

Abstract
FL118 is a novel camptothecin (CPT) analogue that possesses exceptional antitumor efficacy in human tumor animal models. To date, two CPT analogues, irinotecan and topotecan, have been approved by the FDA for cancer treatment. FL118 exhibits superior antitumor activity over irinotecan and topotecan, and effectively overcomes the irinotecan- or topotecan-resistant human tumors in animal models. Accordingly, FL118 selectively inhibits the expression of multiple cancer-associated proteins (survivin, Mcl-1, XIAP, cIAP2, MdmX). However, FL118 has hematopoietic toxicity similar to irinotecan and topotecan, suggesting that FL118’s hematopoietic toxicity may share a mechanism similar to irinotecan and topotecan. It is known that CPTs including irinotecan, SN-38 (active metabolite of irinotecan) and topotecan are topoisomerase I (Top1) inhibitors. However, the evidence from our studies failed to reveal that FL118 is a better Top1 inhibitor than SN-38. It was documented that Top1 expression level is positively associated with CPTs’ sensitivity. Low Top1 expression links to CPTs’ resistance. In contrast to these findings, we found that human colorectal tumor sensitivity to FL118 is irrelevant to the expression level of Top1 protein. FL118 can show high antitumor efficacy in Top1-negative tumors, while Top1 highly positive tumors can exhibit FL118 resistance. This suggests that the presence of Top1 target is not critical for FL118 antitumor activity. In other words, targeting Top1 by FL118 may not play a major role for its antitumor efficacy. However, studies indicate that FL118 can bind to, and inhibit Top1 activity. This raises the possibility that inhibition of Top1 by FL118 may predominantly be involved in hematopoietic toxicity, but not in FL118 antitumor activity. In this article, we will summarize existing observations and provide our up-to-date research results to support our opinion on this important topic.
Keywords: FL118, topoisomerase I, survivin, Mcl-1, XIAP, cIAP2, MdmX, camptothecin, irinotecan, topotecan
Introduction
DNA topoisomerase I (Top1) is a ubiquitously expressed gene that is essential for mammalian cell proliferation during embryo development, as well as adult tissue and cell renewal over a lifetime. Top1 knockout mice die during early embryogenesis [1], and inhibition of Top1 activity interferes with normal tissue renewal and induces severe proliferative tissue toxicity (e.g. hematopoietic cell genesis from bone marrow). This is because the Top1 enzyme plays a critical role in cellular DNA replication and can also facilitate many gene transcriptions [2]. Consistent with these facts, the major adverse events of the Top1 inhibitors from the camptothecin (CPT) analogues irinotecan (CPT-11) and topotecan (TPT) are the hematopoietic toxicity and diarrhea during cancer treatment in the clinic. The hematopoietic toxicity is likely due to the disruption of Top1-mediated DNA replication in normal bone marrow stem/progenitor cells. The diarrhea was proposed to be due to off-target effects related to the bis-piperidine that confers water-solubility in the case of irinotecan [2]. However, diarrhea induced by CPTs is likely through a more general mechanism, since many anticancer drugs, including CPT analogues, without a bis-piperidine moiety have been known to induce diarrhea [3].
Discovery of FL118, a CPT analogue with novel mechanism of actions (MOA)
Using the survivin gene as a target and biomarker in high-throughput screening of small molecule libraries, followed by hit-to-lead analogue characterization, we discovered a smallmolecule (designated FL118) that possesses exceptional antitumor activity against colorectal and head-&-neck cancer in animal models of human tumors [4]. Structurally, FL118 is a CPT analogue with a methylenedioxy group linking to positions 10 and 11 of the A-ring. However, we found that mechanistically, FL118 selectively inhibits the expression of multiple antiapoptotic proteins (survivin, Mcl-1, XIAP, cIAP2) in the inhibitor of apoptosis (IAP) and Bcl-2 families. The inhibition of these proteins by FL118 is independent of the tumor suppressor p53 status either wild type (WT), mutant or null [4]. Importantly, individual genetic overexpression or silencing of these proteins revealed that each of these proteins plays a role in FL118-mediated cancer cell growth inhibition and apoptosis induction [4,5]. Furthermore, in p53 WT colorectal cancer (CRC) cells, FL118 induces p53-dependent senescence by promoting MdmX/Mdm4 ubiquitination and degradation [6]. Intriguingly, in the absence of p53, FL118 exhibits an even stronger ability to inhibit CRC cell growth and induce apoptosis [6]. In our studies, we further demonstrated that forced expression of exogenous MdmX in HCT116 CRC cells further enhances FL118 ability to inhibit cell growth and induce apoptosis [6]. This suggests that MdmX is a unique biomarker and target for FL118: the higher the level of MdmX expression, the better it is for FL118 antitumor activity. Mechanistically, the inhibition of MdmX expression by FL118 is through FL118 switching Mdm2-mediated ubiquitination and degradation of the tumor suppressor p53 (oncogenic effects) to Mdm2-mediated ubiquitination and degradation of the oncogenic protein MdmX (tumor suppression effects) [6]. Intriguingly, the degradation of oncogenic protein MdmX by Mdm2 is independent of the DNA damage signaling regulator ATM and the status of p53 and p21 [6].Additionally, different from irinotecan, SN-38 and topotecan, which are substrates of the efflux pump proteins ABCG2/BCRP [7-11] and P-gp/MDR1 [12-16], FL118 is not a substrate of ABCG2 and P-gp, and can overcome treatment resistance resulting from the expression of ABCG2 [17] or P-gp [18]. Consistent with these observations, FL118 effectively overcomes irinotecan and topotecan resistance [18].
