.4, 500 mM NaCl, 5% glycerol, in the presence of a protease inhibitor cocktail (Pierce). After cell disruption by sonication, the cell debris was removed by centrifugation at 4,000 rpm for 10 minutes at 4 °C, and membranes were pelleted by ultracentrifugation at 45,000 rpm at 4 °C for 1 hour. The membranes were suspended in buffer A containing the protease inhibitor cocktail and homogenized with a glass dounce homogenizer. TRPML1 proteins were extracted by 1% lauryl maltose neopentyl glycol (LMNG, Anatrace) for 1 hour at 4 °C. The solubilized membranes were clarified by ultracentrifugation for 30 minutes and incubated with amylose resin (NEB) for 2 hours at 4 °C with gentle agitation. The resin was collected by low speed spin at 2,000 rpm, transferred into a gravity column, and washed with buffer A containing 0.5 mM LMNG and 0.1 mg/ml soybean lipids (Avanti polar lipids). Twenty mM maltose in the wash buffer was used to elute the MBP-tagged TRPML1 proteins. TEV protease was added to the eluted proteins and incubated overnight at 4 °C to cut off the MBP tag. The proteins were mixed with 3× SDS loading buffer with or without DTT, and analyzed by 10% SDS-PAGE.
Native gel electrophoresis and fluorescence-detection size-exclusion chromatography
The WT and L144K/R146S mutant I-II linker proteins were obtained as described above in the CD experiment. The proteins were loaded on a 6% native acrylamide gel. After electrophoresis, the proteins were transferred onto a PVDF membrane for Western blot analysis. His•Tag® monoclonal antibody (EMD Millipore) was used as the primary antibody. Alexa Fluor 680 goat anti-mouse IgG (Invitrogen) was used as the secondary antibody. Images were scanned and analyzed with the Odyssey Infrared Imaging System (Li-COR).
For native gel electrophoresis and fluorescence-detection size-exclusion chromatography (FSEC), full-length C. elegans TRPML1 was cloned in the pEGFP-C1 vector (Clontech), which was modified by replacing the multiple cloning site with a TEV protease recognition site coding region and SbfI/AscI restriction sites. The DNA fragment encoding the full-length TRPML1 was inserted into the SbfI/AscI site. The expressed protein thus had an EGFP tag on its N-terminus and a TEV protease recognition site in between the EGFP tag and the channel.
HEK 293T (ATCC) cells were transfected with the TRPML1 plasmid by using Lipofectamine 2000 (Invitrogen). Forty-eight hours after transfection, cells were washed and resuspended in solution A containing Halt™ protease inhibitor cocktails (Thermo). DDM was added to a final concentration of 2% and incubated at 4 °C for one hour. The solution was clarified by centrifugation at 16000 rpm for 10 min. This cell lysate was used for blue native PAGE and FSEC57.
Blue native PAGE was performed with the NativePAGE Bis-Tris Gel System (Invitrogen). The solubilized cell lysate in DDM was mixed with NativePAGE sample buffer and Coomassie blue G-250. Electrophoresis was carried out on 3-12% Bis-Tris gel at 4 °C. The proteins were transferred to the PVDF membrane for Western blot. Anti-GFP monoclonal antibody (Santa Cluz) and Alexa Fluor 680 goat anti-mouse IgG (Invitrogen) were used for detecting the GFP-tagged TRPML1 protein. Images were scanned and analyzed with Li-COR.
FSEC was performed by using a spectrofluorometric detector RF-10AXL (Shimadzu) for fluorescence detection, and a Superose 6 10/300 column (GE Healthcare) for the size-exclusion chromatography.
Electrophysiology
Constructs
Human TRPML1 tagged with GFP on the N-terminus and containing the V432P point mutation (TRPML1vp) was cloned in the pEGFP-C1 vector. TRPML1vp-3DQ, TRPML1vp-3C and TRPML1VP-LR were also cloned in this vector. TRPML1VP was also cloned in pcDNA3.1, in which TRPML1vp-L106P, TRPML1vp-C166F and TRPML1vp-T232P were cloned.
