

Abstract
Psychological stress has a pervasive influence on our lives. In many cases adapting to stress strengthens organisms, but chronic or severe stress is usually harmful. One surprising outcome of psychological stress is activation of an inflammatory response, resembling inflammation caused by infection or trauma. Excessive psychological stress and the consequential inflammation in the brain can increase susceptibility to psychiatric diseases, such as depression, and impair learning and memory, including in some patients with cognitive deficits. An emerging target to control detrimental outcomes of stress and inflammation is glycogen synthase kinase-3 (GSK3). GSK3 promotes inflammation, partly by regulating key transcription factors in the inflammation signaling pathway, and GSK3 can impair learning by promoting inflammation and by inhibiting long term potentiation (LTP). Drugs inhibiting GSK3 may prove beneficial for controlling mood and cognitive impairments caused by excessive stress and the associated neuroinflammation.
Keywords: Alzheimer’s disease, depression, glycogen synthase kinase-3, inflammation, learning, stress
Excessive stress and inflammation are detrimental to health
Stress and inflammation are normal processes that help organisms respond and adapt to changes in the environment, to trauma, and to infection (see Glossary). In this article we focus on psychological stress and the ensuing inflammation in the brain (neuroinflammation). If these are severe or prolonged, they are usually detrimental, often leading to a variety of medical problems or worsening of pre-existing health problems. The deleterious effects of psychological stress and inflammation can impact many organs and diseases, including cardiovascular diseases, cancers, and immunological diseases. However, here we focus on how psychological stress and neuroinflammation impact the regulation of mood and cognition.
Along with new revelations of the detrimental effects of psychological stress and neuroinflammation on the central nervous system (CNS), there has been a parallel surge in studies attempting to counteract such effects. One promising therapeutic target found in these studies is glycogen synthase kinase-3 (GSK3), which refers to two homologous serine/threonine kinases, GSK3α and GSK3β. GSK3 is a fascinating kinase with several unconventional characteristics: it is constitutively active, substrate pre-phosphorylation by another kinase is required for GSK3 to phosphorylate most of its substrates, and GSK3 is inhibited, rather than activated like many kinases, by most signaling pathways impinging on GSK3 [6]. Among its actions, GSK3 has pivotal roles in the response to psychological stress and in inflammation, and inhibition of GSK3 can control many detrimental outcomes of psychological stress and inflammation in the CNS.
Psychological stress and Inflammation
Why are psychological stress and inflammation considered together here? Activation of the hypothalamic–pituitary–adrenal (HPA) axis is the most widely recognized response to stress, which involves sequentially increased secretion of corticotrophin-releasing factor, adrenocorticotrophin and cortisol, a hormone that is thought to mediate some of the adverse effects of severe or chronic psychological stress. Another key component of the psychological stress response is inflammation, a response that appears to be particularly important in the damaging effects of psychological stress on the CNS [1]. It is often surprising to learn that inflammation is activated by stress, since inflammation is most often thought of as a response to injury or disease. But psychological stress induces many of the same inflammatory signals that are induced by injury and disease. This raises many questions that are currently receiving much attention by researchers: how and why does psychological stress activate inflammation, is it beneficial or detrimental, and if detrimental, can it be controlled pharmacologically? Research has yet to fully answer these questions, but some tantalizing clues have been uncovered.
Before considering the links among stress, inflammation, GSK3, and CNS functions and diseases, it is useful to briefly review key components of the inflammatory response that are activated by psychological stress. The biochemical signals involved in inflammation were initially identified in studies of the immune system where the regulatory effects of GSK3 were first identified, and several of the same processes have been shown to be involved in psychological stress-induced inflammation as depicted in Figure 1. Responses of the immune system have been divided into innate immunity and adaptive immunity. Innate immunity provides a rapid response to pathogens, trauma and stress [2]. One role of the innate immune system is to stimulate the production and release of cytokines, small proteins, such as tumor necrosis factor-α (TNFα), interleukins (IL), interferons, and chemokines, which stimulate specific receptors on cells to induce intracellular signaling pathways. One mechanism used for this is activation of Toll-like receptors (TLRs). Ten TLRs have identified in humans and 12 in mice, and of these TLR4 has been most closely linked to inflammation caused by psychological stress. Nuclear factor-κB (NF-κB) is one of the main transcription factors activated by TLRs, which mediates the increased production of many cytokines. Cytokine production is only one facet of the complex inflammatory response, which is described in more detail elsewhere [3]. However, cytokines are the focus of many studies of diseases, in part because loss of control of cytokine production can be lethal, such as in sepsis. The cytokine response is mounted in two phases: first the production of proinflammatory cytokines (such as TNFα, IL-1, IL-6), and second the production of anti-inflammatory cytokines (such as IL-10), which functions to counteract inflammation to avoid excessive or prolonged inflammation.
Figure 1.
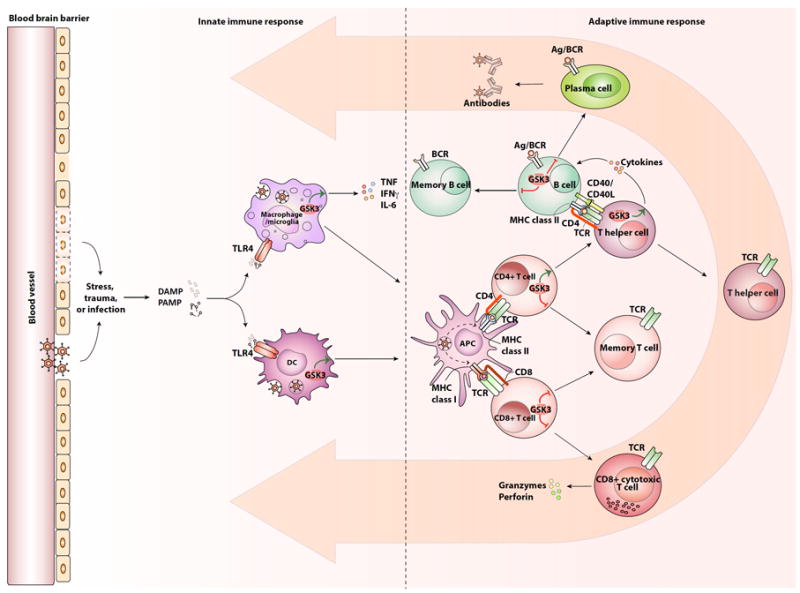
The innate and adaptive immune systems and the involvement of GSK3.
Upon stress, trauma, or infection by microbes, the innate immune system is activated. This occurs in part through the activation of TLR4, either directly by bacteria, by pathogen-associated molecular patterns (PAMPs), or by damage-associated molecular patterns (DAMPs, also called alarmins) that are generated from either the microbes or insults to the tissue. Activated TLR4 leads to the production of cytokines by macrophages and microglia, such as TNFα, IFNγ, and IL-6, and their expression is promoted by GSK3. Activated TLR4 induces immature dendritic cell (DC) development towards mature antigen presenting cells (APCs), which is facilitated by GSK3. Mature APCs present the antigen at the cell surface to activate the adaptive immune system, such as CD8+ T cells via presentation through major histocompatibility complex (MHC) class I or CD4+ T cells via MHC class II. Once activated, CD8+ T cells become cytotoxic CD8+ T cells and they produce granzymes and perforins to eliminate infected cells. GSK3 promotes the expression of PD-1 at the surface of CD8+ cells, which promotes exhaustion of the CD8+ cells and thereby reduces their cytotoxic function. Activated CD4+ T cells differentiate towards T helper cells. GSK3 promotes the differentiation of CD4+ T cells towards Th1 and Th17 subtypes. T helper cells also activate B cells to become plasma cells, which produce the immunoglobulin (antibody) specific to the encountered insult. CD8+, CD4+ and B cells all have the capacity to form a pool of memory cells, which are reduced by GSK3, which can be reactivated upon experiencing the same antigen. Once activated, the cells migrate to the site of the insult to resolve the inflammation, as indicated by the large orange arrows.
Put GSK3 inside macrophages and DCs
Delete NK cells
change “Stress or trauma” to “Stress, trauma, or infection” and lead an arrow from that to DAMP and PAMP
Adaptive immune responses are slower than innate immune responses, and include antibody production and responses generated by two types of T cells, CD8+ T cells and CD4+ T cells. Once activated and differentiated, adaptive immune cells travel to target sites following chemokine gradients generated primarily by innate immune cells. Activation of innate and adaptive immune responses also turns on suppressive systems as a way to limit these responses, such as producing anti-inflammatory IL-10 and regulatory T (Treg) cells to limit the magnitude and duration of the inflammatory response to avoid damage to the host.