Evidence for Top1-independent modulation of gene expression by topotecan
A critical question that we must ask ourselves is whether FL118 can inhibit the expression of multiple antiapoptotic and oncogenic proteins independent of Top1. In May 2016, Mabb, et al. published an interesting study in PLOS ONE [19]. In this study, the authors used multiple approaches to knock down or delete the Top1 gene (TOP1) in neurons to determine the role of Top1 in topotecan-mediated gene modulation. These authors found that in the presence of Top1, topotecan modulates much more gene expression than in the absence of Top1 through both Top1/DNA cleavage complex-dependent and -independent mechanisms [19]. These interesting findings triggered our enthusiasm to discuss the protein-encoding genes that were modulated by topotecan treatment in the neurons with TOP1 knockdown. We analyzed the raw data provided in the study of Mabb, et al. for the 38 downregulated genes and 4 upregulated genes by topotecan in the neurons with conditional knockout (cKO) of TOP1 (Table 1). We then asked ourselves whether the inhibition or induction of these genes by topotecan is Top1-independent or due to the incomplete cKO of Top1. We noticed approximate 75% of neurons with TOP1 cKO (means: 25% is still WT Top1) in the studies. Based on this fact, if Top1-independent downregulation of these genes by topotecan is not involved, even though the 25% of WT Top1 neurons completely wipe out the 38 gene expression, there will be only a 25% downregulation of these genes overall. Thus, the 38 genes shown in Table 1 (inhibition: from 1.97 to 5.17 fold) should be true Top1-independent, topotecan-regulated genes. Furthermore, 16 of the 38 genes were downregulated for ≥ 2-fold after cKO of Top1 itself without topotecan treatment (not shown). Therefore, topotecan downregulation of these 16 genes is on a low baseline expression level as a control (Table 1, blue). Furthermore, the Htr2c gene (HTR2C) was only downregulated in the topotecan-treated TOP1 cKO neurons but not in WT neurons (Table 1, red). On the other hand, we recognized that if there is a ≥ 10 fold gene induction in 25% of neurons, the induction could override the overall gene expression level, even in the 75% of neurons that have no induction of such genes. If this were the case, we would be unable to distinguish whether the induction of the gene by topotecan is Top1-independent or Top1-dependent. However, in the case of the four-upregulated genes shown in the bottom of Table 1, the first three genes were not induced in the WT neurons treated with topotecan, but were induced in the TOP1 cKO neurons treated with topotecan (Table 1). The fourth Nes gene (NES) that was induced in both WT and TOP1 cKO neurons that were treated with topotecan showed similar induction levels of less than 3 folds (2.45 versus 2.4 folds), which suggests that induction of NES is a Top1-independent event.
Table 1.
Gene modulation in mouse neurons by topotecan with topoisomerase I knockout
| Genes inhibited | Top1 cKOveh value* | Top1 cKOtopot value | Fold inhibition |
|
| |||
| Cadm2 | 29.7 | 6.37 | 4.66 |
| Nkain2 | 10.23 | 2.32 | 4.41 |
| Syt1 | 180.8 | 70.4 | 2.57 |
| Nrg3 | 9.91 | 2.8 | 3.54 |
| Nbea | 15.3 | 5.77 | 2.65 |
| Kcnip4 | 11.68 | 3.18 | 3.67 |
| Luzp2 | 23.6 | 7.5 | 3.15 |
| Opcml | 36.4 | 14.43 | 2.52 |
| Negr1 | 60.1 | 26.2 | 2.3 |
| Lphn3 | 9.41 | 3.29 | 2.86 |
| Vps13b | 2.64 | 0.85 | 3.11 |
| Pcdh11x | 5.13 | 1.67 | 3.07 |
| Tenm1 | 3.51 | 1.03 | 3.41 |
| Grm7 | 8.63 | 2.9 | 2.98 |
| Grm5 | 18.6 | 7.24 | 2.57 |
| Nrxn3 | 10.75 | 4.58 | 2.35 |
| Galntl6 | 2.17 | 0.42 | 5.17 |
| Robo2 | 8.32 | 3.24 | 2.57 |
| Dgkb | 12.49 | 4.98 | 2.51 |
| Spock3 | 27.9 | 11.81 | 2.36 |
| Erc2 | 23.7 | 11.18 | 2.12 |
| Pcdh9 | 13.2 | 5.23 | 2.52 |
| Mctp1 | 9.73 | 3.9 | 2.5 |
| Dpp10 | 12.8 | 5.41 | 2.37 |
| March1 | 8.34 | 3.44 | 2.42 |
| Npas3 | 2.43 | 0.51 | 4.77 |
| Htr2c | 11.5 | 4.79 | 2.4 |
| Gria4 | 17 | 7.21 | 2.36 |
| Reln | 10.07 | 2.46 | 4.09 |
| Gucy1a2 | 9.35 | 4.17 | 2.24 |
| Akt3 | 48.7 | 24.6 | 1.98 |
| Ppfia2 | 11.98 | 4.93 | 2.43 |
| Slc4a4 | 18.8 | 4.73 | 3.98 |
| Gphn | 13.4 | 5.7 | 2.35 |
| Ptprd | 10.4 | 4.76 | 2.19 |
| Adarb2 | 1.84 | 0.39 | 4.72 |
| Ctnna2 | 25 | 11.7 | 2.14 |
| Epha5 | 20.7 | 10.5 | 1.97 |
|
| |||
| Genes induced | Top1 cKOveh value | Top1 cKOtopot value | Folds induction |
|
| |||
| Cdkn1a | 92.8 | 210.3 | 2.27 |
| Phlda3 | 48.1 | 99.2 | 2.06 |
| Slc5a3 | 7.16 | 15.8 | 2.21 |
| Nes | 8.7 | 20.9 | 2.4 |
Top1, topoisomerase I;
cKO, conditional knockout; veh, vehicle treated; topot, topotecan treated.
The key point that we want to emphasize is that the Supplemental Data provided in the study from Mabb, et al. [19] gives strong evidence that topotecan can inhibit or induce gene expression independent of Top1 expression/activity. This evidence provides a possibility that some novel CPT analogues (e.g. FL118) may mainly use Top1-independent mechanism to deliver their antitumor activity.
Does FL118 need Top1 as a target for its superior antitumor efficacy?
In the initial FL118 discovery period, we largely used animal models of human tumors to screen the top candidates for antitumor activity. We found that FL118 is the top candidate that exhibits superior antitumor efficacy over other candidates, including irinotecan and topotecan, two FDA-approved Top1 inhibitors for cancer treatment. Since FL118 is structurally similar to irinotecan and topotecan, we thought that FL118 could be a much better Top1 inhibitor than irinotecan and topotecan. Thus, we tested them in the Top1-DNA complex biochemical cleavage assay [4]. Irinotecan is a pro-drug and showed very low activity in the in vitro experiment; we therefore used its active metabolite SN-38 in parallel with FL118 to compare their relative ability to inhibit Top1 enzyme activity. The experiment revealed that even at a 1 0µM concentration, the highest SN-38 dose that can be reached by irinotecan in vivo, FL118 did not exhibit a better ability to inhibit Top1 activity of converting supercoiled DNA into relaxed DNA by nicking the DNA (in fact, FL118 at 1 0µM only exhibited approximately one-half of inhibition that SN-38 showed) [4]. In contrast, FL118 can effectively inhibit cancer cell growth at far below a nM level [4].
