

Abstract
Glioblastoma (GBM) is the most frequent, aggressive and fatal tumor in the central nervous system, while PTEN signaling is frequently deregulated in human GBM. We previously reported the up-regulation of the carboxyl terminal of Hsp70-interacting protein (CHIP) in GBM, however, the causal link between its dysregulation and tumorigenesis has not been established. Using miRNA microarrays and quantitative RT-PCR (qRT-PCR), we found activation of CHIP leads to increased transcription of miR-92b. Further studies in T98G and LN229 cells showed overexpression of miR-92b elicited reduction of PTEN and efficiently rescued glioma development in CHIP knock-down cells. The core pathway, PI3K/Akt pathway, was then upregulated, which promoted GBM cell proliferation. Meanwhile, genetic ablation of miR-92b could restore PTEN expression and inhibit glioma growth. These data demonstrate that the CHIP/miR-92b/PTEN axis serves as a new mechanism underlying GBM tumorigenesis, providing potential new therapeutic targets.
Keywords: Glioblastoma, CHIP, microRNA-92b, PTEN, tumorigenesis
Introduction
Glioblastoma (GBM) accounts for approximately 46 percentage of all primary malignant tumors in the brain [1]. Characterized by the most aggressive phenotype, GBM almost presents recurrence, though undergone invasive surgery and adjuvant chemotherapy combined with radiotherapy [2]. Ultimately, the disease follows a fatal course with the median survival of 12 to 15 months [3]. Despite new biological insights and advances in therapy, the prognosis for GBM has not significantly improved [4]. Therefore, identifying novel molecular mechanisms, which may be candidates to become therapeutic targets, underlying gliomagenesis is urgently needed for developing more effective treatment strategies.
Carboxyl-terminal of Hsp70-interacting protein (CHIP), an E3 ubiquitin ligase, functions to ubiquitinate and degrade proteins presented by chaperones, which plays a major role in maintaining the protein homeostasis in the cytoplasm [5]. Recent studies have shown that CHIP acts as a tumor suppressor through regulating oncogenic proteins such as epidermal growth factor receptor (EGFR) in pancreatic cancer [6], steroid receptor coactivator 3 (SRC3) in breast cancer [7], and androgen receptor in prostate cancer [8]. Moreover, CHIP levels are predominantly downregulated in late stages of various types of cancer, including colorectal cancer and breast cancer [7,9]. Contrarily, we previously reported that CHIP contributed to oncogenesis of glioma and its increased expression was related to high histological grade of glioma [10]. However, the exact mechanisms of CHIP in GBM have not been elucidated to date.
microRNAs (miRNAs), single-stranded non-coding small RNAs of approximately 22 nucleotides in length, regulate gene expression via directly targeting the 3’-untranslated region at the posttranscriptional level, which can play important roles in regulating cell growth, differentiation, proliferation and apoptosis [11,12]. Dysregulation of miRNAs has been found to be a common epigenetic event in the development of various cancers including GBM [13-18].
To search for the downstream of CHIP in the tumorigenic processes of GBM, we determined to identify miRNAs that possibly are regulated by CHIP. In the present study, we found that miRNA-92b expression significantly decreased when CHIP was inhibited and up-regulated miRNA-92b enhanced proliferation of GBM cells. Coincidently, several studies have found over-expression of miRNA-92b in malignant glioma, and it could promote glioma proliferation and invasion [19-21]. However, its role of miR-92b in the initiation and progression of GBM is also not fully explored.
In our bioinformatics analysis, PTEN was found to contain the miR-92b binding site. As a tumor suppressor gene, PTEN primarily functions by negatively regulating PI3K/Akt signaling [22]. Loss of PTEN is a very frequent event in GBM, which finally conduces to the activation of the PI3K/Akt pathway that promotes tumor cell proliferation [23]. Our study proved that PTEN is a direct target of miRNA-92b. Overexpression of miR-92b markedly decreased PTEN and stimulated GBM cell proliferation. What’s more, genetic ablation of miRNA-92b restored PTEN expression and effectively contributed to cell-growth inhibition. Thus, we provided new insight into the interaction between miRNAs and PTEN, and these findings may lay a foundation for novel molecular targeted therapeutic approaches in GBM.
Materials and methods
Tissue samples
All samples for RNA detection were obtained from Changzheng Hospital in Shanghai, China. The study protocol and acquisition of tissue specimens were approved by the Specialty Committee on Ethics of Biomedical Research, Second Military Medical University. Tumor samples and adjacent non-tumor tissues were collected between September 2012 and December 2014 from patients with pathologically confirmed GBM who underwent surgical treatment at Changzheng Hospital. Either the patients or their legal guardians signed the written informed consent following National Regulations on the Use of Clinical Samples in China.