Cell culture and transfection
HEK 293T cells were grown in DMEM (30-2002, ATCC) supplemented with 0.5% penicillin/streptomycin (Sigma, P-0781) and 10% FBS (Standard quality, PAA laboratory, 95025-534) using standard procedures. Transfections were done by using LipoD293™ (SignaGen Laboratories) per manufacturer's instruction.
Whole-cell recording and data analysis
All experiments were performed at room temperature (22-23 °C). Pipettes were fabricated from borosilicate glass (World Precision Instruments) and were fire-polished to resistances of 2-5 MΩ. Whole-cell currents were elicited by 150-ms voltage steps from -120 to +100 mV with 20-mV increments, with a holding potential of -30 or 0 mV. Currents were amplified by Axopatch 200B and digitized by Digidata 1440A (Molecular Devices). Currents were low-pass filtered at 1 kHz and sampled at 10 kHz. pCLAMP software (Molecular Devices) was used for data acquisition and analysis. The pipette solution contained (in mM): 120 cesium methanesulphonate, 4 NaCl, 10 EGTA, 2 MgCl2, 20 HEPES, 2 Na2ATP, pH 7.2 with CsOH. pH 7.4 NDF (nominal divalent ion-free) bath solution contained (in mM): 160 NaCl, 5 KCl, 10 glucose, 20 HEPES, pH 7.4 with NaOH. pH 4.6 NDF bath solution contained (in mM): 160 Na-gluconate, 5 KCl, 10 MES, 10 glucose, 10 HEPES, pH 4.6 with HCl. To obtain different Ca2+ concentrations, an appropriate amount of 1 M CaCl2 stock solution was added to the NDF solutions to achieve a desired final concentration. In the cysteine modification experiment, MTSET was freshly added to the pH 7.4 NDF solution and kept in dark for <30 minutes before use.
Data points represent mean ± S.E.M. Dose-response relationships for calcium inhibition of TRPML1 channels were fitted to the Hill equation in the form of I(X)=Imax-Xn/(Xn+IC50n), where Imax is the current in the absence of calcium, I(X) is the normalized current in the presence of calcium, X the calcium concentration, n the Hill coefficient, and IC50 the calcium concentration producing half maximal inhibition. Statistical differences were determined by the Student's t-test.
Intracellular Ca2+ imaging
TRPML1vp and TRPML1vp-3DQ cloned in pEGFP-C1 were transfected into HEK 293T cells. Forty eight hours after transfection, cells were loaded with Fura-2 AM (10 μM) and Pluronic F-127 (0.02%) (Molecular Probes) at 37 °C for 1 h in a Ca2+-free solution. The fluorescence ratios of F340/F380 were measured by using a fluorescence microscopic system. The Ca2+-free solution contained (in mM) 150 NaCl, 1 MgCl2, and 10 HEPES, pH 7.4 with NaOH. In the Ca2+-containing solution, 3 mM CaCl2 was added to the solution above.
Confocal imaging
HeLa cells (gift of Ron Prywes, Columbia University) were maintained in DMEM (Gibco) containing 10% FBS (Atlanta Biologicals), and 100 μg/mL of penicillin/streptomycin (Invitrogen). Cells were cultured and plated onto poly-D-lysine hydrobromide (Sigma) coated coverslips 18 to 24 hours prior to transfection and grown on the coverslips in DMEM plus 10% FBS supplemented with 100 μg/mL of penicillin/streptomycin at 37°C until they became about 80% confluent. Cells were transfected with WT or mutant full-length TRPML1, all tagged with EGFP on the N-terminus and cloned in the pCDNA3.1 vector, by using LipoD293 In Vitro DNA Transfection Reagent Ver II (SignaGen Laboratories). Seventeen hours after transfection, the cells were loaded with 100 nM of the lysosomal marker LysoTracker® Red DND-99 (Invitrogen) for 50 minutes at 37°C. Cells were gently and briefly washed three times with PBS (Gibco), and live-cell images were acquired in PBS at 20°C. Images were acquired from random cells in an unbiased manner using a Nikon Eclipse TE2000-S microscope with a 60× oil-immersion objective utilizing a spinning disc confocal system (Perkin Elmer). Images were acquired and analyzed by using the Volocity program (Perkin Elmer).