Alternatively, it was reported that mutations of TOP1 in CRC cells result in high CPT-resistance [20]. Actually, plants use mutations in TOP1 as a self-defensive strategy to avoid harming themselves with their own production of CPT [21]. We compared the relative resistance of cancer cells to CPT, SN-38, topotecan and FL118 before and after Top1 mutations, respectively, using the Du145 parental prostate cancer cells (Top1 gene WT) in parallel with the Du145-derived two sub-cell lines (RC0.1, RC1) with Top1 R364H mutations [22]. The data showed that in the parental DU145 cells (Top1 WT), the relative potency (RP) of CPT, SN-38, topotecan and FL118 calculated from their IC50 (set topotecan RP as 1) is in turn 3.17, 4.75, 1.0 and 41.7. In contrast, in the Top1-mutated RC0.1 cells, the RP of CPT, SN-38, topotecan and FL118 (still set topotecan RP as 1) is in turn 0.97, 5.24, 1.0 and 778; and in the Top1-mutated RC1 cells, the RP of CPT, SN-38, topotecan and FL118 is in turn 2.69, 13.2, 1.0, and 572 [23]. Thus, the RP of FL118 in comparison with other CPTs (CPT, SN-38, topotecan) is significantly enhanced after Top1 mutation.
If Top1 inhibition by FL118 does make a small contribution to FL118 antitumor activity, the observation from Top1 mutation may imply important consequences. That is, although all 4 of the compounds (CPT, SN-38, topotecan and FL118) showed increased IC50 values after Top1 mutation, FL118 was much less affected. This resulted in a substantial increase in FL118’s relative potential (RP). This difference could cause CPT, SN-38 and topotecan to be ineffective, while FL118 can still be effective to kill cancer cells after Top1 mutation. On the other hand, Top1 expression level/enzyme catalytic activity has been reported to affect CPT compound sensitivity in killing tumor cells. For example, it was shown that reduced Top1 expression and/or Top1 catalytic activity is associated with increased resistance to CPT compounds [24-26], while increased Top1 in cancer cells sensitizes CPTs (irinotecan and SN-38) [27-29]. Intriguingly, in contrast to these documented findings from the literature, we found that the sensitivity of CRC cell line-established xenograft tumors to FL118 is irrelevant to Top1 expression levels. CRC xenograft tumors with high Top1 expression can be highly resistant to FL118 treatment. For example, RKO tumors highly express Top1 and are also highly resistant to FL118 (Figures 1A and 2 first row left). In contrast, CRC xenograft tumors with low/negative Top1 expression can be sensitive to FL118 treatment. For example, LIM2551 tumors do not express Top1 and are also highly sensitive to FL118 treatment (Figures 1A and 2 second row left). In this regard, we determined the Top1 expression in all of our available 30 CRC cell lines (Figure 1). In parallel, we determined the CRC cell-established xenograft tumor sensitivity to FL118 treatment in SCID mice. While the in vivo experiments haven’t been completely finished, available tumor sensitivity to FL118 from 18 CRC xenograft tumors is presented in Figure 2. The in vivo data indicated that CRC tumors with either high Top1 expression or low/negative Top1 expression can be resistant or sensitive to FL118 treatment (Figure 2). This finding is contrast to the finding in the literature for the relationship of Top1 expression and CPTs’ sensitivity [24-27,29].
Figure 1.
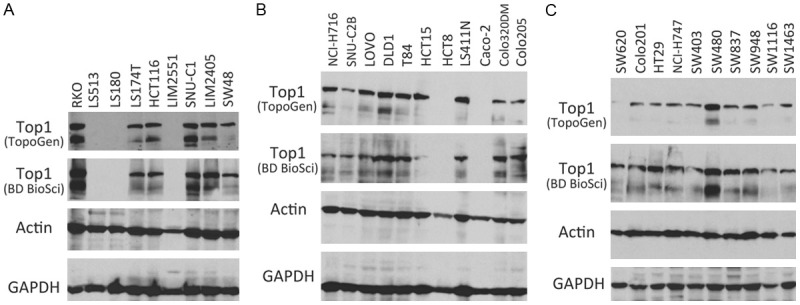
Topoisomerase I (Top1) expression in 30 colorectal cancer (CRC) cell lines: Individual CRC cells grown to 80% confluence were lysed and subjected to western blot analysis to determine the expression of Top1 protein. Two Top1 antibodies were used in this analysis. One is purchased from TopoGen (TG2012-4, Lot 12FB04) and the other is purchased from BD Biosciences (X-21, RUO). Both actin and GAPDH are internal controls for total protein loading evaluation.
Figure 2.

The sensitivity of CRC xenograft tumors to FL118 treatment is not associated with the expression of the CPTs target Top1: Individual xenograft tumors were first established from the corresponding CRC cell lines (RKO, LS513, LS180, LIM2551, SUN-C1, LIM2405, SW48, NCI-H716, LOVO, T84, LS411N, Caco-2, Colo205, Colo201, NCI-H747, SW837, SW1116, SW1463) by subcutaneous injection of 2 million cells at the flank area of SCID mice, respectively. Then the established tumors were inoculated into SCID mice at the flank area for testing FL118 sensitivity. FL118 treatment was initiated at the time when the inoculated individual xenograft tumors reached 100-200 mm3 (designated day 0). FL118 was administrated with the schedule of weekly x 4 (arrowed) via p.o. (per oral) routes at a dose of 10 mg/kg (MTD: maximum tolerated dose). Individual tumor curves were derived from the mean tumor sizes 0± SD from up to five mice. The small image insert within each xenograft tumor histogram is the cut-and-past Top1 expression from Figure 1.
What is the role of FL118 interaction with Top1/DNA complex?