Cell transfection
miR-92b mimic (sense, 5’-UAUUGCACUCGUCCCGGCCUCC-3’; antisense, 5’-GGA GGCCGGGACGAGUGCAAUA-3’), miR-92b control (miR-con, sense, 5’-UUG UACUACACAAAAGUACUG-3’; antisense, 5’-CAGUACUUUUGUGUAGUACAA-3’), miR-92b inhibitor (anti-miR-92b, 2’-O-Me-GGAGGCCGGGACGAGUGCAAUA), miR-92b inhibitor control (anti-miR-con, 2’-O-Me-UUG UACUACACAAAAGUACUG), PTEN short interfering RNA (siRNA) (sense, 5’-AACCCACCACAGCUAGAACtt-3’; antisense, 5’-AAGUUCUAGCUGUGGUGGGtt-3’), PTEN control siRNA (scramble siRNA, sense, 5’-UUCUCCGAACGUGUCACGUtt-3’; antisense, 5’-ACGUGACACGUUCGGAGAAtt-3’), Akt siRNA (sense, 5’-GACGGGCACAUUAAGAUCAtt-3’; antisense, 5’-UGAUCUUAAUGUGCCCGUCtt-3’), and Akt control siRNA (Akt siR-con, sense, 5’-UUCUCCGAACGUGUCACGUtt-3’; antisense, 5’-ACGUGACACGUUCGGAGAAtt-3’) were purchased from GenePharma Corporation (Shanghai, China). CHIP shRNA plasmid and its normal control (NC) have been described previously [10]. The construct that was used for PTEN expression was pcDNA3.1(+)-PTEN. The coding sequence of the PTEN gene was amplified from the cDNA of T98G cells (forward primer, 5’-CCGGAATTCATGACAGCCATCATCAAAGAGATCG-3’; reverse primer, 5’-CCGCTCGAGTCAGACTTTTGTAATTTGTGTATGC-3’) and inserted into the pcDNA3.1(+) vector between the EcoR I and Xho I restriction sites (Invitrogen, USA).
miRNAs and siRNAs, as well as PTEN expression vector, were transfected into cultured glioma cells including U251, U87, LN229 and T98G (Type Culture Collection of Chinese Academy of Science, Shanghai, China) with the use of Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Lentivirus carrying the shRNA against CHIP was then used to infect glioma cells in the presence of 4 μg/ml polybrene (Sigma-Aldrich, USA) as previously described [24].
Quantitative RT-PCR
Glioma cells were harvested for total RNA extracting using the Trizol reagent (Invitrogen) according to the manufacturer’s instructions. Subsequently, cDNA was prepared with M-MLV kit (Promega, USA). Quantitative RT-PCR was performed in triplicates using SYBR Green Master Mix (Takara, Japan) on 7500 Fast Real-Time PCR system (Applied Biosystems, Life Technology, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and small nuclear RNA U6 was used as internal controls of mRNA and miRNA, respectively. The sequences of reverse transcription primer were miR-92b: 5’-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACGGAGGCC-3’ and U6: 5’-AACGCTTCACGAATTTGCGT-3’. The primers used are as follows: CHIP forward primer, 5’-AGCAGGGCAATCGTCTGTTC-3’ and reverse primer, 5’-CAAGGCCCGGTTGGTGTAATA-3’; miR-92b forward primer, 5’-TATTGCACTCGTCCCGGCCTCC-3’ and reverse primer, 5’-GTGCAGGGTCCGAGGT-3’; GAPDH forward primer, 5’-AACGGATTTGGTCGTATTG-3’ and reverse primer, 5’-GGAAGATGGTGATGGGATT-3’; U6 forward primer, 5’-CTCGCTYCGGCAGCAC-3’ and reverse primer, 5’-AACGCTTCACGAATTTGCGT-3’.
miRNA microarray
T98G cells were treated with shCHIP (CHIP knockdown) or control shRNA and cultured for 48 h. Then, total miRNA was extracted using the mirVana miRNA Isolation Kit (Ambion, USA). miRNA microarray analysis was performed at KangChen Bio-tech Corporation (Shanghai, China) by an Agilent human miRNA microarray (Version 19.0). Data acquisition and analysis followed standard Agilent protocols. miRNAs with fold change >2 were considered to be differentially expressed between CHIP knockdown and control.
Western blot
Whole cell lysates were extracted from glioma cells with radio-immunoprecipitation assay buffer. The lysates were resolved by SDS-PAGE and blotted with CHIP (C3B6) Rabbit mAb (Cell Signaling Technology, USA), PTEN (138G6) Rabbit mAb (Cell Signaling Technology), Phospho-Akt (Ser473) Antibody (Cell Signaling Technology), Akt Antibody (Cell Signaling Technology), Phospho-mTOR (Ser2448) Antibody (Cell Signaling Technology), Phospho-p70 S6 Kinase (Thr389) Antibody (Cell Signaling Technology), Phospho-4E-BP1 (Ser65) Antibody (Cell Signaling Technology), and Survivin (71G4B7) Rabbit mAb (Cell Signaling Technology). GAPDH (14C10) Rabbit mAb (Cell Signaling Technology) was used as an internal control. The blots were visualized by an enhanced chemiluminescence detection system (Thermo Scientific, USA).