Cryo-EM sample preparation, data acquisition, and image processing
C. elegans TRPML1 was expressed in Hi5 insect cells and purified by amylose resin using the same procedures as described above in the crosslinking experiment. Eluted TRPML1 protein from amylose resin was mixed with amphipol A8-35 (Anatrace) at 1:6 (w/w), and TEV protease was added at 1:20 (w/w) to cleave the MBP tag. The mixture was incubated with gentle agitation at 4°C overnight, and detergent was removed by Bio-Beads SM-2 (Bio-rad) at 4 °C. After Bio-Beads was removed, the TRPML1 protein was concentrated and further purified on a Superose 6 10/300 column in a buffer composed of 20 mM HEPES-NaOH, 150 mM NaCl, pH7.4. The peak corresponding to the tetrameric TRPML1 protein was collected for cryo-EM analysis.
A drop of 4 μl the amphipol-solubilized protein of 0.6 mg/ml concentration was loaded to a glow-discharged Quantifoil R1.2/1.3 holey carbon grid, incubated for 3s, blotted, and then plunged into liquid ethane cooled by liquid-nitrogen using a Vitrobot (FEI Company) at 100% humidity and 8 °C. The grids were imaged using a Titan Krios microscope operated at 300 kV, and equipped with a K2 Summit electron counting camera (Gatan Company). UCSFImage458 was used for data collection with nominal magnification of 22,500×, corresponding to an image pixel size of 0.66 Å under super-resolution counting mode. Images were recorded with defocus range from 2.1 to 3.1 μm. The dose rate was set to 8.2 counts/physical pixel/s on the camera plane. The total exposure time was 8 s, resulting in a total accumulated dose of ∼50 e-/Å2. Each micrograph was fractionated into 32 frames, each with an exposure time of 0.25 s.
All micrographs were first 2×2 binned, generating a pixel size of 1.32 Å. Motion correction was performed using MotionCorr59. The aligned frames were integrated and used for further processing. Defocus of all images was determined using CTFFIND360. Particle picking and all two-dimensional (2D) and three-dimensional (3D) classification and refinement steps were performed using RELION61. Approximately 200,000 auto-picked particles were screened by several rounds of 2D classifications to remove most of the junk and bad particles. The particles were 4×4 binned to a pixel size of 2.64 Å for further image analysis. A star-shaped density map with C4 symmetry was made and used as initial reference for 3D classification. 71,052 good particles were selected by 3D classification and subjected to the first round 3D refinement with C4 symmetry imposed, which yielded a reconstruction at a resolution of 8.12 Å. The transmembrane region showed much worse quality than the soluble region, consistent with the asymmetric features shown in 2D class averages. To improve the resolution of the soluble region, we applied a soft-edged mask to remove the possible transmembrane region. Further 3D refinement of the soluble region generated a reconstruction at a resolution of 5.28 Å. The resolutions were estimated based on the gold-standard FSC=0.143 criterion. ResMap62 was used to calculate the local resolution map. The crystal structure of the human I-II linker at pH 6.0 was fit into this cryo-EM density map by using Chimera63.
Supplementary Material
Acknowledgments
We thank Y. Chen and M. Chalfie for providing the C. elegans cDNA library, T. Patel and S. Banta for help with the circular dichroism experiment, and the staff at X29 of NSLS, Brookhaven National Laboratory, for synchrotron support. This work was supported by grants to J.Y. from the National Key Basic Research Program of China (2014CB910301), the National Institutes of Health (R01GM085234 and RO1NS053494), the National Natural Science Foundation of China (31370821), the Top Talents Program of Yunnan Province (2011HA012), and the High-level Overseas Talents of Yunnan Province; to X.L. from the China Youth 1000-Talent Program of the State Council of China, Beijing Advanced Innovation Center for Structural Biology, Tsinghua-Peking Joint Center for Life Sciences, and the National Natural Science Foundation of China (31570730).