The data presented in Figures 1 and 2, showing FL118 antitumor efficacy is not associated with Top1 expression, suggest that FL118 may not use Top1 as a target for its antitumor activity. However, several studies using the in vitro Top1 biochemical cleavage assay found that most CPTs, including FL118 [10,11-MD-20(S)-CPT]and closely related compounds (RS racemic), exhibit an association of antitumor activity with inhibition of Top1-mediated DNA cleavage and re-ligation activity in in vitro Top1 biochemical cleavage assays [30,31]. In this regard, we have analyzed 29 CPT analogues including CPT, SN-38, topotecan, FL118 and many FL118 analogues using molecular docking and the molecular mechanics generalized Born model with solvent accessible surface area (MMGBSA) [32] rescoring to compare poses in the Top1/DNA complex. Our in-house studies and published work reveal that MMGBSA rescoring of top-ranked (10-20) docking poses (protein/DNA-inhibitor complex configurations) allows discrimination of the physiologically relevant pose/configuration ~83% of the time in large test assessments (e.g. PDB-BIND) [33] with improved affinity correlation for the lowest MMGBSA rescored pose. We conducted this study using two different docking approaches, Autodock VINA and Schrodinger GLIDE SP/XP. In both cases, we started with the crystal structure of the topotecan-Top1-DNA complex [34](PDB 1K4T). Protein preparation for GLIDE docking followed typical work flows and the use of OPLS3 refinement (sidechain completion/hydrogen addition followed by steepest descents and conjugate gradient energy minimization) of the crystal structure components prior to docking grid computation. To prepare for Autodock VINA docking, we initially prepared the protein/DNA complex using AMBER14 [34] libraries and using GAMEES-UK [35] computation of potential derived charges for 5’-thio-2’-deoxy-guanosine phosphonic acid (TGP) which were incorporated into an XLEAP prepared representation of the modified base. Docking with standard GLIDE SP [36] protocol (5000 poses for initial docking phase/keeping 400 initial poses for energy minimization and best scoring assessment wherein only poses within 100 of the top pose are considered) or Autodock VINA [37] with exhaustiveness = 80 and collection of 20 poses for each ligand was conducted. In both cases we performed MMGBSA rescoring of the docked poses. In the case of GLIDE poses, the Pime-MMGBSA (OPLS3/VSGP) approach was used and in the case of VINA, AMBER14-MMGBSA rescoring was performed allowing for full protein/DNA/docked ligand minimization. The lowest MMGBSA poses for each ligand were then collected to examine the probable ‘bound’ configuration for the putative ligands of the protein-DNA complex.
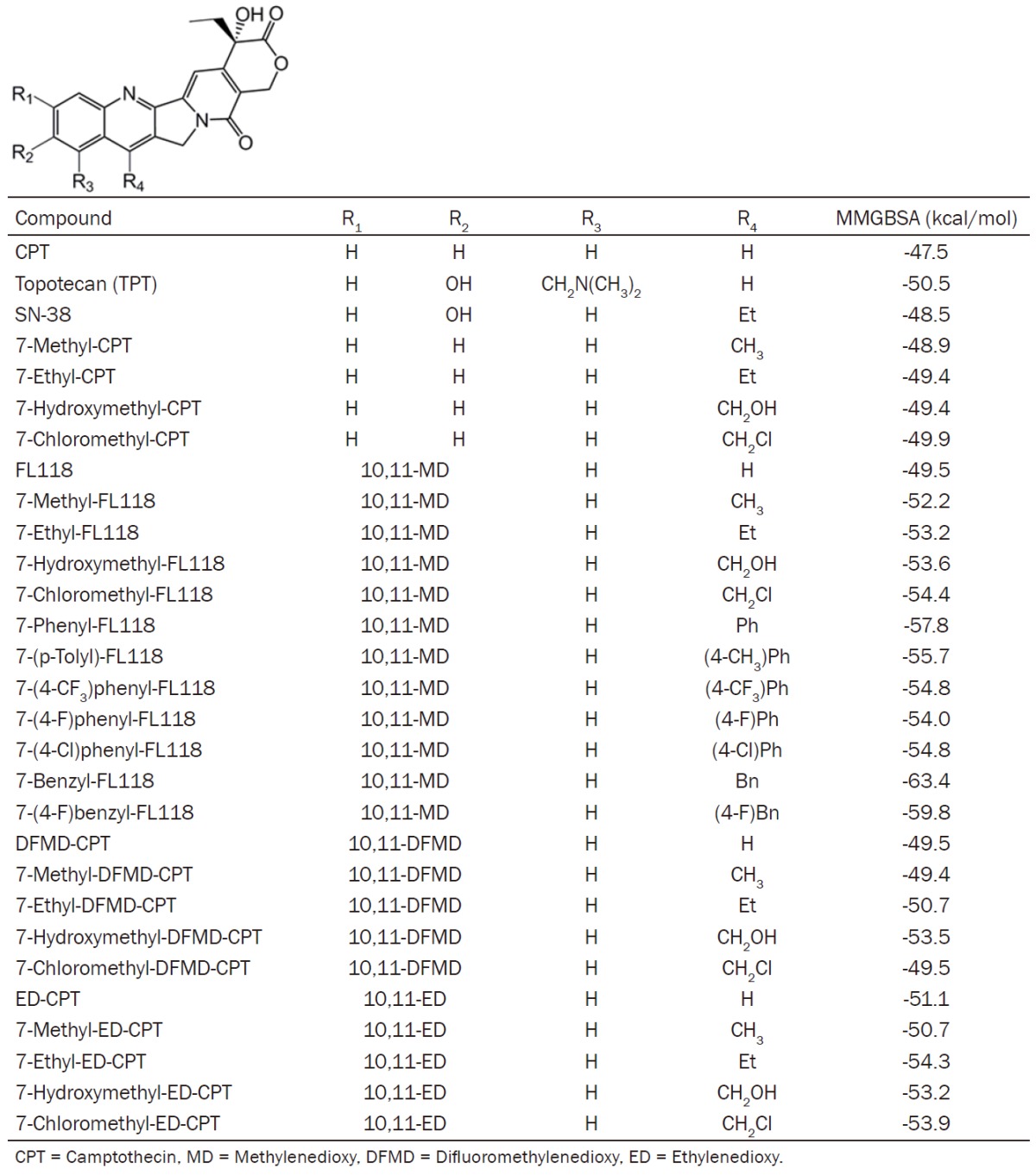
Figure 3A shows the lowest MMGBSA energy minimized VINA/MMGBSA pose for topotecan (TPT, green, ball and stick) with a stacking interaction between TGP on one face of the ligand and an adenine/thymine base pair on the opposite side of the ligand. As seen in Figure 3, the root-mean-square deviation (RMSD) fit of the docked/free-energy rescored minimized pose (green) compared to the crystal structure solution (cyan) is less than an angstrom with similar interactions with surrounding residues. The TPT ligand in this docked pose interacts with Asp533 of Top1 (not shown in Figure 3A) through a hydrogen bonding interaction and has π-π stacking interactions with the TGP on one side and adenine on the other. The ligand in this intimate intercalated fashion is bordered by Glu356 and Asn352 of Top1 at the bottom of this panel. As the lowest MMGBSA docked pose of TPT resembles the crystal structure configuration of TPT, we next examined the predicted pose for FL118 in Figure 2B where it shows the overlap with the docked pose of TPT. The ligands are shown to have a similar disposition with respect to the interacting residues. Table 2 reports the VINA/MMGBSA scores for 29 CPT derivatives. While FL118 has a slightly less negative MMGBSA score than topotecan (-49.5 vs -50.5), several FL118 derivatives tested are seen to have more negative MMGBSA scores (i.e., predicted to be better Top1 inhibitors) than topotecan (Table 2). In consonance with the MMGBSA scores, we found that compared to FL118, most FL118 analogues with more negative MMGBSA scores (i.e. predicted to be better Top1 inhibitors) exhibit greater toxicity (body weight loss) than FL118. This is likely due to their stronger inhibition of Top1 than FL118 in animal models (Figure 4).