MicroRNA target prediction
The target genes of miR-92b were predicted in the following databases through computer-aided algorithms: miRTarBase (http://mirtarbase.mbc.nctu.edu.tw), miRDB (http://mirdb.org) and microRNA (http://www.microrna.org).
TCGA data processing
TCGA (www. cancergenome.nih.gov) provides multimodal data of more than 500 GBM cases. Level 3 microRNA expression data based on the Agilent microarrays (UNC H-miRNA 8 × 15 K) and clinical follow-up information were collected for further analysis. The expression of miR-92b expression was classified as either High (expression value ≥7.70) or Low (expression value <7.70). OS and progression-free survival (PFS) were calculated in days from the date of diagnosis to the time of death and to the time of tumor progression or recurrence, or death of the patient from GBM, respectively.
Luciferase reporter assay
The wild-type and mutated putative miR-92b target on PTEN 3’UTR were cloned into the pMIR-Report-Luc vector (Ambion) and transfected into T98G and LN229 cells. After 24 hours of culture, the cells were further transfected with miR-92b mimic or negative control and incubated for another 48 hours. The cells were then lysed and luciferase activity was detected using the dual Luciferase Report Assay System (Promega).
Cell proliferation assay
Cells in the logarithmic phase of growth were seeded and cultured in 96-well plates at 3 × 103 cells per well in 100 μl media and were allowed to grow for 24, 48, 72, and 96 hours. Cell proliferation assay was analyzed by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide). A diluted MTT (5 mg/mL, 20 μl) (Sigma-Aldrich) was added to each well, and the cells were incubated at 37°C. After 4 hours, the cells were resuspended in 150 μl of Dimethyl sulfoxide (DMSO) and shaken for several minutes. The optical density was measured at 570 nm wavelength using a microplate reader (EL × 800, BIO-TEK, USA).
Colony formation assay
Colony formation was assessed by plating 1000 cells onto 6-well tissue culture plates. After 10-14 days, visible colonies were fixed with 100% methanol and stained with Giemsa’s stain (Sigma-Aldrich). Colony-forming ability was evaluated by counting the number of colonies (defined as cell cluster consisting of at least 50 cells).
Cell cycle analysis
Cells in the logarithmic phase of growth were harvested, washed with phosphate-buffered saline (PBS), and fixed with 70% ice-cold ethanol. After extensive washing, cells were re-suspended in PBS containing 25 mg/mL propidium iodide (Sigma-Aldrich), 10 mg/mL RANse (Sigma-Aldrich) and 0.1% Triton (Sigma-Aldrich), followed by incubation for 30 minutes in the dark. The fractions of viable cells in G0/G1, S and G2 phases of the cell cycle were analyzed by flow cytometry using a FACScan instrument (Becton Dickinson, USA).
Cell apoptosis analysis
Cells were plated into 6-well plates at 6 × 104 cells per well. Forty-eight hours after transfection, the cells were harvested and washed with PBS. Cellular apoptosis was evaluated by flow cytometry using annexin V-FITC apoptosis detection kit (BD Biosciences, USA) as described by the manufacturer’s instructions.
Subcutaneous xenograft models
Immuno-deficient male/female nude mice (4-5 weeks old) (BALB/c nu/nu; Slac Laboratory Animal) were used for subcutaneous tumor growth experiments. For each group four mice were used. All mice were bred in a suitable environment according to standard guidelines under a protocol approved by Second Military Medical University.
Briefly, shCHIP-infected and normal LN229 cells (5 × 106 in 200 μl PBS) were injected subcutaneously into the right flank of 2 groups of 8 mice. After the xenografts had become visible, the size of tumors was measured every week with digital caliper. And the simplified formula: volume = 1/2 × length × width2 was used to calculated approximate tumor volumes. After 35 days, the mice were sacrificed by decapitation under adequate anesthesia, and the tumors were partially fixed for immunohistochemical analysis.
Immunohistochemistry
The xenograft tumors were fixed in formalin, and then embedded in paraffin wax. Tissue sections (5 μm) on poly-L-lysine-coated slides were deparaffinized in xylol and rehydrated using graded ethanol. Peroxidase was quenched with methanol and 3% H2O2. Slides were then placed in 10 mM citric acid (pH 6.0) for 12 minutes at 100°C. After incubation with a 1:500 dilution of Ki67 antibody (Abcam, USA) overnight at 4°C, sections were subject to secondary antibody incubation with linked reagent at room temperature for 1 hour. For the negative control, the primary antibody was replaced with PBS. The antigen-antibody complex was detected by using diaminobenzidine (DAB) substrate. The sections were then counterstained with hematoxylin and eosin (H&E). The evaluation of Ki67 expression was based on both staining intensity and percentage of positive cells in the total number of tumor cells.
Statistical analysis
All statistical tests were conducted using the SPSS software (Version 17.0, Chicago, IL, USA). The significance of the differences between groups was evaluated by the Student t-test and two-way ANOVA. Data were presented as the mean ± SD unless otherwise indicated. The expression level of miR-92b related to cumulative survival of GBM patients was described by the Kaplan-Meier method and analyzed by log-rank test. A level of P<0.05 was considered to be statistically significant.