Footnotes
Author Contributions: M.L. and J.Y. conceived and initiated the project. M.L. obtained the first crystal structure at pH 6.0 and contributed to most other experiments. W.K.Z. performed most electrophysiology experiments. N.M.B. obtained the crystal structures at pH 4.5 and 7.5 and performed the imaging experiments. L.T. helped supervise X-ray crystallography data collection and atomic model building. X.Z. and X.L. performed the cryo-EM experiments and data processing. D.S., H.L., S.W. and I.E.M. performed experiments or analysis. All authors contributed to manuscript preparation and editing. M.L., W.K.Z., N.M.B. and J.Y. wrote the paper.
Competing Financial Interests: The authors declare no competing financial interests.
References
- 1.Xu H, Martinoia E, Szabo I. Organellar channels and transporters. Cell Calcium. 2015;58:1–10. doi: 10.1016/j.ceca.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leanza L, et al. Intracellular ion channels and cancer. Front Physiol. 2013;4:227. doi: 10.3389/fphys.2013.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiselyov KK, Ahuja M, Rybalchenko V, Patel S, Muallem S. The intracellular Ca(2)(+) channels of membrane traffic. Channels (Austin) 2012;6:344–351. doi: 10.4161/chan.21723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stauber T, Jentsch TJ. Chloride in vesicular trafficking and function. Annu Rev Physiol. 2013;75:453–477. doi: 10.1146/annurev-physiol-030212-183702. [DOI] [PubMed] [Google Scholar]
- 5.Xu H, Ren D. Lysosomal physiology. Annu Rev Physiol. 2015;77:57–80. doi: 10.1146/annurev-physiol-021014-071649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Venkatachalam K, Wong CO, Zhu MX. The role of TRPMLs in endolysosomal trafficking and function. Cell Calcium. 2015;58:48–56. doi: 10.1016/j.ceca.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel S, Cai X. Evolution of acidic Ca(2)(+) stores and their resident Ca(2)(+)-permeable channels. Cell Calcium. 2015;57:222–230. doi: 10.1016/j.ceca.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 8.Thrower EC, Hagar RE, Ehrlich BE. Regulation of Ins(1,4,5)P3 receptor isoforms by endogenous modulators. Trends Pharmacol Sci. 2001;22:580–586. doi: 10.1016/s0165-6147(00)01809-5. [DOI] [PubMed] [Google Scholar]
- 9.Meissner G. Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annu Rev Physiol. 1994;56:485–508. doi: 10.1146/annurev.ph.56.030194.002413. [DOI] [PubMed] [Google Scholar]
- 10.Pitt SJ, et al. TPC2 is a novel NAADP-sensitive Ca2+ release channel, operating as a dual sensor of luminal pH and Ca2+ J Biol Chem. 2010;285:35039–35046. doi: 10.1074/jbc.M110.156927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo J, et al. Structure of the voltage-gated two-pore channel TPC1 from Arabidopsis thaliana. Nature. 2016;531:196–201. doi: 10.1038/nature16446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jha A, Ahuja M, Patel S, Brailoiu E, Muallem S. Convergent regulation of the lysosomal two-pore channel-2 by Mg(2)(+), NAADP, PI(3,5)P(2) and multiple protein kinases. EMBO J. 2014;33:501–511. doi: 10.1002/embj.201387035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun M, et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Human molecular genetics. 2000;9:2471–2478. doi: 10.1093/hmg/9.17.2471. [DOI] [PubMed] [Google Scholar]
- 14.Bargal R, et al. Identification of the gene causing mucolipidosis type IV. Nature genetics. 2000;26:118–123. doi: 10.1038/79095. [DOI] [PubMed] [Google Scholar]
- 15.Bassi MT, et al. Cloning of the gene encoding a novel integral membrane protein, mucolipidin-and identification of the two major founder mutations causing mucolipidosis type IV. American journal of human genetics. 2000;67:1110–1120. doi: 10.1016/s0002-9297(07)62941-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weitz R, Kohn G. Clinical spectrum of mucolipidosis type IV. Pediatrics. 1988;81:602–603. [PubMed] [Google Scholar]
- 17.Bach G. Mucolipidosis type IV. Molecular genetics and metabolism. 2001;73:197–203. doi: 10.1006/mgme.2001.3195. [DOI] [PubMed] [Google Scholar]