Inflammatory signaling by Toll-like receptor-4 (TLR4)
A prevalent signaling pathway that activates inflammation, including after psychological stress, is mediated by TLR4 (Figure 2). Besides being expressed by immune cells, in the CNS TLR4 is expressed by microglia, astrocytes and neurons. During infections, TLR4 can be activated by lipopolysaccharide (LPS), the major component of the outer membranes of Gram-negative bacteria, and uncontrolled activation of TLR4 leads to the potentially fatal condition of sepsis. Activated TLR4 induces the production of cytokines by Myd88-dependent and Myd88-independent pathways, and GSK3 enhances inflammatory signaling by both pathways (Figure 2).
Figure 2.
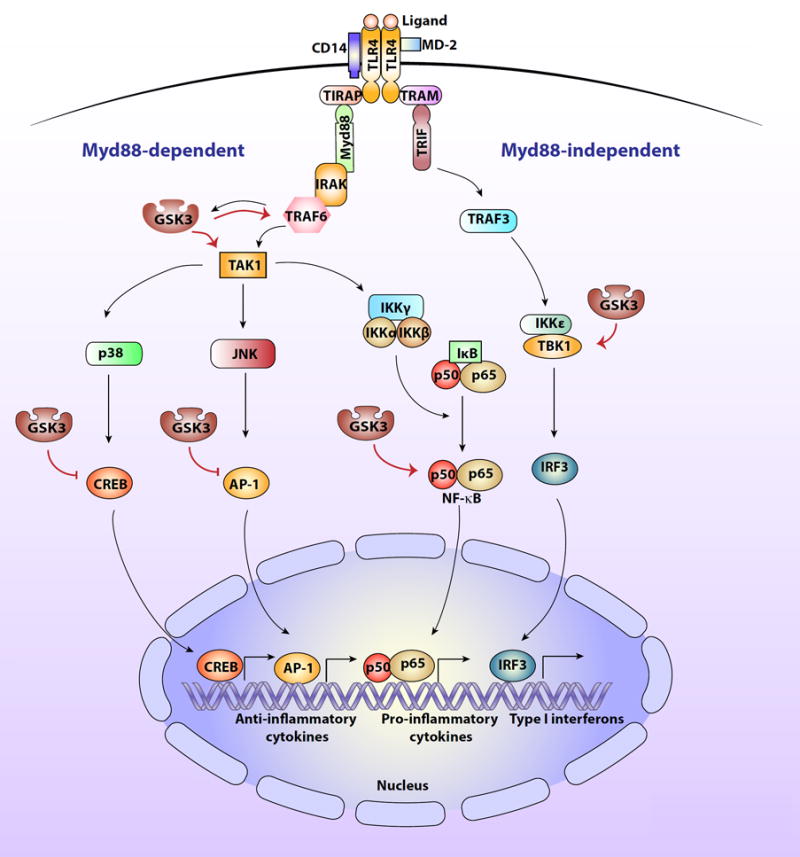
The involvement of GSK3 in TLR4 signaling pathways.
Activation of TLR4 by LPS or alarmins (called ligand in the figure) involves dimers of TLR4 binding to CD14 and MD-2. This induces Myd88 (myeloid differentiation factor 88)-dependent signaling to two serine-threonine kinases, IRAK4 (IL-1-receptor associated kinase 4) and IRAK1, which in turn bind to the E3 ubiquitin ligase TRAF6 (TNF receptor-associated factor 6) leading to activation of the serine-threonine kinase TAK1 (transforming growth factor beta-activated kinase 1), which is a site of regulation by GSK3. TAK1 phosphorylates the IκB kinase complex (IKK), which activates IKK. IKK in turn phosphorylates IκB, which releases active NF-κB. NF-κB, activation of which is promoted by GSK3, then migrates to the nucleus and activates the transcription of proinflammatory cytokines. TAK1 also activates the kinases p38 and JNK, which activate other transcription factors, such as AP-1 (activator protein 1), and CREB (cyclic AMP response element binding protein) that are involved in the production of anti-inflammatory cytokines and are inhibited by GSK3. Activated TLR4 also signals through a Myd88-independent pathway via TRAM (TRIF-related adaptor molecule) and TRIF (TIR domain-containing adaptor-inducing IFN-β) to activate TRAF3 (TNF receptor-associated factor 3), which in turn activates the serine/threonine kinase, TBK1 (TANK Binding kinase 1), which is regulated by GSK3. TBK1 promotes activation of the transcription factor IRF3 (interferon regulatory factor-3), which translocates to the nucleus where it induces the production of type I interferons.
Remarkably, psychological stress also causes activation of TLR4 to induce the production of cytokines. Research is only now identifying how this occurs, and TLR4 activation is likely only one of several means by which stress induces an inflammatory response. The mechanism for this appears to be that stress increases the extracellular levels of proteins that are called alarmins, or danger-associated molecular patterns (DAMPs). Just like LPS from infecting bacteria, a variety of different alarmins can directly activate TLR4, including some that are normally stored intracellularly and released by insults (e.g., certain heat shock proteins), proteolytic products of the extracellular matrix (e.g., hyaluronic acid), and some that are normally secreted by cells (e.g., HMGB1: high-mobility group box-1 protein) [4]. Still unclear in many cases is how stress is able to increase the levels of extracellular alarmins. One example of stress-induced alarmins are epitopes caused by oxidative stress, a condition many believe is associated with CNS disorders, such as depression and Alzheimer’s disease. Alarmins also can arise from the atrophy and loss of neurons and glia that occur with chronic stress in animals and in brains of patients with neurodegenerative diseases. This extensive pool of structurally diverse alarmins can cause the shared outcome of TLR4 activation, demonstrating that there are multiple potential sources of alarmins that could activate TLRs following stress. Thus, one pathway by which stress induces inflammation is by increasing levels of alarmins that activate TLR4, which then induces intracellular signaling cascades that increase the production of cytokines. However, this is only one mechanism by which stress increases inflammation and others are also being identified, such as activation of other TLRs and of inflammasomes (Box 1).
Box 1. Stress, GSK3, and the NLRP3 inflammasome.
Inflammasomes are part of the innate immune response that contribute to inflammation. After exposure to microbes or stress, inflammasomes are activated to induce inflammation, which includes activation of caspase-1. In the brain, the cytosolic inflammasome complex containing NLRP3 has particularly received attention because it is activated in microglia, macrophages and astrocytes in response to psychological stress. Activation of NLRP3 after psychological stress has been associated with depression-like behaviors [15,47-49]. For example, NLRP3 in the brain was increased by treatments that can increase depression-like behaviors, including inescapable tail shocks [50] and by chronic unpredictable mild stress, which was blocked by treatment with the antidepressant fluoxetine [51]. NLRP3 activation leads to the cleavage of pro-caspase-1 into active capase-1, which is responsible for activation of the inflammatory cytokine IL-1β. Caspase-1 deficiency decreased chronic restraint stress-induced depression-like behaviors [52]. GSK3β activates the NLRP3/IL-1β pathway in lupus nephritis [53]. Psychological stress-induced activation of GSK3β leads to activation of the NLRP3/IL-1β pathway [15,54]. Thus, NLRP3 inflammasome activation is another mechanism, in addition to TLR-mediated signaling, that is stimulated by psychological stress, utilizes activated GSK3, and increases inflammatory cytokines in the brain.
Chronic increases of cytokines in the CNS can have a variety of different outcomes depending in part on the types of cells that produce the cytokines and their localization. Although the CNS was previously considered isolated and protected from immune responses, it is now apparent that neuroinflammation occurs in the brain and has similar features as inflammation in the periphery and that the blood brain barrier (BBB) is not impermeable to inflammation. Cytokine levels increase in the brain after peripheral exposure or administration of LPS and they can cause sickness behavior in mice, which is characterized by the same symptoms as the flu except for the fever, and has been used as a model of depression [5]. In neurodegenerative diseases, such as Alzheimer’s disease or multiple sclerosis, immune cells damage neurons, which contributes to loss of memory in Alzheimer’s disease and loss of locomotor function in multiple sclerosis. Thus, cytokines in the CNS exacerbate both psychiatric and neurological diseases, effects that have led to studies aimed at finding mechanisms to control neuroinflammation.