Results
miR-92b expression is decreased in shCHIP glioma cell lines
Our previous study has demonstrated that CHIP could contribute to human glioma tumorigenesis, overexpression of CHIP significantly increased proliferation of glioma cells [10]. To explore the role of miRNAs in glioma formation caused by variant expression of CHIP, we first constructed Agilent microarrays and compared miRNA expression profiles between shCHIP and controlled T98G cells (Figure 1A). Among all deregulated miRNAs, miR-92b showed a dramatic decrease for 26 folds after CHIP inhibition, which was further confirmed by real-time quantitative PCR (RT-PCR) in four glioma cell lines (U251, U87, LN229 and T98G) (Figure 1B). As LN229 and T98G cells showed the highest rate of inhibition of miR-92b expression induced by shCHIP treatment, and ideal restoration of the expression of miR-92b when treated with shCHIP and miR-92b mimic (Figure 1C), the two cell lines were then used for future experiments.
Figure 1.

miR-92b is regulated by CHIP in GBM cells. A. Clustering map of differentially expressed miRNAs between normal T98G cells (n = 2) and shRNA treated T98G cells (n = 2). Upregulated miRNAs are shown in red and downregulated miRNAs in green. B. Comparison of relative expression of miR-92b in four glioma cell lines treated with shCHIP. C. Detection of relative expression of miR-92b in T98G and LN229 with different treatments, respectively. RT-PCR shows high rate of inhibition of miR-92b expression, and miR-92b mimic can ideally restore downregulation of miR-92b induced by shCHIP. D. Western blot analysis of CHIP in T98G and LN229 cells transfected with NC or shRNA or shRNA and miR-92b mimic after 48 h; GAPDH was used as an internal control.
Further, overexpression of miR-92b was performed in shCHIP glioma cell lines (LN229 and T98G), and the expression pattern of CHIP was detected by western blot. However, no changes were found in CHIP protein level after increasing miR-92b (Figure 1D). These results indicated that miR-92b might be involved in the downstream and regulated by CHIP.
miR-92b is highly expressed in glioma samples and associated with patient survival
The expression of miR-92b in patient samples was studied by RT-PCR in paired human GBM samples and adjacent non-tumor tissues from 6 patients. As a result, a six-fold higher expression of miR-92b was found in GBM samples compared with adjacent non-tumor tissues (P<0.01) (Figure 2A). We further studied the relationship between miR-92b expression and clinical characteristics. By searching TCGA database, we have collected 519 GBM cases with available microRNA expression data plus matching clinical follow-up information. Kaplan-Meier analysis (log-rank) found that high expression of miR-92b was significantly related to short OS of GBM patients (median OS, 357 vs. 393 days, P = 0.029) (Figure 2B). Though no statistically significant result was got for PFS, there was still a moderate difference between high and low miR-92b subgroups (median PFS: 195 days vs. 214 days, P = 0.100) (Figure 2C). Together, these showed the potential impact of miR-92b on malignant behaviors of human glioma.
Figure 2.

miR-92b expression is increased in GBM tissues and correlated to patient survival. A. Compared with the normal brain tissues, GBM samples exhibits high expression of miR-92b (P<0.01). B, C. Based on TCGA database, Kaplan-Meier survival analysis in 519 GBM cases stratified by miR-92b expression shows high level of miR-92b predicts a significantly short OS (357 vs. 393 days, P = 0.029) and a short PFS without statistical significance (195 days vs. 214 days, P = 0.100).
miR-92b enhances proliferation and colony formation and inhibits cell cycle arrest and apoptosis in shCHIP glioma cells
To assess the role of miR-92b in the growth of human glioma cells after CHIP inhibition, we performed MTT assays to examine the cell proliferation of LN229 and T98G. As shown in Figure 3A and 3B, shCHIP treatment significantly suppressed both LN229 and T98G cells growth by 30-50% compared to negative controls. However, when overexpressing miR-92b, shCHIP treatment glioma cells displayed an evident increase in cell numbers. Consistently, a soft agar colony formation assay revealed that down-regulation of miR-92b by shCHIP resulted in a ~80% reduction in colony numbers, while stable overexpression of miR-92b restored the colony formation ability in both LN229 and T98G cell lines (Figure 3C). Flow cytometric analysis showed that miR-92b could inhibit both G1 arrest (Figure 3D) and cell apoptosis induced by shCHIP treatment (Figure 3E).
Figure 3.