- 18.Wakabayashi K, Gustafson AM, Sidransky E, Goldin E. Mucolipidosis type IV: an update. Molecular genetics and metabolism. 2011;104:206–213. doi: 10.1016/j.ymgme.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kiselyov K, et al. TRP-ML1 is a lysosomal monovalent cation channel that undergoes proteolytic cleavage. J Biol Chem. 2005;280:43218–43223. doi: 10.1074/jbc.M508210200. [DOI] [PubMed] [Google Scholar]
- 20.Dong XP, et al. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature. 2008;455:992–996. doi: 10.1038/nature07311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong XP, et al. Activating mutations of the TRPML1 channel revealed by proline-scanning mutagenesis. J Biol Chem. 2009;284:32040–32052. doi: 10.1074/jbc.M109.037184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong XP, et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nature communications. 2010;1:38. doi: 10.1038/ncomms1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong XP, Wang X, Xu H. TRP channels of intracellular membranes. Journal of neurochemistry. 2010;113:313–328. doi: 10.1111/j.1471-4159.2010.06626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.LaPlante JM, et al. Lysosomal exocytosis is impaired in mucolipidosis type IV. Molecular genetics and metabolism. 2006;89:339–348. doi: 10.1016/j.ymgme.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 25.Samie M, et al. A TRP Channel in the Lysosome Regulates Large Particle Phagocytosis via Focal Exocytosis. Dev Cell. 2013;26:511–524. doi: 10.1016/j.devcel.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miedel MT, et al. Membrane traffic and turnover in TRP-ML1-deficient cells: a revised model for mucolipidosis type IV pathogenesis. The Journal of experimental medicine. 2008;205:1477–1490. doi: 10.1084/jem.20072194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, et al. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat Cell Biol. 2016;18:404–417. doi: 10.1038/ncb3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Venkatachalam K, et al. Motor deficit in a Drosophila model of mucolipidosis type IV due to defective clearance of apoptotic cells. Cell. 2008;135:838–851. doi: 10.1016/j.cell.2008.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vergarajauregui S, Connelly PS, Daniels MP, Puertollano R. Autophagic dysfunction in mucolipidosis type IV patients. Human molecular genetics. 2008;17:2723–2737. doi: 10.1093/hmg/ddn174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nature communications. 2016;7:12109. doi: 10.1038/ncomms12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raychowdhury MK, et al. Molecular pathophysiology of mucolipidosis type IV: pH dysregulation of the mucolipin-1 cation channel. Human molecular genetics. 2004;13:617–627. doi: 10.1093/hmg/ddh067. [DOI] [PubMed] [Google Scholar]
- 32.Cantiello HF, et al. Cation channel activity of mucolipin-1: the effect of calcium. Pflugers Archiv : European journal of physiology. 2005;451:304–312. doi: 10.1007/s00424-005-1448-9. [DOI] [PubMed] [Google Scholar]
- 33.Zhang X, Li X, Xu H. Phosphoinositide isoforms determine compartment-specific ion channel activity. Proc Natl Acad Sci U S A. 2012;109:11384–11389. doi: 10.1073/pnas.1202194109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Appelqvist H, Waster P, Kagedal K, Ollinger K. The lysosome: from waste bag to potential therapeutic target. J Mol Cell Biol. 2013;5:214–226. doi: 10.1093/jmcb/mjt022. [DOI] [PubMed] [Google Scholar]
- 35.Everett KV. Transient receptor potential genes and human inherited disease. Adv Exp Med Biol. 2011;704:1011–1032. doi: 10.1007/978-94-007-0265-3_52. [DOI] [PubMed] [Google Scholar]
- 36.Bargal R, et al. Mucolipidosis type IV: novel MCOLN1 mutations in Jewish and non-Jewish patients and the frequency of the disease in the Ashkenazi Jewish population. Human mutation. 2001;17:397–402. doi: 10.1002/humu.1115. [DOI] [PubMed] [Google Scholar]