Figure 3.
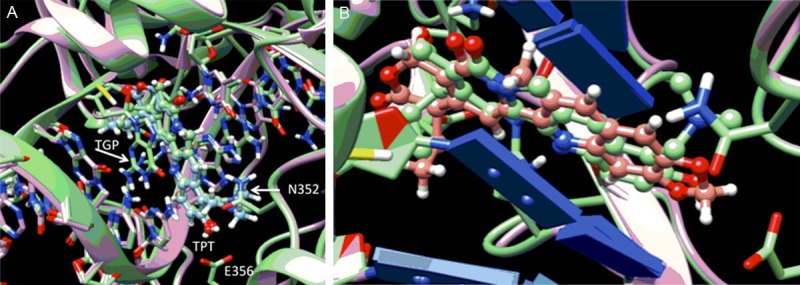
A. Overlap of lowest MMGBSA docked pose of topotecan (TPT, green, ball and stick) with crystallographic TPT (cyan, ball and stick) in Top1/DNA complex. B. Lowest MMGBSA docked pose of TPT (green carbons) and FL118 (brown carbons) show analogous amino acid and base pair interactions. Heteroatoms are colored by normal conventions (red = oxygen, blue = nitrogen).
Table 2.
CPT analogues structure and VINA/MMGBSA results
|
Figure 4.
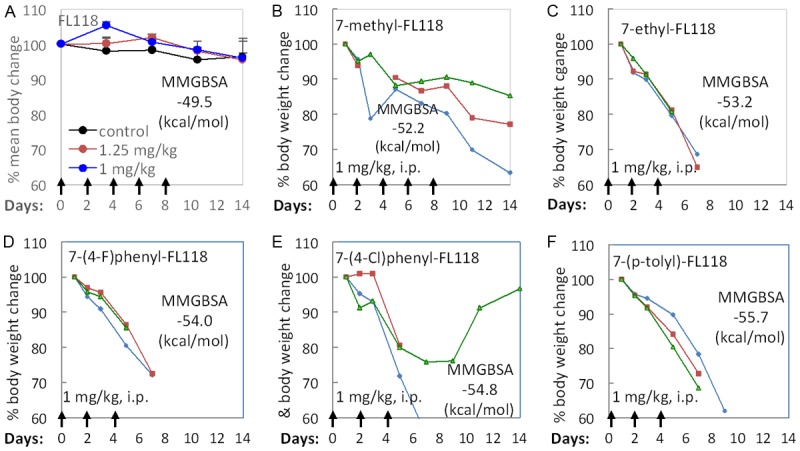
Body weight changes (a marker of toxicity) of mice after treatment with FL118 or a FL118 analogue: A. SCID mice were treated with FL118 (q2dx5). Individual body weight change curves were derived from the mean 0± SE from five mice. B. Individual SCID mice were treated with 7-methyl-FL118 (q2dx5). Two mice were euthanized on day 14 and one on day 16. C. Individual SCID mice were treated with 7-ethyl-FL118 (q2dx3). One mouse was euthanized on day 5 and the other two on day 7. D. Individual SCID mice were treated with 7-(4-F)phenyl-FL118 (q2dx3). One mouse was euthanized on day 5 and the other two on day 7. E. Individual SCID mice were treated with 7-(4-Cl)phenyl-FL118 (q2dx3). One mouse was euthanized on day 5 and one on day 7. F. Individual SCID mice were treated with 7-(p-tolyl)-FL118 (q2dx3). Two mice were euthanized on day 7 and one on day 9. The reason for euthanization of mice is because body weight loss for ≥ 20% and/or mice in a moribund state.
While MMGBSA rescoring of docking poses improves relative Top1/DNA complex binding affinity prediction, correlations rarely give Pearson r > 0.8. The method is semi-quantitative due to theoretical approximations (e.g. neglect of explicit water, lack of inclusion of conformational entropies), as well as the challenges of predicting flexible ensemble poses that are free-energy rescored. Nevertheless, the results from Table 2 suggest that favorable interactions with the Top1/DNA complex (more negative MMGBSA score) link to a greater toxicity potential of FL118 analogues tested in vivo (Figure 4).
Based on the literature-documented Top1 biochemical cleavage data and our molecular modeling results with relevant animal model studies, further discussion of some critical points may benefit future research in the pertinent field.