miR-92b is involved in the downstream of CHIP, contributes to GBM cell growth. A, B. The effects of miR-92b on glioma cells are measured via a MTT assay. shCHIP significantly inhibits T98G and LN229 cell proliferation, which can be reversed by miR-92b. The results are presented as the mean ± SD of the values obtained in three independent experiments (*P<0.05, shCHIP vs. NC; shCHIP vs. shCHIP+miR-92b). C. The proliferation ability of miR-92b overexpression in shCHIP glioma cells is examined by clonogenic cell survival assay. D. Overexpression of miR-92b can inhibit G1 arrest of glioma cells induced by shCHIP. E. The apoptosis of T98G and LN229 cells induced by shCHIP is significantly inhibited by the miR-92b mimic.
miR-92b targets PTEN in glioma and re-introduction of PTEN abrogates the miR-92b-induced effects on cell proliferation
The previous results showed that CHIP regulated miR-92b expression during glioma tumorigenesis. To dissect the potential mechanism and identify the specific gene targets of miR-92b, we searched public miRNA target prediction databases (miRTarBase, miRDB, and microRNA). Bioinformatics analysis revealed that the 3’-UTRs of PTEN contain evolutionarily conserved miR-92b-binding sites (Figure 4A). As a result, the miR-92b mimic significantly inhibited the luciferase reporter activities of the wild-type 3’-UTR constructs of PTEN in both LN229 and T98G cells, but not the mutant 3’-UTR constructs containing mutations in the complementary seed site (Figure 4A and 4B). Also, overexpression of miR-92b markedly decreased the expression level of PTEN, while anti-miR-92b clearly increased the concentration of PTEN proteins (Figure 4C). Furthermore, knockdown of PTEN using siRNA greatly induced T98G cell growth. Anti-miR-92b rescued the PTEN-overexpressing phenotype of cell growth inhibition (Figure 3D), supporting the dominant role of PTEN in miR-92b-mediated proliferation enhancement in glioma cells. Taken together, these data demonstrated that miR-92b promoted glioma cell proliferation by inhibiting its target PTEN.
Figure 4.

miR-92b targets PTEN. A. The predicted miR-92b binding sequence in the 3’UTR of PTEN mRNA is shown. Mutations are generated in the PTEN 3’UTR sequence at the complementary sites for the seed regions in miR-92b. B. Luciferase activity assays are performed in T98G and LN229 cells transfected with pGL3-wild-PTEN 3’UTR or pGL3-mutant-PTEN 3’UTR tighter with miR-92b or miRNA control or anti-miR-92b or anti-miRNA control (*P<0.05; **P<0.01; ***P<0.001). C. Western bolt analysis of PTEN expression in T98G and LN229 cells transfected with miR-92b or miRNA control or anti-miR-92b or anti-miRNA control after 48 h; GAPDH was used as an internal control. D. MTT assay is used to measure proliferation ability of T98G cells treated with miR-92b or PTEN siRNA or miR-92b and PTEN or their respective control. PTEN inhibition significantly decreases the rate of T98G cell growth, and miR-92b promotes glioma cell growth, while PTEN overexpression abrogates miR-92b-induced glioma cell rapid growth (*P<0.05, PTEN siRNA vs. scramble siRNA; miR-92b vs. miR-con; miR-92b+PTEN vs. miR-92b+vector).
CHIP/miR-92b/PTEN axis promotes proliferation and inhibits apoptosis of glioma cell through regulating the activity of PI3K/AKT pathway
It has been established that PTEN is the core molecular which negatively modulates the activity of PI3K/AKT pathway, one of the most important pathways in the oncogenesis of GBM. We wonder the changes of the current studied CHIP/miR-92b/PTEN axis might influent the activity of PI3K/AKT pathway. As expected, Western blot revealed a consistent activation of the PI3K/AKT pathway after PTEN deficiency caused by miR-92b overexpression or direct PTEN RNAi (Figure 5A). Then some key factors involved in the PI3K/AKT pathway including p-AKT, p-mTOR, p-p70S6K, p-4E-BP1 and survivin were chosen as downstream candidates. Since AKT acts as a main downstream effector of PTEN in this pathway, we would like to detect further whether the phenomena was directly caused by the activation of AKT by PTEN deficiency. We changed the expression pattern of AKT in LN229 cells using siRNA (Figure 5B), and then MTT and flow cytometric analysis were applied to detect the proliferation and apoptosis of this cell line (Figure 5C and 5D). After AKT inhibition, the CHIP/miR-92b/PTEN axis could no longer maintain the proliferative and anti-apoptotic abilities, which supported that AKT was the direct effector of this axis and contributed to the oncogenesis of human glioma.
Figure 5.

PI3K/AKT pathway activity is associated with CHIP/miR-92b/PTEN axis. A. Western blot analysis is performed to detect activity of several proteins involved in PI3K/AKT pathway when CHIP or PTEN inhibition or miR-92b overexpression; GAPDH is used as an internal control. B. AKT expression and its activity in LN229 cells are analyzed by Western blot when AKT and miR-92b expression are changed together or separately. C, D. In different conditions of AKT and miR-92b expression pattern, proliferation ability and apoptosis of LN229 cells are measured by MTT assay and flow cytometry. AKT inhibition offsets the effect on glioma cell growth induced by miR-92b overexpression, and re-introduction of miR-92b can abrogate the AKT inhibition induced effect on glioma cell apoptosis (*P<0.05, miR-92b+Akt siRNA vs. miR-92b+siR-con; miR-92b+Akt siRNA vs. miR-con+Akt siRNA).