Glycogen synthase kinase-3 (GSK3) is activated by stress and promotes inflammation
Recent findings indicate that one way to reduce detrimental effects of psychological stress and to limit inflammation is by administering drugs that inhibit GSK3. GSK3α and GSK3β are primarily regulated by inhibitory phosphorylation on ser-21-GSK3α and ser-9-GSK3β, which can be mediated by several kinases, such as Akt (protein kinase B), cyclic AMP-dependent protein kinase, or others (Figure 3) [6]. This serine-phosphorylation causes the N-terminal tail of GSK3 to act as a pre-phosphorylated substrate, or pseudosubstrate, that fits into the substrate-binding pocket of GSK3, hindering the binding of primed substrates, and thus diminishing the ability of GSK3 to phosphorylate substrates. Functions of the inhibitory phosphorylation of GSK3 can be studied by using GSK3 knockin mice in which the serines are mutated to alanines to prohibit this major mechanism of GSK3 inhibition, leaving GSK3 constitutively active but not overexpressed [7]. These mice provide a means to test the actions of abnormally active GSK3 expressed at normal levels and without associated pathology.
Figure 3.
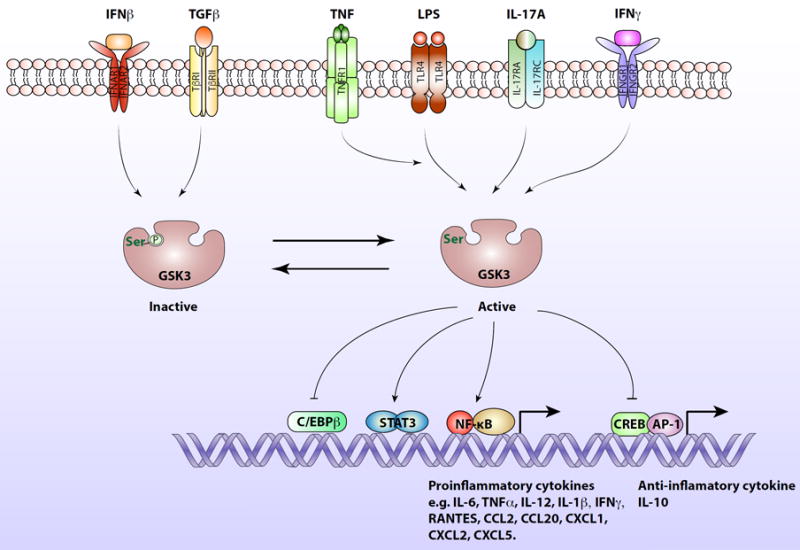
GSK3 is a central kinase in the inflammatory response.
Intracellular signaling induced by IFNβ and TGFβ promotes the inhibitory serine-phosphorylation of GSK3 on serine-9 of GSB3β and serine-21 on GSK3α, allowing the induction of IL-10 production by blocking the inhibitory effect of GSK3 on the CREB and AP-1 transcription factors that induce IL-10 expression. In contrast, signaling induced by LPS activates GSK3, which promotes NF-κB activation and translocation to the nucleus where it activates the expression of proinflammatory cytokines. Activated GSK3 also inhibits CREB and AP-1 transcriptional activities in response to LPS, diminishing production of the anti-inflammatory cytokine, IL-10. TNFα promotes the LPS-induced activation of GSK3. Several cytokines stimulate intracellular signaling pathways that regulate GSK3. For example, IL-17A stimulates signaling that activates GSK3, which causes inhibition of C/EBPβ transcriptional activity. GSK3 is also activated by IFNγ-stimulated signaling, and in response to IFNγ active GSK3 promotes STAT3 phosphorylation and activation, leading to its translocation to the nucleus where dimerized STAT3 induces the expression of proinflammatory cytokines.
In addition to activating the inflammatory response, psychological stress also activates GSK3 in the CNS. GSK3 is activated in rodent brains by several stressors that are widely used to induce depression-like behaviors in rodents, such as inescapable foot shocks [9], social defeat stress [10] restraint stress [11] and social isolation [12]. However, one must keep in mind that depression as occurs in the human disease is not thought to occur in rodents, which has led to measurements of behaviors (depression-like behaviors) that model various components of depression [13]. Psychological stress-induced activation of GSK3 occurs by reduction of the inhibitory serine-phosphorylation on GSK3, either by activation of protein phosphatases or by inhibition of the kinases mediating this modification, such as Akt. One mechanism for stress to achieve activation of GSK3 is by stress-induced increases in corticosterone (cortisol in humans) [14], although the signaling mechanism for corticosterone-induced activation of GSK3 remains unclear. Recently it was found that psychological stress-induced activation of GSK3 is intimately linked to inflammation, as blocking signaling through TLR4 blocked the activation of GSK3 by stress. Blocking this signaling through TLR4 and GSK3 also decreased stress-induced inflammation, and nearly eliminated the development of depressive symptoms in mice [15]. Thus, susceptibility to depressive characteristics in rodents, and perhaps depression in humans, is promoted by psychological stress-induced signaling through TLR4 that activates GSK3 and neuroinflammation.
In 2005 it was discovered that GSK3 is a component of the inflammatory signaling pathway [16], and since then it has been found that the anti-inflammatory effects of GSK3 inhibitors are particularly remarkable. The inflammatory response was first found to require GSK3 in studies of several subtypes of TLRs in human monocytes [16], demonstrating that GSK3 is necessary for the stimulated production of many inflammatory cytokines (Figure 1). Importantly, GSK3 inhibitors administered to mice in vivo protected them from otherwise lethal sepsis induced by LPS, demonstrating the potential for disrupting inflammation in vivo using GSK3 inhibitors [16]. Remarkably, GSK3 also inhibits production of the anti-inflammatory cytokine IL-10, which is reversed by inhibiting GSK3. Thus, GSK3 inhibitors have the powerful ability to shift the balance of the inflammatory response from pro-inflammatory to anti-inflammatory, demonstrating the potential for therapeutic use of these drugs to control inflammation.
Inhibition of GSK3 was subsequently found to reduce inflammation in the CNS as well as in the periphery. Using isolated cells in vitro, GSK3 inhibitors were found to inhibit cytokines produced by the two main sources in the CNS, microglia and astrocytes. Thus, GSK3 inhibition effectively reduces inflammation throughout the periphery and in the CNS [17]. Several mechanisms were found to contribute to the regulation of cytokine production by GSK3 (Figure 3), such as promoting the activation of transcription factors in the inflammatory response, including NF-κB and signal transducer and activator of transcription-3 (STAT3). Oppositely, GSK3 inhibits production of the anti-inflammatory cytokine IL-10 by inhibiting the transcription factors CREB and AP-1. Thus, inhibitors of GSK3 act on both arms of the inflammatory response, reducing inflammatory cytokines and increasing anti-inflammatory cytokines. These anti-inflammatory actions of GSK3 inhibitors have been shown to produce beneficial effects in animal models of multiple inflammatory diseases, such as endotoxic shock, arthritis, peritonitis, and colitis [17]. Increasing evidence suggests that GSK3 inhibitors also may relieve inflammation in diseases of the CNS, as discussed later.
In addition to regulating cytokine production, GSK3 also regulates T cells, affecting their proliferation, survival and differentiation [17]. For example, GSK3 promotes the differentiation of pro-inflammatory Th1 and Th17 cells, whereas GSK3 blocks the suppressive activity of Tregs [18-20]. Promotion of the formation of Th17 cells by GSK3 may be particularly important because Th17 cells are known to be pathogenic to the CNS (Box 2), in part by releasing the inflammatory cytokine IL-17, which can be controlled by GSK3 inhibitors. Altogether, GSK3 is a critical regulator of both the innate and adaptive immune responses and GSK3 inhibitors provide a means to control these processes when they are prolonged or severe and cause damage.
Box 2. Interleukin-17 (IL-17) production and actions.
IL-17 is produced mainly by Th17 cells, but also by other cell types. GSK3 is regulatory in both the genesis of cells that produce IL-17 and the signaling induced by IL-17. \GSK3 is required for the differentiation of Th17 cells, as CD4+ T cells deficient in GSK3β are unable to differentiate towards Th17 cells. Blocking Th17 cell production by inhibition or depletion of GSK3 confers resistance to the mouse model of multiple sclerosis, experimental autoimmune encephalomyelitis (EAE) [19,20]. Also, IL-13 blocks the production of IL-17A by inhibiting GSK3β [55]. There is also evidence that GSK3β is itself regulated as part of the Th17 differentiation program: GSK3β expression, usually relatively stable in most cells, increased 10-fold during the differentiation of Th17 cells [19].
