

Abstract
Protein degradation has many critical functions in the nervous system such as refinement of synaptic connections during development and synaptic plasticity and memory in the adult organisms. A major cellular machinery of proteolysis is the ubiquitin-proteasome pathway (UPP). The UPP precisely regulates proteolysis by covalently attaching ubiquitin, a small protein, to substrates through sequential enzymatic reactions and the proteins marked with the ubiquitin tag are degraded by complex containing many subunits called the proteasome. Research over the years has shown a role for the UPP in regulating presynaptic and postsynaptic proteins critical for neurotransmission and synaptic plasticity. Studies have also revealed a role for the UPP in various forms of memory. Mechanistic investigations suggest that the function of the UPP in neurons is not homogenous and is subject to local regulation in different neuronal sub-compartments. In both invertebrate and vertebrate model systems, local roles have been found for enzymes that attach ubiquitin to substrate proteins as well as for enzymes that remove ubiquitin from substrates. The proteasome also has disparate functions in different parts of the neuron. In addition to the UPP, proteolysis by the lysosome and autophagy play a role in synaptic plasticity and memory. This review details the functions of proteolysis in synaptic plasticity and summarizes the findings on the connection between proteolysis and memory mainly focusing on the UPP including its local roles.
1. Introduction
The quest for understanding how the nervous system stores information has led to the exploration of synaptic plasticity and memory in several model systems: from worms to human beings. Many decades of research in the 20th century focused on the role of protein synthesis in long-term synaptic plasticity and memory. Research that began in the 1990s revealed a role for regulated proteolysis in long-term synaptic plasticity. Protein degradation that functions to sculpt synapses and thus in aiding memory formation occurs mainly through the ubiquitin-proteasome pathway. Evidence over the last few years has also indicated a role for other types of proteolysis that occur through the lysosome and autophagy. This review mainly focuses on ubiquitin-proteasome-mediated degradation and provides brief descriptions of the functions of the lysosome and autophagy.
2. The ubiquitin-proteasome pathway
In the ubiquitin-proteasome pathway (UPP), covalent attachment of ubiquitin, a highly conserved 76-amino acid protein, to substrate proteins marks them for degradation by a proteolytic complex called the proteasome. The attachment of ubiquitin (ubiquitination) to proteins requires sequential activity of three enzymes (E1, E2, and E3) (Fig. 1). There are two E1s in many organisms but multiple genes encoding E2s exist.
Fig. 1.
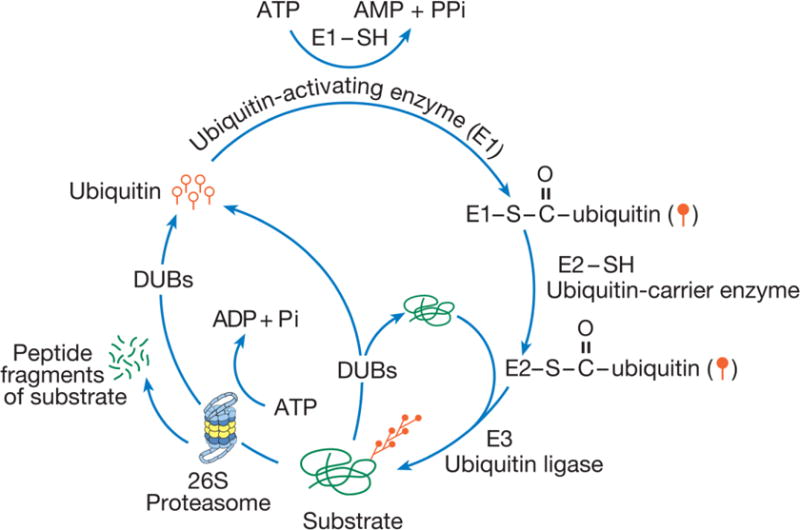
The ubiquitin-proteasome pathway. In this proteolytic pathway, ubiquitin (single ubiquitin molecule is represented by open circles with straight tails) is selectively and covalently attached to the substrate. The enzymatic process of attaching ubiquitin to substrates depends on the action of three different classes of enzymes E1, E2 and E3. First, ubiquitin is activated by E1 to form a ubiquitin-AMP intermediate. Activated ubiquitin (closed circles with straight tails) is passed on to E2 (ubiquitin carrier enzymes). E2s transfers ubiquitin to an E3 (ubiquitin ligase) which ligates the activated ubiquitin to the substrate. To the ubiquitin attached to substrate another ubiquitin is attached and thus through successive linkages of ubiquitin a polyubiquitin chain forms. Polyubiquitinated substrates are degraded by a multi-subunit proteolytic complex called the 26S proteasome in an ATP-dependent reaction. Ubiquitin is not degraded but the polyubiquitin chain is disassembled and ubiquitin is recycled by deubiquitinating enzymes (DUBs). Before being committed to be degraded by the proteasome, ubiquitination is reversible. DUBs can disassemble the polyubiquitin chain if a substrate is ubiquitinated erroneously and prevent the degradation of the substrate.