CHIP/miR-92b/PTEN axis enhances tumor growth in vivo
Given the fact that CHIP/miR-92b/PTEN axis could improve proliferation and colony formation, we examined the effect of this axis on glioma cell growth in vivo. LN229 cells with or without shCHIP were injected subcutaneously into nude mice, and xenograft tumors were taken out and measured 35 days later. The expressions of CHIP, miR-92b, PTEN in tumors were detected using RT-PCR or western blot. As shown in Figure 6A, shCHIP significantly slowed tumor growth speed in mice. Meanwhile, shCHIP treatment also led to downregulation of miR-92b and upregulation of PTEN (Figure 6B and 6C). Compared to the control, immunohistochemistry revealed a lower Ki67 index in shCHIP tumors (Figure 6D). These results indicated that CHIP/miR-92b/PTEN could efficiency inhibit tumor growth in a nude mouse model.
Figure 6.
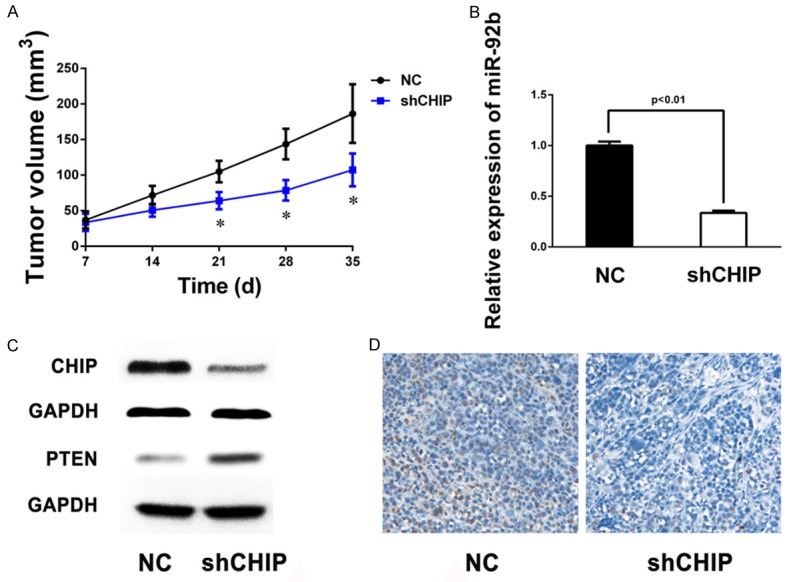
miR-92b and PTEN are involved in CHIP-mediated glioma growth in vivo. A. The in vivo glioma cell growth of LN229 was determined by xenograft model assay, shCHIP and control cells are transplanted into nude mice. Tumor growth curves represented by tumor weight are shown. Each experiment was performed in triplicate, and the results are presented as the mean ± SD of the values (*P<0.05, shCHIP vs. NC). B. RT-PCR shows miR-92b expression in two different groups of xenograft glioma samples. C. Western blot analysis reveals low expression of CHIP and high PTEN expression in shCHIP tumors; GAPDH is used as an internal control. D. Representative immunohistochemical staining of Ki-67-stained cells from the indicated tumors. Lower level of Ki-67 is observed in tumors from the shCHIP group compared with those from control group.
Discussion
Extensive studies have revealed that CHIP may play multi-faceted roles in cancer depending upon its types and status of progression [25]. In our previous study, we found that CHIP was accumulated in malignant glioma and contributed to the oncogenesis [10], but the detailed mechanism was not very clear. Here, we showed that CHIP could positively regulate miR-92b expression, which was a direct negative regulator of PTEN, the most important tumor suppressor in the process of glioblastoma onset, and thereby activate the PI3K/Akt pathway, thus promote cancer cell growth [26]. The consistent results could be found both in vitro and in vivo. To our knowledge, this CHIP/miR-92b/PTEN axis involved in gliomagenesis has never been reported before.
miR-92b was frequently reported as significantly upregulated in GBM compared with normal brain tissues, and overexpression of miR-92b increased cell proliferation, which was consistent with our findings [19-21,27]. Moreover, miR-92b expression was positively correlated with the degree of glioma infiltration and negatively associated with overall survival in high-grade glioma [20]. Several miRNA database algorithms predicted that miR-92b had multiple potential targets among which were many tumor suppressors such as NLK, smad3, DDK3 and PTEN (http://mirdb.org, http://mirtarbase.mbc.nctu.edu.tw, http://cm.jefferson.edu/rna22/). It was just as NLK, smad3 and DDK3 were confirmed as direct targets of miR-92b in glioma [19-21], the interaction between PTEN and miR-92b was also verified by a dual luciferase reporting assay in our study. With a critical role in the regulation of diverse cellular processes, such as proliferation, survival, growth, DNA repair, and genomic stability, PTEN demands tight regulation [28-31]. It has previously been shown that PTEN protein could transiently associate with the molecular chaperones and thereby got diverted to the degradation pathway through its interaction with CHIP, and overexpression of CHIP contributed to elevated ubiquitination and a shortened half-life of endogenous PTEN [32]. Our study elaborated an additional mode of molecular interaction between CHIP and PTEN. Thus it could be seen that PTEN was strictly regulated by CHIP both at the protein and mRNA levels.