In the Top1 biochemical cleavage assay, the concentration of the drug used in these studies was usually in the range of 1-100 0µM and drug concentrations below 1 0µM showed no significant Top1 inhibition activity [30,31]. A key concern here is whether the in vitro result obtained from any concentration higher than 0.5-1 0µM from the Top1 biochemical cleavage assay experiment could mimic the in vivo activity of FL118 to inhibit Top1. Our view on this issue is negative in terms of our findings from the experimental studies and molecular docking of FL118 and FL118 core structure-based analogues. This view is based on two facts: (a) FL118 effectively inhibits cancer cell growth in the concentration range of 0.01 to 10 nM, depending on cancer cell types and/or genetic backgrounds [4]; and (b) our pharmacokinetics (PK) studies with animal models of human tumors indicated that the peak of FL118’s concentration in blood is under 100 nM with a short half-life (< 1.8 hours), while the peak of FL118’s concentration in tumors can be around 300 nM (rapidly accumulated in tumor) with a much longer half-life (6.8-12.8 hours) [18]. This means that FL118 may not even be able to reach a sustained 1 0µM concentration in the in vivo situation. Furthermore, molecular docking results also indicated that FL118 is not a better Top1 inhibitor than topotecan (Table 2). However, FL118 has much better antitumor activity than topotecan [4] and can overcome topotecan-resistant tumors [18].
The critical questionis whether the inhibition of Top1 activity by FL118 plays a major role in FL118’s exceptional antitumor activity or is mainly involved in its side effects of hematopoietic toxicity. FL118 possesses exceptional antitumor activity in animal models of human tumors, and inhibits multiple antiapoptotic proteins (survivin, Mcl-1, XIAP, cIAP2), oncogenic protein MdmX [4-6,18], and overcomes both ABCG2 and P-gp resistance in cancer cells [17,18,38]. However, we failed to find that FL118 is a better Top1 inhibitor than SN-38/irinotecan and topotecan, in either the Top1 biochemical cleavage assay [4] or after Top1 mutation in cancer cells [23]. Furthermore, the data from Figures 1 and 2 demonstrate that FL118 antitumor efficacy is not associated with Top1 expression. In other words, CRC xenograft tumors with either high or low/negative Top1 expression can be sensitive or resistant to FL118 treatment, suggesting Top1 expression is irrelevant to FL118 antitumor efficacy.
Although use of a range of 1-100 0µM for drug concentrations in Top1 biochemical cleavage assay may not well mimic the FL118 in vivo situation (1-100 0µM in vitro versus ≤ 1 0µM in vivo), it would be safe to propose that FL118 at its in vivo therapeutic doses may still have the ability to negatively affect Top1 enzyme activity.However, this may not make a major contribution to FL118’s antitumor efficacy, especially when considering the data shown in Figures 1 and 2. The fact is that FL118 possesses muchbetter antitumor activity than irinotecan and topotecan [4], and could overcome irinotecan- or topotecan-resistant human tumors in animal models [18]. However, FL118 exhibits hematopoietic toxicity (as well as diarrhea) similar to irinotecan and topotecan. Practically, due to the much higher in vivo potency of FL118 in comparison with irinotecan and topotecan as we previously found [4], the side effects (hematopoietic toxicity and diarrhea) for FL118 appear less severe. Nevertheless, previous studies have shown that CPTs-induced Top1-mediated lesions and DNA damage signaling in primitive hematopoietic cells confer significant oncogenic potential [39]. Therefore, in order to decrease irinotecan or topotecan-induced hematopoietic toxicity, clinical chemotherapy including irinotecan or topotecan has been used in parallel with peripheral-blood stem cell or bone marrow transplantation [40-42]. Thus, the hematopoietic toxicity resulting from FL118 treatment is likely due to the inhibition of Top1-mediated DNA replication by FL118 during hematopoietic cell renewal from bone marrow stem/progenitor cells.
Concluding remarks
There is no doubt that Top1 is not an ideal target for cancer treatment since normal tissue/cell renewal needs Top1 for DNA replication and cell proliferation. Blocking Top1 function would induce serious toxicity in renewing tissues (e.g. hematopoietic toxicity). Intriguingly, examples of potent CPT compounds that are not dependent on Top1 expression level were reported previously. Jaxel et al. showed that while CPT analogues 10-NH2-(RS)-CPT and 11-CN-(RS)-CPT showed very poor Top1 inhibition, these two CPT compounds extended survival time much longer than other CPTs with strong Top1 inhibition in the L1210 leukemia metastatic mouse model [30]. The disagreement between antitumor activity and the potential inhibition of Top1 enzyme activity argues 10-NH2-(RS)-CPT and 11-CN-(RS)-CPT may use alternative targets instead of Top1 for their anti-leukemia activity. Based on this observation and in consideration of the recent finding in neurons (Table 1), as well as our data shown in Figures 1 and 2, we propose that inhibition of Top1 activity by FL118 may not play a major role in FL118’s exceptional antitumor activity; instead, inhibition of Top1 by FL118 may mainly be involved in the side effects of hematopoietic toxicity and possibly in diarrhea as well. Therefore, we believe that the next generation of low toxicity and high efficacy anticancer agents that are structurally relevant to FL118 should focus on the development of compounds having weaker affinity to the Top1-DNA complex, while keeping their strong ability to inhibit the expression of survivin, Mcl-1, XIAP, cIAP2 and MdmX.
Acknowledgements
The authors thank Dr. Yves Pommier (Laboratory of Molecular Pharmacology, Center for Cancer Research, National Cancer Institute, USA) and the lab manager Keli Agama for kindly providing their Du145-derived two cell lines (RC.1 and RC0.1) that have Top1 mutations and resistance to SN-38 and topotecan. We also thank Ms. Amanda Hess for editorial check of this manuscript before submission. The research currently conducted in The Li laboratory is supported in part by grants from USA National Cancer Institute (NCI, R21CA180764, R03CA182552 and a subcontract from R44CA176937). The animal model of human tumor in vivo work was performed in Roswell Park Cancer Institute Animal Center and the work was supported in part by the Institute NCI Core grant (P30CA016056).
Disclosure of conflict of interest
FL118 and FL118 core structure-based analogues will be further developed in Canget BioTekpharma LLC (www.canget-biotek.com), a Roswell Park Cancer Institute-spinoff company. FL, XL and YW are initial investors of Canget for development of FL118 and FL118 core structure-relevant anticancer agents.