GSK3 is also a component of the IL-17-induced signaling pathway. IL-17 activates mainly non-haematopoeitic cells and it is considered to be an inflammatory cytokine. In neutrophils, IL-17 induces the genes encoding G-CSF, IL-6 and CXC chemokines (CXCL1, CXCL2 and CXCL5), which promote neutrophil chemotaxis. IL-17 also induces the expression of antimicrobial peptides, such as β-defensins and lipocalin 2. The signaling pathway induced by IL-17 has unique properties. The receptor for IL-17, IL-17R, lacks similarity with other known receptor families. There are 5 homologous subunits of IL-17R (IL-17RA-E), and 5 ligands (IL-17A, IL-17B, IL-17C, IL-17E, IL-17F), which all bind IL-17RA except for IL-17B. IL-17-induced signaling activates NF-κB and C/EBPδ and β, and both transcription factors are regulated by GSK3β and other mechanisms. GSK3β negatively regulates IL-17 signaling by phosphorylating both the downstream effector C/EBPβ (thereby decreasing downstream gene expression) [56-58] and the IL-17RA receptor, which triggers its degradation [59]. Altogether, GSK3β is central in the regulation of IL-17 by controlling both its production and its effectors. This has tremendous consequences for diseases with an inflammatory component, such as multiple sclerosis, as inhibiting GSK3 even after the first episode is sufficient to reduce the relapse and the severity of EAE in mice [19,20]. Although controlling Th17 and IL-17 signaling may be one of the mechanisms whereby GSK3 promotes EAE, other actions of GSK3 may be involved in disease progression, such as promoting infiltration of inflammatory cells through the weakened BBB [14,32,47] and the production of cytokines [16], and decreasing PD-1 expression [60]. Altogether, inhibiting GSK3 may provide therapeutic benefits for multiple sclerosis and other inflammatory diseases.
CNS diseases involving psychological stress, inflammation and GSK3
Psychological stress and inflammation contribute to many psychiatric and neurological diseases. The recent revelations that psychological stress increases neuroinflammation, and that this is mediated by a pathway involving GSK3, raise the notion that stress-induced GSK3 activation and inflammation may contribute to psychiatric and neurological diseases and provide targets for developing new therapeutic interventions. Here we discuss these connections in relation to mood disorders and Alzheimer’s disease as exemplary psychiatric and neurodegenerative diseases.
Stress, inflammation and GSK3 in the regulation of mood: mood disorders
Mood disorders include bipolar disorder (previously called manic-depression) and major depressive disorder, commonly called depression, although depression also occurs in other conditions. Mood disorders afflict ~17% of people in the United States at some time during their lives, and mood disorders are chronic, recurring conditions that are often not adequately treated with currently available medications [21]. Thus there is a critical need to better understand the causes of mood disorders in order to develop more effective treatments.
Psychological stress is the most common precipitating factor of mood disorders [22]. Now that it is evident that stress activates GSK3 and causes neuroinflammation, a current goal of research is to decipher the interactions among stress, GSK3 activation and inflammation to determine if these may provide new avenues for intervening in mood disorders. Increases in many cytokines, particularly IL-6 and TNFα, have been found in the serum of patients with depression. Inflammation in patients with depression is associated with resistance to treatment with classical antidepressant drugs, and there is some evidence that anti-inflammatory drugs can improve antidepressant actions [23,24]. Administration of LPS, the agonist of TLR4, induces symptoms of depression in humans [25], and administration of interferon to bolster immunity induces depression in many patients [26]. Most investigators feel that aberrant activation of inflammation, in combination with genetic and environmental factors, contributes to susceptibility and onset of mood disorders, rather than inflammation arising as a consequence of mood disorder episodes [23], and this is supported by evidence from studies in animals. In rodents, administration of inflammatory cytokines or LPS causes depression-like behaviors that are attenuated by antidepressants [24]. In rodents, aberrant innate (microglia) and adaptive (Th17 cells) immune system responses have been associated with depression-like behaviors [20,27,28]. Intriguing emerging studies also demonstrate that the gut microbiota has a strong influence on inflammation, stress responses, and their effects on mood regulation and cognition (Box 3). This and much additional evidence has demonstrated that inflammation can precipitate depression and impair therapeutic responses [29].
Box 3. The microbiome modulates functions of the CNS.
One of the most intriguing concepts that is currently receiving widespread attention is that microbes in the gut (i.e., gut microbiota) substantially influence human behavior and health. The composition of the gut microbiota can influence both the development and the exacerbation of mental disorders. The human microbiome is composed of ~100 trillion bacteria that are crucial for health, which process complex polysaccharides from the diet and are required for a healthy immune system. The composition of the gut microbiota varies among individuals, but it is estimated to consist of ~1000 species and >7000 strains. Besides being modified by diet, drugs (e.g., antibiotics) and other environmental factors, the composition of the gut microbiota can be altered by exposure to stress (e.g., sleep disruption) and mice displaying depression-like behaviors have different gut microbiota than non-depressed mice [61]. Conversely, the gut microbiota modulates the immune system [62], including the inflammatory response to stress [63]. The composition of the gut microbiota also affects many facets of behavior and cognition [64]. For example, modification of the microbiome using treatment with either antibiotics or probiotics alters depressive-like behaviors in rodents [61]. Of particular interest for the topics of this review, the inhibitory serine-phosphorylation of GSK3 in rodent brains is increased after treatments with several antibiotics (e.g., salinomycin, minocycline), indicating that changes of the microbiota also influences the activity of GSK3, which may impact the influence of GSK3 on mood regulation, including depression, and learning and memory, including Alzheimer’s disease [65]. Thus, the gut microbiota is an important modulator of CNS functions that affects behavior, mood regulation, inflammation, and cognition.
There is also substantial evidence that abnormally active GSK3 due to reduced inhibitory serine-phosphorylation contributes to susceptibility to mood disorders and is inhibited by therapeutic agents, as reviewed [30]. Increased activation of GSK3 was detected in postmortem brain regions from patients with major depression [31] and in brains of rodents displaying depression-like behaviors, such as learned helplessness [9], social defeat stress [10], and anhedonia [32]. Expression of constitutively active GSK3 in knockin mice increases susceptibility to depression-like behaviors [9]. Conversely, mice expressing diminished levels of GSK3β [33,34] or treated with GSK3 inhibitors (reviewed in [30]) display resistance to all measures of depression-like behavior that have been studied, such as learned helplessness, forced swim test, tail suspension test, novelty suppressed feeding, anhedonia, and social defeat-induced social avoidance. Many of the neuromodulators that may be deficient in depression, such as serotonin, and BDNF (brain-derived growth factor), normally stimulate signaling pathways that maintain inhibition of GSK3 [6], suggesting deficiencies in these signals in mood disorders leave GSK3 inadequately inhibited and that inhibition of GSK3 may be therapeutic for mood disorders, perhaps in part by reducing neuroinflammation. The first identified inhibitor of GSK3 is lithium, a mood stabilizing drug used to treat bipolar mood disorder, and many new GSK3 inhibitors have been developed due to their promising therapeutic potential in several diseases [8]. Besides lithium, other therapeutics used for mood disorders also inhibit GSK3, including valproate, classical antidepressants, ketamine, and atypical antipsychotics [30]. Treatment with each of these drugs alters intracellular signaling pathways that increases the inhibitory serine-phosphorylation of GSK3. Lithium has the additional property of directly binding to GSK3 to inhibit its activity as a competitive inhibitor with magnesium, so lithium both indirectly and directly inhibits GSK3. Thus, therapeutics that inhibit GSK3 are safe and effective in many patients with mood disorders. However, side effects can limit their tolerability in some patients and only a few clinical trials have been carried out with direct inhibitors of GSK3, other than lithium, so it remains to be seem if these are safe, tolerated, and effective.
Altogether, psychological stress, inflammation, and abnormally active GSK3 due to reduced inhibitory serine-phosphorylation all coincide in mood disorders. Substantial evidence indicates that a signaling pathway involving psychological stress-induced activation of GSK3 that promotes inflammation is a central facet of mood disorders and is a feasible target for therapeutic intervention.
Stress, inflammation and GSK3 in the regulation of learning and memory: Alzheimer’s disease
Besides impacting mood regulation, psychological stress, GSK3 and inflammation also have major influences on learning and memory, and contribute to cognitive impairments in neurodegenerative diseases, for which we use Alzheimer’s disease as an example.