- 37.Zeevi DA, Frumkin A, Bach G. TRPML and lysosomal function. Biochimica et biophysica acta. 2007;1772:851–858. doi: 10.1016/j.bbadis.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 38.Xu H, Delling M, Li L, Dong X, Clapham DE. Activating mutation in a mucolipin transient receptor potential channel leads to melanocyte loss in varitint-waddler mice. Proc Natl Acad Sci U S A. 2007;104:18321–18326. doi: 10.1073/pnas.0709096104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grimm C, et al. A helix-breaking mutation in TRPML3 leads to constitutive activity underlying deafness in the varitint-waddler mouse. Proc Natl Acad Sci U S A. 2007;104:19583–19588. doi: 10.1073/pnas.0709846104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim HJ, et al. Gain-of-function mutation in TRPML3 causes the mouse Varitint-Waddler phenotype. J Biol Chem. 2007;282:36138–36142. doi: 10.1074/jbc.C700190200. [DOI] [PubMed] [Google Scholar]
- 41.Di Palma F, et al. Mutations in Mcoln3 associated with deafness and pigmentation defects in varitint-waddler (Va) mice. Proc Natl Acad Sci U S A. 2002;99:14994–14999. doi: 10.1073/pnas.222425399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liao M, Cao E, Julius D, Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504:107–112. doi: 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pryor PR, Reimann F, Gribble FM, Luzio JP. Mucolipin-1 is a lysosomal membrane protein required for intracellular lactosylceramide traffic. Traffic. 2006;7:1388–1398. doi: 10.1111/j.1600-0854.2006.00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manzoni M, et al. Overexpression of wild-type and mutant mucolipin proteins in mammalian cells: effects on the late endocytic compartment organization. FEBS letters. 2004;567:219–224. doi: 10.1016/j.febslet.2004.04.080. [DOI] [PubMed] [Google Scholar]
- 45.Venkatachalam K, Hofmann T, Montell C. Lysosomal localization of TRPML3 depends on TRPML2 and the mucolipidosis-associated protein TRPML1. J Biol Chem. 2006;281:17517–17527. doi: 10.1074/jbc.M600807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vergarajauregui S, Puertollano R. Two di-leucine motifs regulate trafficking of mucolipin-1 to lysosomes. Traffic. 2006;7:337–353. doi: 10.1111/j.1600-0854.2006.00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Altarescu G, et al. The neurogenetics of mucolipidosis type IV. Neurology. 2002;59:306–313. doi: 10.1212/wnl.59.3.306. [DOI] [PubMed] [Google Scholar]
- 48.Geer JS, Skinner SA, Goldin E, Holden KR. Mucolipidosis type IV: a subtle pediatric neurodegenerative disorder. Pediatric neurology. 2010;42:223–226. doi: 10.1016/j.pediatrneurol.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen PS, et al. The Structure of the Polycystic Kidney Disease Channel PKD2 in Lipid Nanodiscs. Cell. 2016;167:763–773 e711. doi: 10.1016/j.cell.2016.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 51.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 52.Weeks CM, Miller R. The design and implementation of SnB version 2.0. J Appl Crystallogr. 1999;32:120–124. [Google Scholar]
- 53.Terwilliger TC. SOLVE and RESOLVE: automated structure solution and density modification. Methods Enzymol. 2003;374:22–37. doi: 10.1016/S0076-6879(03)74002-6. [DOI] [PubMed] [Google Scholar]
- 54.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 55.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 56.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kawate T, Gouaux E. Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure. 2006;14:673–681. doi: 10.1016/j.str.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 58.Li XM, Zheng S, Agard DA, Cheng YF. Asynchronous data acquisition and on-the-fly analysis of dose fractionated cryoEM images by UCSFImage. J Struct Biol. 2015;192:174–178. doi: 10.1016/j.jsb.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li X, et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol. 2003;142:334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 61.Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kucukelbir A, Sigworth FJ, Tagare HD. Quantifying the local resolution of cryo-EM density maps. Nat Methods. 2014;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for Figures 1, 3, 4, 5 and 6 and for Supplementary Figures 5 and 6 are provided with the paper. Other data supporting the findings of this study are available from the corresponding author upon reasonable request.