References
- 1.Morham SG, Kluckman KD, Voulomanos N, Smithies O. Targeted disruption of the mouse topoisomerase I gene by camptothecin selection. Mol Cell Biol. 1996;16:6804–6809. doi: 10.1128/mcb.16.12.6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 3.Dy GK, Adjei AA. Understanding, recognizing, and managing toxicities of targeted anticancer therapies. CA Cancer J Clin. 2013;63:249–279. doi: 10.3322/caac.21184. [DOI] [PubMed] [Google Scholar]
- 4.Ling X, Cao S, Cheng Q, Keefe JT, Rustum YM, Li F. A Novel Small Molecule FL118 That Selectively Inhibits Survivin, Mcl-1, XIAP and cIAP2 in a p53-Independent Manner, Shows Superior Antitumor Activity. PLoS One. 2012;7:e45571. doi: 10.1371/journal.pone.0045571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao J, Ling X, Cao S, Liu X, Wan S, Jiang T, Li F. Antitumor activity of FL118, a survivin, Mcl-1, XIAP, cIAP2 selective inhibitor, is highly dependent on its primary structure and steric configuration. Mol Pharm. 2014;11:457–467. doi: 10.1021/mp4004282. [DOI] [PubMed] [Google Scholar]
- 6.Ling X, Xu C, Fan C, Zhong K, Li F, Wang X. FL118 Induces p53-Dependent Senescence in Colorectal Cancer Cells by Promoting Degradation of MdmX. Cancer Res. 2014;74:7487–7497. doi: 10.1158/0008-5472.CAN-14-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Houghton PJ, Germain GS, Harwood FC, Schuetz JD, Stewart CF, Buchdunger E, Traxler P. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer Res. 2004;64:2333–2337. doi: 10.1158/0008-5472.can-03-3344. [DOI] [PubMed] [Google Scholar]
- 8.Su Y, Hu P, Lee SH, Sinko PJ. Using novobiocin as a specific inhibitor of breast cancer resistant protein to assess the role of transporter in the absorption and disposition of topotecan. J Pharm Pharm Sci. 2007;10:519–536. doi: 10.18433/j3qp4w. [DOI] [PubMed] [Google Scholar]
- 9.Su Y, Lee SH, Sinko PJ. Inhibition of efflux transporter ABCG2/BCRP does not restore mitoxantrone sensitivity in irinotecan-selected human leukemia CPT-K5 cells: evidence for multifactorial multidrug resistance. Eur J Pharm Sci. 2006;29:102–110. doi: 10.1016/j.ejps.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 10.Yoshikawa M, Ikegami Y, Sano K, Yoshida H, Mitomo H, Sawada S, Ishikawa T. Transport of SN-38 by the wild type of human ABC transporter ABCG2 and its inhibition by quercetin, a natural flavonoid. J Exp Ther Oncol. 2004;4:25–35. [PubMed] [Google Scholar]
- 11.Shishido Y, Ueno S, Yamazaki R, Nagaoka M, Matsuzaki T. ABCG2 inhibitor YHO-13351 sensitizes cancer stem/initiating-like side population cells to irinotecan. Anticancer Res. 2013;33:1379–1386. [PubMed] [Google Scholar]
- 12.Kruijtzer CM, Beijnen JH, Rosing H, ten Bokkel Huinink WW, Schot M, Jewell RC, Paul EM, Schellens JH. Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-glycoprotein inhibitor GF120918. J. Clin. Oncol. 2002;20:2943–2950. doi: 10.1200/JCO.2002.12.116. [DOI] [PubMed] [Google Scholar]
- 13.de Vries NA, Zhao J, Kroon E, Buckle T, Beijnen JH, van Tellingen O. P-glycoprotein and breast cancer resistance protein: two dominant transporters working together in limiting the brain penetration of topotecan. Clin Cancer Res. 2007;13:6440–6449. doi: 10.1158/1078-0432.CCR-07-1335. [DOI] [PubMed] [Google Scholar]
- 14.Takeba Y, Sekine S, Kumai T, Matsumoto N, Nakaya S, Tsuzuki Y, Yanagida Y, Nakano H, Asakura T, Ohtsubo T, Kobayashi S. Irinotecan-induced apoptosis is inhibited by increased P-glycoprotein expression and decreased p53 in human hepatocellular carcinoma cells. Biol Pharm Bull. 2007;30:1400–1406. doi: 10.1248/bpb.30.1400. [DOI] [PubMed] [Google Scholar]
- 15.Tagen M, Zhuang Y, Zhang F, Harstead KE, Shen J, Schaiquevich P, Fraga CH, Panetta JC, Waters CM, Stewart CF. P-glycoprotein, but not multidrug resistance protein 4, plays a role in the systemic clearance of irinotecan and SN-38 in mice. Drug Metab Lett. 2010;4:195–201. doi: 10.2174/187231210792928251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Filipski E, Berland E, Ozturk N, Guettier C, van der Horst GT, Levi F, Okyar A. Optimization of irinotecan chronotherapy with P-glycoprotein inhibition. Toxicol Appl Pharmacol. 2014;274:471–479. doi: 10.1016/j.taap.2013.12.018. [DOI] [PubMed] [Google Scholar]
- 17.Westover D, Ling X, Lam H, Welch J, Jin C, Gongora C, Del Rio M, Wani M, Li F. FL118, a novel camptothecin derivative, is insensitive to ABCG2 expression and shows improved efficacy in comparison with irinotecan in colon and lung cancer models with ABCG2-induced resistance. Mol Cancer. 2015;14:92. doi: 10.1186/s12943-015-0362-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ling X, Liu XJ, Zhong K, Smith N, Prey J, Li F. FL118, a novel camptothecin analogue, overcomes irinotecan and topotecan resistance in human tumor xenograft models. Am J Transl Res. 2015;7:1765–1781. [PMC free article] [PubMed] [Google Scholar]