First of all, it is important to note that in the absence of induced inflammation, learning and memory are surprisingly dependent on molecules such as cytokines that are usually thought of as mediators of inflammation. Learning is mediated in part by experience-dependent strengthening of synapses that can be measured electrophysiologically as a process called long-term potentiation (LTP). There is ample evidence that several cytokines, including IL-1, IL-6, and TNFα, among others, produced locally by microglia, astrocytes and neurons are supportive of LTP under basal conditions [35]. These are often observational measurements and questions remain about the underlying mechanisms, although there are some clues. IL-6, which signals through the IL-6 receptor complexed with two gp130 subunits, triggers activation of members of the Janus kinase (JAK) family of tyrosine kinases that are intracellularly associated with gp130. This leads to phosphorylation-mediated activation of signal transducer and activator of transcription-3 (STAT3). JAK2 and STAT3 are found within postsynaptic densities of hippocampal synapses and inhibition of JAK2 impairs hippocampal-dependent memory [66]. The induction of LTP at the Schaffer collateral to CA1 pyramidal neuron synapse in thr hippocampus increases hippocampal IL-6 levels, and IL-6 negatively regulates LTP maintenane, which allows new memory formation [87]. The JAK2/STAT3 pathway also is essential in hippocampal NMDA receptor-dependent long-term depression (LTD) [68], but the role of IL-6 in this process remains to be firmly established. Thus, as with many biological molecules, “normal” cytokine levels support basic cellular functions, whereas excessive levels can be harmful, so basal levels of cytokines are present to support learning, but excessive or prolonged up-regulation of cytokines can be detrimental for learning. Thus, cytokines are not inherently “bad”, but unregulated increases in cytokines may become pathological.
In contrast to the actions of cytokines without stimulated inflammation, cytokines can impair learning and memory after treatments that induce or mimic inflammation. Administration of LPS, IL-6, TNFα, IL-1β and other cytokines impair learning and memory. For example, treatment with LPS or IL-1β impaired hippocampal-dependent memory consolidation [36]. One mechanism for this appears to involve IL-1β-induced impaired neurogenesis mediated in part by activation of c-Jun-N-terminal kinase (JNK), although precise signals have yet to be determined [69]. However, for many of the reported impairments in learning following inflammation it is difficult to attribute to a specific cytokine since multiple cytokines are increased during inflammation. Not surprisingly, psychological stress-induced inflammation can contribute to cognitive impairments. Additionally, impaired resolution of inflammation or persistence of stimulated inflammatory signaling are thought to be important mechanisms that maintain inflammation in neurodegenerative diseases and promote the impairments associated with the primary disease lesion in many neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, as well as multiple sclerosis, which is generally considered an inflammatory autoimmune disease [37].
Impaired learning and memory are particularly crucial in Alzheimer’s disease, which is characterized by neurofibrillary tangles (intraneuronal aggregates of hyperphosphorylated tau and other proteins), amyloid plaques (extracellular aggregates of amyloid-β peptides [Aβ]), synaptic degeneration, and neuronal loss. Chronic psychological stress promotes cognitive impairments in Alzheimer’s disease, both in patients and rodent models [37,38]. For example, several epidemiological studies have reported relationships between chronic stress and increased risk for developing cognitive impairments and Alzheimer’s disease [39-41]. Stress also accelerates the disease pathologies in rodent Alzheimer’s disease models, including increased Aβ production, increased tau phosphorylation, and impaired cognition [37,38]. Although the psychological stress response is multifaceted, there is substantial evidence that inflammation promotes deleterious consequences in patients with Alzheimer’s disease [42]. Many cytokines are elevated in Alzheimer’s disease patients and in transgenic mouse models of Alzheimer’s disease, such as IL-1β, IL-6, and TNFα, and chemokines [37]. Like stress, inflammatory cytokines promote Aβ production, tau phosphorylation and cognitive impairments in rodents, suggesting that inflammation contributes to the deleterious effects of stress. Plaques and tangles, and likely other causes, promote continued inflammation by activation of inflammatory microglia and astrocytes in the brain. These inflammatory microglia and astrocytes, often found in the vicinity of plaques and tangles, continue to produce more cytokines and other inflammatory molecules. Perhaps particularly pernicious in this regard is the capacity of Aβ itself to directly activate TLR4 and receptor for advanced glycation end-products (RAGE) to further activate microglia and promote inflammation [43].
GSK3 is well-established to promote tau phosphorylation and Aβ production in Alzheimer’s disease, and since it promotes inflammation it also likely contributes to the elevated cytokines that occur in Alzheimer’s disease. GSK3β is also activated after overexpression of IL-1β in a mouse model of Alzheimer’s disease, which was proposed to contribute to exacerbation of tangles [44]. Furthermore, GSK3 inhibits LTP [45], likely contributing to the cognitive deficits in Alzheimer’s disease. Altogether, it is thought that any insult that activates GSK3, such as psychological stress and certain cytokines, will increase the pathological characteristics of Alzheimer’s disease [46]. It may be that a vicious cycle is established as the initial formation of plaques and tangles associated with early Alzheimer’s disease in turn induces local inflammation that is augmented by psychological stress and by active GSK3, which further amplify plaque and tangle formation [37,38]. A challenge for future research is to identify the extent to which psychological stress-induced inflammation, as opposed to other components of the stress response, contributes to the progression of Alzheimer’s disease and cognitive deficits in general. Although we focus here on inflammation, it is important to note that stress-induced glucocorticoids also display level-dependent regulatory effects on cognition. Thus, it is evident that psychological stress, GSK3, and inflammatory cytokines are capable of promoting many facets of Alzheimer’s disease, including cognitive impairments, suggesting that controlling inflammation and/or GSK3 may provide a beneficial effect, but this would likely be limited to slowing progression of the disease rather than reversing existing pathology.
Altogether, it is evident that stress, inflammation, and GSK3 each can impair learning and memory. These mechanisms may act in concert to amplify degenerative changes in Alzheimer’s disease.
Conclusions
In summary, psychological stress induces inflammation in part through a pathway encompassing TLR4 and GSK3. The stress-induced inflammation appears to contribute to some of the medically detrimental effects of severe or prolonged stress, such as mood dysregulation and cognitive problems, as well as amplifying pathological processes in depression and Alzheimer’s disease. Research in the near future should further define the contributions of inflammation caused by stress to these and other disorders, and test if interfering with stress-induced inflammation and activation of TLR4 and of GSK3 provide means for developing new therapeutics (see Outstanding Questions).
Outstanding Questions.
What are the mechanisms by which stress is able to increase the levels of extracellular alarmins that lead to inflammation?
To what extent does inflammation contribute to the susceptibility, severity, and treatment response of mood disorders?
To what extents do stress-induced activation of GSK3 and stress-induced inflammation contribute to mood disorders and the inflammation associated with mood disorders, and are interventions that disrupt these responses to stress therapeutic?
To what extent do stress-induced activation of GSK3 and inflammation contribute to cognitive deficits and the progression of Alzheimer’s disease?
Are drugs that inhibit GSK3 therapeutic for treating inflammatory disorders, mood disorders, and Alzheimer’s disease, or would it be more efficacious to target signaling molecules that regulate GSK3 or downstream substrates of GSK3?
Would diminishing peripheral inflammation, controlling peripheral immune cells (such as Th17 cells) or altering the gut microbiota be sufficient to provide therapeutic effects in mood disorders or Alzheimer’s disease, or is it be necessary to directly regulate inflammation in the CNS?
Which inflammatory molecules, types of cytokines, or types of cells (e.g., subtype of T cells) are the major causes of impaired mood regulation and impaired learning and memory, and contribute to depression and Alzheimer’s disease?
What are the mechanisms underlying individual differences in the ability to cope with stress and control inflammation that determine resilience or susceptibility to mood dysregulation and cognitive impairments?
Are there potential detrimental consequences of pharmacologically regulating the stress and inflammatory responses, such as impairing the healthy actions of these responses in mood regulation and learning and memory?
Rather than interventions causing global inhibition of inflammation or of GSK3, are there specific cell types that can be targeted to minimize side effects while still being therapeutic?
Trends Box.
Psychological stress and inflammation can generate healthy responses, but if they are chronic or excessive, produce deleterious consequences on mood regulation and learning and memory.
Inflammation is induced by psychological stress by many of the same signaling pathways that activate inflammation after infection or trauma, including activating TLRs and GSK3.
A signaling pathway involving stress-induced activation of GSK3 that promotes inflammation may be a central facet of mood disorders and a potential therapeutic target.
Stress-induced activation of GSK3 and inflammation impair LTP and some facets of learning and memory.
A vicious cycle may be established in Alzheimer’s disease as the initial formation of plaques and tangles associated with early Alzheimer’s disease induces local inflammation that is augmented by stress and by active GSK3, which further amplify plaque and tangle formation.
Acknowledgments
Research in the authors’ laboratories was supported by grants from the NIMH (MH038752, MH095380, MH104656).