However, we found that in our study overexpression of miR-92b markedly decreased PTEN, while miR-92b inhabitation could dramatically increase the concentration of PTEN protein, which implied that CHIP/miR-92b/PTEN axis might be the predominant pattern regulating PTEN triggered by CHIP in glioma. Recently, CHIP was identified to play a novel role in cardio-protection through ERK5 activation regulating CHIP-mediated inducible cAMP early repressor (ICER) ubiquitination and degradation [33]. As known, ERK5 associated with CHIP via CHIP-Fr2 and CHIP-Fr3 fragments, among which the latter one contains a U-box domain that is essential for ubiquitination activity [5]. They thought that ERK5 could bind to this region of CHIP and alter its protein conformation and consequently increased its ubiquitin ligase activity for ICER ubiquitination [33]. So it’s reasonable to speculate that in glioma one or more specific molecules binding to CHIP may affect CHIP enzyme activity through a conformational change, and then prevents CHIP-mediated ubiquitination and degradation of PTEN protein. Nevertheless, this protein-protein interaction does not have much effect on the expression of miR-92b. Further investigations are needed to demonstrate the exact mechanism.
Collectively, our study determined that CHIP acted as an oncogene and contributed to gliomagenesis, and identified the unusual CHIP/miR-92b/PTEN regulatory network, in which CHIP controlled glioma proliferation and growth through PTEN/PI3K/AKT signaling via up-regulation of miR-92b. We believe that therapeutics targeting the CHIP/miR-92b/PTEN axis will expand strategies for the treatment of glioma.
Acknowledgements
This work was supported by the National Natural Science Foundation (81101907, 81472354, 81272781 and 81572501), Program for Academic Leaders of Shanghai (No. 043), and “Pu Jiang Talent” Project of Shanghai (PJ[2014]0002617).
Disclosure of conflict of interest
None.
References
- 1.Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro Oncol. 2015;17(Suppl 4):iv1–iv62. doi: 10.1093/neuonc/nov189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310:1842–1850. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 3.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 4.Wang H, Xu T, Jiang Y, Xu H, Yan Y, Fu D, Chen J. The challenges and the promise of molecular targeted therapy in malignant gliomas. Neoplasia. 2015;17:239–255. doi: 10.1016/j.neo.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Hohfeld J, Patterson C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol. 2001;3:93–96. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- 6.Wang TX, Yang JX, Xu JW, Li J, Cao Z, Zhou L, You L, Shu H, Lu ZH, Li HH, Li M, Zhang TP, Zhao YP. CHIP is a novel tumor suppressor in pancreatic cancer and inhibits tumor growth through targeting EGFR. Oncotarget. 2014;5:1969–1986. doi: 10.18632/oncotarget.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kajiro M, Hirota R, Nakajima Y, Kawanowa K, So-Ma K, Ito I, Yamaguchi Y, Ohie S, Kobayashi Y, Seino Y, Kawano M, Kawabe Y, Takei H, Hayashi S, Kurosumi M, Murayama A, Kimura K, Yanagisawa J. The ubiquitin ligase CHIP acts as an upstream regulator of oncogenic pathways. Nat Cell Biol. 2009;11:312–9. doi: 10.1038/ncb1839. [DOI] [PubMed] [Google Scholar]
- 8.Sarkar S, Brautigan DL, Parsons SJ, Larner JM. Androgen receptor degradation by the E3 ligase CHIP modulates mitotic arrest in prostate cancer cells. Oncogene. 2014;33:26–33. doi: 10.1038/onc.2012.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Ren F, Wang Y, Feng Y, Wang D, Jia B, Qiu Y, Wang S, Yu J, Sung JJ, Xu J, Zeps N, Chang Z. CHIP/Stub1 functions as a tumor suppressor and represses NF-kappa B-mediated signaling in colorectal cancer. Carcinogenesis. 2014;35:983–991. doi: 10.1093/carcin/bgt393. [DOI] [PubMed] [Google Scholar]
- 10.Xu T, Zhou Q, Zhou J, Huang Y, Yan Y, Li W, Wang C, Hu G, Lu Y, Chen J. Carboxyl terminus of Hsp70-interacting protein (CHIP) contributes to human glioma oncogenesis. Cancer Sci. 2011;102:959–966. doi: 10.1111/j.1349-7006.2011.01888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 12.Esquela-Kerscher A, Slack FJ. Oncomirs-microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 13.Wang H, Xu T, Jiang Y, Yan Y, Qin R, Chen J. MicroRNAs in human glioblastoma: from bench to beside. Front Biosci (Landmark Ed) 2015;20:105–118. doi: 10.2741/4300. [DOI] [PubMed] [Google Scholar]