- 19.Mabb AM, Simon JM, King IF, Lee HM, An LK, Philpot BD, Zylka MJ. Topoisomerase 1 Regulates Gene Expression in Neurons through Cleavage Complex-Dependent and -Independent Mechanisms. PLoS One. 2016;11:e0156439. doi: 10.1371/journal.pone.0156439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arakawa Y, Ozaki K, Okawa Y, Yamada H. Three missense mutations of DNA topoisomerase I in highly camptothecin-resistant colon cancer cell sublines. Oncol Rep. 2013;30:1053–1058. doi: 10.3892/or.2013.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sirikantaramas S, Yamazaki M, Saito K. Mutations in topoisomerase I as a self-resistance mechanism coevolved with the production of the anticancer alkaloid camptothecin in plants. Proc Natl Acad Sci U S A. 2008;105:6782–6786. doi: 10.1073/pnas.0801038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urasaki Y, Laco GS, Pourquier P, Takebayashi Y, Kohlhagen G, Gioffre C, Zhang H, Chatterjee D, Pantazis P, Pommier Y. Characterization of a novel topoisomerase I mutation from a camptothecin-resistant human prostate cancer cell line. Cancer Res. 2001;61:1964–1969. [PubMed] [Google Scholar]
- 23.Li F. Anticancer drug FL118 is more than a survivin inhibitor: Where is the Achilles’ heel of cancer? Am J Cancer Res. 2014;4:304–311. [PMC free article] [PubMed] [Google Scholar]
- 24.Sugimoto Y, Tsukahara S, Oh-hara T, Isoe T, Tsuruo T. Decreased expression of DNA topoisomerase I in camptothecin-resistant tumor cell lines as determined by a monoclonal antibody. Cancer Res. 1990;50:6925–6930. [PubMed] [Google Scholar]
- 25.Kapoor R, Slade DL, Fujimori A, Pommier Y, Harker WG. Altered topoisomerase I expression in two subclones of human CEM leukemia selected for resistance to camptothecin. Oncol Res. 1995;7:83–95. [PubMed] [Google Scholar]
- 26.Liao Z, Robey RW, Guirouilh-Barbat J, To KK, Polgar O, Bates SE, Pommier Y. Reduced expression of DNA topoisomerase I in SF295 human glioblastoma cells selected for resistance to homocamptothecin and diflomotecan. Mol Pharmacol. 2008;73:490–497. doi: 10.1124/mol.107.041178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kotoh S, Naito S, Yokomizo A, Kumazawa J, Asakuno K, Kohno K, Kuwano M. Increased expression of DNA topoisomerase I gene and collateral sensitivity to camptothecin in human cisplatin-resistant bladder cancer cells. Cancer Res. 1994;54:3248–3252. [PubMed] [Google Scholar]
- 28.Sakai A, Kasahara K, Ohmori T, Kimura H, Sone T, Fujimura M, Nakao S. MET increases the sensitivity of gefitinib-resistant cells to SN-38, an active metabolite of irinotecan, by up-regulating the topoisomerase I activity. J Thorac Oncol. 2012;7:1337–1344. doi: 10.1097/JTO.0b013e31825cca4c. [DOI] [PubMed] [Google Scholar]
- 29.Smith PJ, Makinson TA, Watson JV. Enhanced sensitivity to camptothecin in ataxia-telangiectasia cells and its relationship with the expression of DNA topoisomerase I. Int J Radiat Biol. 1989;55:217–231. doi: 10.1080/09553008914550271. [DOI] [PubMed] [Google Scholar]
- 30.Jaxel C, Kohn KW, Wani MC, Wall ME, Pommier Y. Structure-activity study of the actions of camptothecin derivatives on mammalian topoisomerase I: evidence for a specific receptor site and a relation to antitumor activity. Cancer Res. 1989;49:1465–1469. [PubMed] [Google Scholar]
- 31.Vladu B, Woynarowski JM, Manikumar G, Wani MC, Wall ME, Von Hoff DD, Wadkins RM. 7- and 10-substituted camptothecins: dependence of topoisomerase I-DNA cleavable complex formation and stability on the 7- and 10-substituents. Mol Pharmacol. 2000;57:243–251. [PubMed] [Google Scholar]
- 32.Hou T, Wang J, Li Y, Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model. 2011;51:69–82. doi: 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenidge PA, Kramer C, Mozziconacci JC, Wolf RM. MM/GBSA binding energy prediction on the PDBbind data set: successes, failures, and directions for further improvement. J Chem Inf Model. 2013;53:201–209. doi: 10.1021/ci300425v. [DOI] [PubMed] [Google Scholar]
- 34.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB Jr, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci U S A. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guest MF, Bush IJ, van Dam HJJ, Sherwood P, Thomas JMH, van Lenthe JH, Havenith RWA, Kendrick J. The GAMESS-UK electronic structure package: algorithms, developments and applications. Molecular Physics. 2005;103:719–747. [Google Scholar]
- 36.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 37.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Westover D, Li F. New trends for overcoming ABCG2/BCRP-mediated resistance to cancer therapies. J Exp Clin Cancer Res. 2015;34:159. doi: 10.1186/s13046-015-0275-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morales M, Liu Y, Laiakis EC, Morgan WF, Nimer SD, Petrini JH. DNA damage signaling in hematopoietic cells: a role for Mre11 complex repair of topoisomerase lesions. Cancer Res. 2008;68:2186–2193. doi: 10.1158/0008-5472.CAN-07-2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Egusa Y, Fujiwara Y, Syaharuddin E, Sumiyoshi H, Isobe T, Yamakido M. Mobilization of peripheral blood stem cells in patients with advanced thoracic malignancies after irinotecan (CPT-11) administration. Anticancer Res. 1998;18:481–487. [PubMed] [Google Scholar]
- 41.Cacciari N, Zamagni C, Martoni A. The addition of topotecan to carboplatin and paclitaxel as first-line therapy for advanced ovarian cancer; is it possible only with peripheral blood stem cell support? Eur J Gynaecol Oncol. 2000;21:84–85. [PubMed] [Google Scholar]
- 42.Schilder RJ, Gallo JM, Millenson MM, Bookman MA, Weiner LM, Rogatko A, Rogers B, Padavic-Shallers K, Boente M, Rosenblum N, Adams AL, Ciccotto S, Ozols RF. Phase I trial of multiple cycles of high-dose carboplatin, paclitaxel, and topotecan with peripheral-blood stem-cell support as front-line therapy. J. Clin. Oncol. 2001;19:1183–1194. doi: 10.1200/JCO.2001.19.4.1183. [DOI] [PubMed] [Google Scholar]