Glossary
- Amyloid plaques
Extracellular aggregates of misfolded proteins, one of the defining characteristics of Alzheimer’s disease with the plaques primarily containing amyloid beta (Aβ) peptides derived from cleavage of the amyloid precursor protein.
- AP-1
A transcription factor composed of heterodimers of Fos and Jun family proteins that is rapidly activated by many stimuli, such as stress, injury, and infection, that regulates gene expression influencing proliferation, differentiation, apoptosis, and other cell processes.
- Astrocyte
Glia cell in the CNS characterized by star-like projections that interacts with neurons, and is involved in responses to trauma and stress, including production of inflammatory molecules.
- BBB
Blood-brain-barrier. A barrier formed by endothelial cells, connected via tight adhesive junctions, which provides highly selective permeability for molecules passing from the blood to the extracellular space in the brain.
- CD4
Cluster of differentiation 4. CD4 is a glycoprotein present on the surface of immune cells, such as T helper cells, that serves as a co-receptor for the T cell receptor.
- CD8
Cluster of differentiation 8. CD8 is a transmembrane glycoprotein serving as a co-receptor for the T cell receptor and is expressed by cytotoxic T cells.
- Chemokine
A family of a subtype of cytokines that are most well-known to serve as guides for migrating cells, including in immunology chemotaxis of competent cells.
- CREB
cyclic AMP response element-binding protein, a transcription factor that, among its functions, regulates learning and memory, and may be deficient in Alzheimer’s disease and Major Depressive Disorder.
- Cytokine
A class of small signaling proteins secreted by cells that generally act through cell surface receptors and are particularly important in the immune response.
- DAMP
Damage-associated molecular patterns, or danger-associated molecular patterns, also called alarmins, are proteins in the host that act on receptors on immune cells, which can initiate a noninfectious, or sterile, inflammatory response.
- Depressive-like behavior
Rodent models of depression in which they exhibit behavior with characteristics similar to some of the symptoms of major depression in humans. Used as an experimental method to identify regulators of components of depression, such as despair, and social isolation.
- GSK3
Glycogen synthase kinase-3, refers to two homologous paralogs, GSK3α and GSK3β, ubiquitous serine/threonine protein kinases with over 100 known substrates that have been implicated in many prevalent disorders, including psychiatric and neurological diseases, inflammatory diseases, cancer, cardiovascular diseases, diabetes and others.
- Inflammation
A coordinated biological response to infection, trauma or stress that involves the production of signaling molecules, including but not limited to cytokines.
- LTP
Long-term potentiation, is a long-lasting increase in the amplitude of excitatory post-synaptic potentials measured electrophysiologically in response to high frequency stimulation. LTP is a critical phenomenon underlying synaptic plasticity and is a necessary cellular process leading to learning and memory.
- Microglia
Act as resident macrophages of the CNS that can produce inflammatory molecules, and are mobilized to present antigens and become phagocytes during infection or injury.
- Neurofibrillary tangles
Abnormal intracellular aggregates made up of paired, helical filaments primarily composed of the microtubule binding protein, Tau, which can accumulate in neuronal cell bodies, dendrites, and axons, disrupting normal neuronal function, and are present in Alzheimer’s disease and other tauopathies.
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells, a transcription factor that controls the expression of many cytokines and other cellular responses.
- PAMP
Pathogen-associated molecular patterns. PAMPs are molecules associated with pathogens that are recognized by the innate immune system.
- RAGE
Receptor for advanced glycation end-products. RAGE is a pattern recognition receptor involved in pro-inflammatory gene activation.
- Sepsis
A potentially lethal condition characterized by high risk of damage to tissues and organs resulting from an overreaction of the immune system’s response to infection.
- STAT3
Signal transducer and activator of transcription 3, a transcription factor that is phosphorylated and activated by receptor-associated Janus kinases (JAK) in response to cytokines and growth factors.
- Stress
There are many types of stress, however this review focuses on psychological stress, which involves exposure to, or perception of, threats or challenges that are especially difficult to cope with, that may or may not involve physical stressors.
- TLR
Toll-like receptors are receptors found in sentinel cells of the immune system, and in microglia, astrocytes and neurons in the CNS, that induce intracellular signaling pathways after activation, often leading to inflammation.
Abbreviations
- AP-1
Activator Protein-1
- BBB
Blood Brain Barrier
- BCR
B Cell Receptor
- C/EBP
CAAT/Enhancer Binding Protein
- CCL20
Chemokine (C-C motif) ligand 20
- CD14
Cluster of differentiation 14
- CD28
Cluster of differentiation 28
- CD4
Cluster of differentiation 4
- CD8
Cluster of differentiation 8
- CNS
Central Nervous System
- CREB
cAMP Response Element Binding
- CXCL
chemokine (C-X-C motif) ligand
- DAMP
Damage-associated molecular patterns
- EAE
experimental autoimmune encephalomyelitis
- G-CSF
Granulocyte-Colony Stimulating Factor
- GSK3
Glycogen Synthase Kinase-3
- HMGB1
High Mobility Group Box 1 protein
- HPA
Hypothalamic–Pituitary–Adrenal axis
- IFN
Interferon
- IKK
IKB kinase
- IL-1β
Interleukin-1β
- IL-6
Interleukin-6
- IL-10
Interleukin-10
- IL-13
Interleukin-13
- IL-17
Interleukin-17
- IL-17R
Interleukin-17 Receptor
- IRAK
interleukin-1 receptor-associated kinase-1
- IRF3
Interferon Regulatory Factor 3
- JNK
C-Jun N terminal Kinase
- LPS
Lipopolysaccharide
- LTP
long-term potentiation
- MHC
Major Histocompatibility Complex
- MS
Multiple Sclerosis
- Myd88
Myeloid differentiation primary response gene 88
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NLRP3
NLR Family, Pyrin Domain Containing 3)
- PAMP
Pathogen-associated molecular patterns
- STAT3
signal transducer and activator of transcription-3
- RAGE
Receptor for advanced glycation end-products
- TAK1
TGFβ Activated Kinase-1
- TBK1
TANK Binding Kinase-1
- TCR
T Cell Receptor
- Th17
T helper 17
- TIRAP
Toll-Interleukin 1 Receptor Domain Containing Adaptor Protein
- TGFβ
Transforming Growth Factor β
- TLR4
Toll-like Receptor-4
- TNF
Tumor Necrosis Factor
- TRAF6
TNF Receptor Associated Factor-6
- TRAM
TRIF-Related Adaptor Molecule
- TRIF
TIR domain-containing adaptor-inducing Interferon β
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lyman M, Lloyd DG, Ji X, Vizcaychipi MP, Ma D. Neuroinflammation: the role and consequences. Neurosci Res. 2014;79:1–12. doi: 10.1016/j.neures.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 2.Gasteiger G, Rudensky AY. Interactions between innate and adaptive lymphocytes. Nat Rev Immunol. 2014;14:631–639. doi: 10.1038/nri3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 4.Schaefer L. Complexity of danger: the diverse nature of damage-associated molecular patterns. J Biol Chem. 2014;289:35237–35245. doi: 10.1074/jbc.R114.619304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. 2015;148:114–131. doi: 10.1016/j.pharmthera.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eldar-Finkelman H, Martinez A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front Mol Neurosci. 2011;4:32. doi: 10.3389/fnmol.2011.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Polter A, Beurel E, Yang S, Garner R, Song L, Miller CA, Sweatt JD, McMahon L, Bartolucci AA, Li X, Jope RS. Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacology. 2010;35:1761–1774. doi: 10.1038/npp.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilkinson MB, Dias C, Magida J, Mazei-Robison M, Lobo M, Kennedy P, Dietz D, Covington H, 3rd, Russo S, Neve R, Ghose S, Tamminga C, Nestler EJ. A novel role of the WNT-dishevelled-GSK3beta signaling cascade in the mouse nucleus accumbens in a social defeat model of depression. J Neurosci. 2011;31:9084–9092. doi: 10.1523/JNEUROSCI.0039-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanno T, Tanaka A, Nishizaki T. Linoleic acid derivative DCP-LA ameliorates stress-induced depression-related behavior by promoting cell surface 5-HT1A receptor translocation, stimulating serotonin release, and inactivating GSK-3beta. Mol Neurobiol. 2015;51:523–532. doi: 10.1007/s12035-014-8718-5. [DOI] [PubMed] [Google Scholar]