- 14.Cortinovis D, Monica V, Pietrantonio F, Ceresoli GL, La Spina CM, Wannesson L. MicroRNAs in non-small cell lung cancer: current status and future therapeutic promises. Curr Pharm Des. 2014;20:3982–3990. doi: 10.2174/13816128113196660755. [DOI] [PubMed] [Google Scholar]
- 15.Serpico D, Molino L, Di Cosimo S. microRNAs in breast cancer development and treatment. Cancer Treat Rev. 2014;40:595–604. doi: 10.1016/j.ctrv.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 16.Song S, Ajani JA. The role of microRNAs in cancers of the upper gastrointestinal tract. Nat Rev Gastroenterol Hepatol. 2013;10:109–118. doi: 10.1038/nrgastro.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xuan Y, Yang H, Zhao L, Lau W, Lau B, Ren N, Hu Y, Yi T, Zhao X, Zhou S, Wei Y. MicroRNAs in colorectal cancer: Small molecules with big functions. Cancer Lett. 2015;360:89–105. doi: 10.1016/j.canlet.2014.11.051. [DOI] [PubMed] [Google Scholar]
- 18.Li F, Mahato RI. MicroRNAs and drug resistance in prostate cancers. Mol Pharm. 2014;11:2539–2552. doi: 10.1021/mp500099g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang K, Wang X, Zou J, Zhang A, Wan Y, Pu P, Song Z, Qian C, Chen Y, Yang S, Wang Y. miR-92b controls glioma proliferation and invasion through regulating Wnt/beta-catenin signaling via Nemo-like kinase. Neuro Oncol. 2013;15:578–588. doi: 10.1093/neuonc/not004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Q, Shen K, Zhao Y, Ma C, Liu J, Ma J. MiR-92b inhibitor promoted glioma cell apoptosis via targeting DKK3 and blocking the Wnt/beta-catenin signaling pathway. J Transl Med. 2013;11:302. doi: 10.1186/1479-5876-11-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu ZB, Cai L, Lin SJ, Lu JL, Yao Y, Zhou LF. The miR-92b functions as a potential oncogene by targeting on Smad3 in glioblastomas. Brain Res. 2013;1529:16–25. doi: 10.1016/j.brainres.2013.07.031. [DOI] [PubMed] [Google Scholar]
- 22.Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JF. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets. 2008;8:187–198. doi: 10.2174/156800908784293659. [DOI] [PubMed] [Google Scholar]
- 23.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meng D, Chen Y, Yun D, Zhao Y, Wang J, Xu T, Li X, Wang Y, Yuan L, Sun R, Song X, Huai C, Hu L, Yang S, Min T, Chen J, Chen H, Lu D. High expression of N-myc (and STAT) interactor predicts poor prognosis and promotes tumor growth in human glioblastoma. Oncotarget. 2015;6:4901–4919. doi: 10.18632/oncotarget.3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun C, Li HL, Shi ML, Liu QH, Bai J, Zheng JN. Diverse roles of C-terminal Hsp70-interacting protein (CHIP) in tumorigenesis. J Cancer Res Clin Oncol. 2014;140:189–197. doi: 10.1007/s00432-013-1571-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwon CH, Zhao D, Chen J, Alcantara S, Li Y, Burns DK, Mason RP, Lee EY, Wu H, Parada LF. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res. 2008;68:3286–3294. doi: 10.1158/0008-5472.CAN-07-6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hua D, Mo F, Ding D, Li L, Han X, Zhao N, Foltz G, Lin B, Lan Q, Huang Q. A catalogue of glioblastoma and brain microRNAs Identified by deep sequencing. OMICS. 2012;16:690–699. doi: 10.1089/omi.2012.0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sulis ML, Parsons R. PTEN: from pathology to biology. Trends Cell Biol. 2003;13:478–483. doi: 10.1016/s0962-8924(03)00175-2. [DOI] [PubMed] [Google Scholar]
- 29.Maehama T, Taylor GS, Dixon JE. PTEN and myotubularin: novel phosphoinositide phosphatases. Annu Rev Biochem. 2001;70:247–279. doi: 10.1146/annurev.biochem.70.1.247. [DOI] [PubMed] [Google Scholar]
- 30.Planchon SM, Waite KA, Eng C. The nuclear affairs of PTEN. J Cell Sci. 2008;121:249–253. doi: 10.1242/jcs.022459. [DOI] [PubMed] [Google Scholar]
- 31.Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–170. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 32.Ahmed SF, Deb S, Paul I, Chatterjee A, Mandal T, Chatterjee U, Ghosh MK. The Chaperone-assisted E3 ligase C terminus of Hsc70-interacting protein (CHIP) targets PTEN for proteasomal degradation. J Biol Chem. 2012;287:15996–16006. doi: 10.1074/jbc.M111.321083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woo CH, Le NT, Shishido T, Chang E, Lee H, Heo KS, Mickelsen DM, Lu Y, McClain C, Spangenberg T, Yan C, Molina CA, Yang J, Patterson C, Abe J. Novel role of C terminus of Hsc70-interacting protein (CHIP) ubiquitin ligase on inhibiting cardiac apoptosis and dysfunction via regulating ERK5-mediated degradation of inducible cAMP early repressor. FASEB J. 2010;24:4917–4928. doi: 10.1096/fj.10-162636. [DOI] [PMC free article] [PubMed] [Google Scholar]