- 12.Ren QG, Gong WG, Wang YJ, Zhou QD, Zhang ZJ. Citalopram attenuates tau hyperphosphorylation and spatial memory deficit induced by social isolation rearing in middle-aged rats. J Mol Neurosci. 2015;56:145–153. doi: 10.1007/s12031-014-0475-4. [DOI] [PubMed] [Google Scholar]
- 13.Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci. 2010;13:1161–1169. doi: 10.1038/nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobarro M, Orejana L, Aguirre N, Ramirez MJ. Propranolol reduces cognitive deficits, amyloid beta levels, tau phosphorylation and insulin resistance in response to chronic corticosterone administration. Int J Neuropsychopharmacol. 2013;16:1351–1360. doi: 10.1017/S1461145712001393. [DOI] [PubMed] [Google Scholar]
- 15.Cheng Y, Pardo M, Armini Rde S, Martinez A, Mouhsine H, Zagury JF, Jope RS, Beurel E. Stress-induced neuroinflammation is mediated by GSK3-dependent TLR4 signaling that promotes susceptibility to depression-like behavior. Brain Behav Immun. 2016;53:207–222. doi: 10.1016/j.bbi.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beurel E, Michalek SM, Jope RS. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3) Trends Immunol. 2010;31:24–31. doi: 10.1016/j.it.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graham JA, Fray M, de Haseth S, Lee KM, Lian MM, Chase CM, Madsen JC, Markmann J, Benichou G, Colvin RB, Cosimi AB, Deng S, Kim J, Alessandrini A. Suppressive regulatory T cell activity is potentiated by glycogen synthase kinase 3{beta} inhibition. J Biol Chem. 2010;285:32852–32859. doi: 10.1074/jbc.M110.150904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beurel E, Yeh WI, Michalek SM, Harrington LE, Jope RS. Glycogen synthase kinase-3 is an early determinant in the differentiation of pathogenic Th17 cells. J Immunol. 2011;186:1391–1398. doi: 10.4049/jimmunol.1003511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beurel E, Harrington LE, Jope RS. Inflammatory T helper 17 cells promote depression-like behavior in mice. Biol Psychiatry. 2013;73:622–630. doi: 10.1016/j.biopsych.2012.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kupfer DJ, Frank E, Phillips ML. Major depressive disorder: new clinical, neurobiological, and treatment perspectives. Lancet. 2012;379:1045–1055. doi: 10.1016/S0140-6736(11)60602-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McEwen BS, Bowles NP, Gray JD, Hill MN, Hunter RG, Karatsoreos IN, Nasca C. Mechanisms of stress in the brain. Nat Neurosci. 2015;18:1353–1363. doi: 10.1038/nn.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 2016;16:22–34. doi: 10.1038/nri.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Remus JL, Dantzer R. Inflammation Models of Depression in Rodents: Relevance to Psychotropic Drug Discovery. Int J Neuropsychopharmacol. 2016 doi: 10.1093/ijnp/pyw028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kullmann JS, Grigoleit JS, Lichte P, Kobbe P, Rosenberger C, Banner C, Wolf OT, Engler H, Oberbeck R, Elsenbruch S, Bingel U, Forsting M, Gizewski ER, Schedlowski M. Neural response to emotional stimuli during experimental human endotoxemia. Hum Brain Mapp. 2013;34:2217–2227. doi: 10.1002/hbm.22063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoyo-Becerra C, Schlaak JF, Hermann DM. Insights from interferon-alpha-related depression for the pathogenesis of depression associated with inflammation. Brain Behav Immun. 2014;42:222–231. doi: 10.1016/j.bbi.2014.06.200. [DOI] [PubMed] [Google Scholar]
- 27.Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9:429–439. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- 28.Wohleb ES, Franklin T, Iwata M, Duman RS. Integrating neuroimmune systems in the neurobiology of depression. Nat Rev Neurosci. 2016;17:497–511. doi: 10.1038/nrn.2016.69. [DOI] [PubMed] [Google Scholar]
- 29.Hodes GE, Kana V, Menard C, Merad M, Russo SJ. Neuroimmune mechanisms of depression. Nat Neurosci. 2015;18:1386–1393. doi: 10.1038/nn.4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jope RS. Glycogen synthase kinase-3 in the etiology and treatment of mood disorders. Front Mol Neurosci. 2011;4:16. doi: 10.3389/fnmol.2011.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karege F, Perroud N, Burkhardt S, Fernandez R, Ballmann E, La Harpe R, Malafosse A. Protein levels of beta-catenin and activation state of glycogen synthase kinase-3beta in major depression. A study with postmortem prefrontal cortex. J Affect Disord. 2012;136:185–188. doi: 10.1016/j.jad.2011.09.024. [DOI] [PubMed] [Google Scholar]
- 32.Cline BH, Costa-Nunes JP, Cespuglio R, Markova N, Santos AI, Bukhman YV, Kubatiev A, Steinbusch HW, Lesch KP, Strekalova T. Dicholine succinate, the neuronal insulin sensitizer, normalizes behavior, REM sleep, hippocampal pGSK3 beta and mRNAs of NMDA receptor subunits in mouse models of depression. Front Behav Neurosci. 2015;9:37. doi: 10.3389/fnbeh.2015.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Brien WT, Harper AD, Jove F, Woodgett JR, Maretto S, Piccolo S, Klein PS. Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci. 2004;24:6791–6798. doi: 10.1523/JNEUROSCI.4753-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beaulieu JM, Marion S, Rodriguiz RM, Medvedev IO, Sotnikova TD, Ghisi V, Wetsel WC, Lefkowitz RJ, Gainetdinov RR, Caron MG. A beta-arrestin 2 signaling complex mediates lithium action on behavior. Cell. 2008;132:125–136. doi: 10.1016/j.cell.2007.11.041. [DOI] [PubMed] [Google Scholar]
- 35.Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 36.Rachal Pugh C, Fleshner M, Watkins LR, Maier SF, Rudy JW. The immune system and memory consolidation: a role for the cytokine IL-1beta. Neurosci Biobehav Rev. 2001;25:29–41. doi: 10.1016/s0149-7634(00)00048-8. [DOI] [PubMed] [Google Scholar]
- 37.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Machado A, Herrera AJ, de Pablos RM, Espinosa-Oliva AM, Sarmiento M, Ayala A, Venero JL, Santiago M, Villaran RF, Delgado-Cortes MJ, Arguelles S, Cano J. Chronic stress as a risk factor for Alzheimer’s disease. Rev Neurosci. 2014;25:785–804. doi: 10.1515/revneuro-2014-0035. [DOI] [PubMed] [Google Scholar]
- 39.Wilson RS, Evans DA, Bienias JL, Mendes de Leon CF, Schneider JA, Bennett DA. Proneness to psychological distress is associated with risk of Alzheimer’s disease. Neurology. 2003;61:1479–1485. doi: 10.1212/01.wnl.0000096167.56734.59. [DOI] [PubMed] [Google Scholar]
- 40.Johansson L, Guo X, Waern M, Ostling S, Gustafson D, Bengtsson C, Skoog I. Midlife psychological stress and risk of dementia: a 35-year longitudinal population study. Brain. 2010;133:2217–2224. doi: 10.1093/brain/awq116. [DOI] [PubMed] [Google Scholar]
- 41.Tsolaki M, Papaliagkas V, Kounti F, Messini C, Boziki M, Anogianakis G, Vlaikidis N. Severely stressful events and dementia: a study of an elderly Greek demented population. Psychiatry Res. 2010;176:51–54. doi: 10.1016/j.psychres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 42.Ricci S, Fuso A, Ippoliti F, Businaro R. Stress-induced cytokines and neuronal dysfunction in Alzheimer’s disease. J Alzheimers Dis. 2012;28:11–24. doi: 10.3233/JAD-2011-110821. [DOI] [PubMed] [Google Scholar]
- 43.Han SH, Park JC, Mook-Jung I. Amyloid beta-interacting partners in Alzheimer’s disease: From accomplices to possible therapeutic targets. Prog Neurobiol. 2016;137:17–38. doi: 10.1016/j.pneurobio.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 44.Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA, O’Banion MK. Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci. 2013;33:5053–5064. doi: 10.1523/JNEUROSCI.4361-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bradley CA, Peineau S, Taghibiglou C, Nicolas CS, Whitcomb DJ, Bortolotto ZA, Kaang BK, Cho K, Wang YT, Collingridge GL. A pivotal role of GSK-3 in synaptic plasticity. Front Mol Neurosci. 2012;5:13. doi: 10.3389/fnmol.2012.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sy M, Kitazawa M, Medeiros R, Whitman L, Cheng D, Lane TE, Laferla FM. Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am J Pathol. 2011;178:2811–2822. doi: 10.1016/j.ajpath.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Liu L, Liu YZ, Shen XL, Wu TY, Zhang T, Wang W, Wang YX, Jiang CL. NLRP3 Inflammasome Mediates Chronic Mild Stress-Induced Depression in Mice via Neuroinflammation. Int J Neuropsychopharmacol. 2015;18 doi: 10.1093/ijnp/pyv006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alcocer-Gomez E, Ulecia-Moron C, Marin-Aguilar F, Rybkina T, Casas-Barquero N, Ruiz-Cabello J, Ryffel B, Apetoh L, Ghiringhelli F, Bullon P, Sanchez-Alcazar JA, Carrion AM, Cordero MD. Stress-Induced Depressive Behaviors Require a Functional NLRP3 Inflammasome. Mol Neurobiol. 2015 doi: 10.1007/s12035-015-9408-7. [DOI] [PubMed] [Google Scholar]
- 49.Iwata M, Ota KT, Li XY, Sakaue F, Li N, Dutheil S, Banasr M, Duric V, Yamanashi T, Kaneko K, Rasmussen K, Glasebrook A, Koester A, Song D, Jones KA, Zorn S, Smagin G, Duman RS. Psychological Stress Activates the Inflammasome via Release of Adenosine Triphosphate and Stimulation of the Purinergic Type 2X7 Receptor. Biol Psychiatry. 2016;80:12–22. doi: 10.1016/j.biopsych.2015.11.026. [DOI] [PubMed] [Google Scholar]
- 50.Weber MD, Frank MG, Tracey KJ, Watkins LR, Maier SF. Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male Sprague Dawley rats: a priming stimulus of microglia and the NLRP3 inflammasome. J Neurosci. 2015;35:316–324. doi: 10.1523/JNEUROSCI.3561-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pan Y, Chen XY, Zhang QY, Kong LD. Microglial NLRP3 inflammasome activation mediates IL-1beta-related inflammation in prefrontal cortex of depressive rats. Brain Behav Immun. 2014;41:90–100. doi: 10.1016/j.bbi.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 52.Wong ML, Inserra A, Lewis MD, Mastronardi CA, Leong L, Choo J, Kentish S, Xie P, Morrison M, Wesselingh SL, Rogers GB, Licinio J. Inflammasome signaling affects anxiety- and depressive-like behavior and gut microbiome composition. Mol Psychiatry. 2016;21:797–805. doi: 10.1038/mp.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao J, Wang H, Huang Y, Zhang H, Wang S, Gaskin F, Yang N, Fu SM. Lupus nephritis: glycogen synthase kinase 3beta promotion of renal damage through activation of the NLRP3 inflammasome in lupus-prone mice. Arthritis Rheumatol. 2015;67:1036–1044. doi: 10.1002/art.38993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu W, Wang H, Wang Y, Li H, Ji L. Metabolic factors-triggered inflammatory response drives antidepressant effects of exercise in CUMS rats. Psychiatry Res. 2015;228:257–264. doi: 10.1016/j.psychres.2015.05.102. [DOI] [PubMed] [Google Scholar]
- 55.Fichtner-Feigl S, Kesselring R, Martin M, Obermeier F, Ruemmele P, Kitani A, Brunner SM, Haimerl M, Geissler EK, Strober W, Schlitt HJ. IL-13 orchestrates resolution of chronic intestinal inflammation via phosphorylation of glycogen synthase kinase-3beta. J Immunol. 2014;192:3969–3980. doi: 10.4049/jimmunol.1301072. [DOI] [PubMed] [Google Scholar]
- 56.Shen F, Li N, Gade P, Kalvakolanu DV, Weibley T, Doble B, Woodgett JR, Wood TD, Gaffen SL. IL-17 receptor signaling inhibits C/EBPbeta by sequential phosphorylation of the regulatory 2 domain. Sci Signal. 2009;2:ra8. doi: 10.1126/scisignal.2000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ge D, Dauchy RT, Liu S, Zhang Q, Mao L, Dauchy EM, Blask DE, Hill SM, Rowan BG, Brainard GC, Hanifin JP, Cecil KS, Xiong Z, Myers L, You Z. Insulin and IGF1 enhance IL-17-induced chemokine expression through a GSK3B-dependent mechanism: a new target for melatonin’s anti-inflammatory action. J Pineal Res. 2013;55:377–387. doi: 10.1111/jpi.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maekawa T, Hosur K, Abe T, Kantarci A, Ziogas A, Wang B, Van Dyke TE, Chavakis T, Hajishengallis G. Antagonistic effects of IL-17 and D-resolvins on endothelial Del-1 expression through a GSK-3beta-C/EBPbeta pathway. Nat Commun. 2015;6:8272. doi: 10.1038/ncomms9272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu S, Zhang Q, Chen C, Ge D, Qu Y, Chen R, Fan YM, Li N, Tang WW, Zhang W, Zhang K, Wang AR, Rowan BG, Hill SM, Sartor O, Abdel-Mageed AB, Myers L, Lin Q, You Z. Hyperinsulinemia enhances interleukin-17-induced inflammation to promote prostate cancer development in obese mice through inhibiting glycogen synthase kinase 3-mediated phosphorylation and degradation of interleukin-17 receptor. Oncotarget. 2016;7:13651–13666. doi: 10.18632/oncotarget.7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taylor A, Harker JA, Chanthong K, Stevenson PG, Zuniga EI, Rudd CE. Glycogen Synthase Kinase 3 Inactivation Drives T-bet-Mediated Downregulation of Co-receptor PD-1 to Enhance CD8(+) Cytolytic T Cell Responses. Immunity. 2016;44:274–286. doi: 10.1016/j.immuni.2016.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mayer EA, Knight R, Mazmanian SK, Cryan JF, Tillisch K. Gut microbes and the brain: paradigm shift in neuroscience. J Neurosci. 2014;34:15490–15496. doi: 10.1523/JNEUROSCI.3299-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Macpherson AJ, Harris NL. Interactions between commensal intestinal bacteria and the immune system. Nat Rev Immunol. 2004;4:478–485. doi: 10.1038/nri1373. [DOI] [PubMed] [Google Scholar]
- 63.Reber SO, Siebler PH, Donner NC, Morton JT, Smith DG, Kopelman JM, Lowe KR, Wheeler KJ, Fox JH, Hassell JE, Jr, Greenwood BN, Jansch C, Lechner A, Schmidt D, Uschold-Schmidt N, Fuchsl AM, Langgartner D, Walker FR, Hale MW, Lopez Perez G, Van Treuren W, Gonzalez A, Halweg-Edwards AL, Fleshner M, Raison CL, Rook GA, Peddada SD, Knight R, Lowry CA. Immunization with a heat-killed preparation of the environmental bacterium Mycobacterium vaccae promotes stress resilience in mice. Proc Natl Acad Sci U S A. 2016;113:E3130–3139. doi: 10.1073/pnas.1600324113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rogers GB, Keating DJ, Young RL, Wong ML, Licinio J, Wesselingh S. From gut dysbiosis to altered brain function and mental illness: mechanisms and pathways. Mol Psychiatry. 2016;21:738–748. doi: 10.1038/mp.2016.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang H, Kumar A, Lamont RJ, Scott DA. GSK3beta and the control of infectious bacterial diseases. Trends Microbiol. 2014;22:208–217. doi: 10.1016/j.tim.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chiba T, Yamada M, Aiso S. Targeting the JAK2/STAT3 axis in Alzheimer’s disease. Expert Opin Ther Targets. 2009;13:1155–1167. doi: 10.1517/14728220903213426. [DOI] [PubMed] [Google Scholar]
- 67.Balschun D, Wetzel W, Del Rey A, Pitossi F, Schneider H, Zuschratter W, Besedovsky HO. Interleukin-6: a cytokine to forget. FASEB J. 2004;18:1788–1790. doi: 10.1096/fj.04-1625fje. [DOI] [PubMed] [Google Scholar]
- 68.Nicolas CS, Peineau S, Amici M, Csaba Z, Fafouri A, Javalet C, Collett VJ, Hildebrandt L, Seaton G, Choi SL, Sim SE, Bradley C, Lee K, Zhuo M, Kaang BK, Gressens P, Dournaud P, Fitzjohn SM, Bortolotto ZA, Cho K, Collingridge GL. The Jak/STAT pathway is involved in synaptic plasticity. Neuron. 2012;73:374–390. doi: 10.1016/j.neuron.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Donzis EJ, Tronson NC. Modulation of learning and memory by cytokines: signaling mechanisms and long term consequences. Neurobiol Learn Mem. 2014;115:68–77. doi: 10.1016/j.nlm.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]