In the UPP, an E1 activates ubiquitin and passes it onto an E2 which can transfer ubiquitin to the substrates directly or through generation of E3~ubiquitin thioester intermediates. The substrate-specificity of ubiquitin ligation is largely determined by E3s. The first ubiquitin is covalently attached to the e amino group of lysine residues in the substrate. After these enzymes attach the first ubiquitin to the substrate protein, to an internal lysine residue a second ubiquitin is attached and thus several ubiquitin molecules are attached to the growing chain which is termed “polyubiquitin”. Substrates that are destined for degradation by the proteasome carry a specific polyubiquitin linkage. Every successive ubiquitin is attached to the 48th lysine residue in the previous ubiquitin (Glickman and Ciechanover, 2002; Hegde, 2010a). It must be noted, however, that ubiquitin attachment to other ubiquitin molecules could occur through any of the seven lysine residues in ubiquitin. For marking the substrate for ubiquitin-proteasome-mediated degradation, additional ubiquitin are attached to the first ubiquitin at its 11th or 48th Lys residue. Lys-63 linked polyubiquitin chains modulate protein function such as NFκ-B activation (Deng et al., 2000). There are instances when polyubiquitin chains are formed through second ubiquitin linkage to Lys-6, Lys-27, Lys-29 and Lys-33 of the first ubiquitin attached to the substrate are known to occur (Komander, 2009; Ye and Rape, 2009). Polyubiquitin chains contain mixed type of linkage between ubiquitin molecules such as through Lys-11 and Lys-48 in the same chain. Furthermore, ubiquitin itself can be posttranslationally modified through acetylation and phosphorylation (Ohtake et al., 2015; Swaney et al., 2015).
The E3 enzymes that ligate ubiquitin to substrate proteins are the most diverse in the UPP. There are two major classes of E3s: (1) HECT (homologous to E6-AP carboxyl-terminus) domain E3s, (2) RING (really interesting new gene) finger E3s. The RING finger E3s in turn can be divided into two classes SCF (SKP1-cullin-F-Box protein) and APC (Anaphase promoting complex). The specificity of the ubiquitin conjugation reaction, although largely occurs at the E3 ligation step, specific interactions between E2s and E3s and the type of ubiquitin linkage (Lys-48, Lys-63 and so on as described above) all add to the “combinatorial coding” of specificity in the ubiquitin conjugation reaction (Hegde, 2010b).
The protein substrate marked by polyubiquitin attachment is then degraded by the proteasome to small peptides and amino acids (Fig. 1). The polyubiquitin chains are not degraded but disassembled by deubiquitinating enzymes (DUBs) and the free ubiquitin molecules are recycled (Fig. 1). There are two types of DUBs. The category called ubiquitin C-terminal hydrolases (UCHs) is characterized by low molecular weight. The second class is that of high molecular weight DUBs which are called ubiquitin-specific proteases (UBPs or USPs). Apart of structural differences, UCHs and UBPs functionally differ with respect to substrates on which they act (Wilkinson, 2000).
The proteasome that functions to degrade the substrate proteins marked by polyubiquitin chain attachment is called the 26S proteasome based on its sedimentation coefficient during ultracentrifugation. It comprises a cylindrical catalytic 20S core and two regulatory complexes (RC) that are attached to either end of the 20S. The 20S consists of two outer rings with seven a subunits (α1 to α7) in each ring and two inner rings consisting of seven β subunits (β1 to β7). The catalytic activity of the proteasome is conferred by three of the seven β subunits (β1, β2 & β5). The catalytic sites in these β subunits are located at their N-termini which are inside the catalytic cavity which has a narrow opening of 13Ǻ in diameter (Cheng, 2009). Because of this, only an unfolded substrate can enter the catalytic core. It is thought that the unfolding activity is provided by the ATPases that are present in the base of the 19S RC which contains six ATPase subunits Rpt1–Rpt6 (Regulatory particle ATPase 1–6) and four non-ATPase subunits Rpn1, Rpn2, Rpn10 & Rpn13 (Regulatory particle non-ATPases 1, 2, 10 & 13). The 19S RC also consists of the ‘lid’ which includes only non-ATPase subunits (Rpn3, Rpn5, Rpn6–9, Rpn11, Rpn12, & Rpn15) (Hegde, 2010a; Marques et al., 2009).
Among the Rpn subunits, Rpn11 (also called Poh1) and Rpn13 (also called Uch37) are DUBs that are integral part of the 19S RC that assist in deubiquitination of the substrate as it is unfolded and threaded into the catalytic chamber of the 20S core. Another DUB called Usp14 (also known as Ubp6) reversibly associates with the Rpn1 and stimulates substrate degradation through deubiquitination (Leggett et al., 2002; Peth et al., 2009). Two Rpn subunits, Rpn10 (S5) and Rpn13, have a role in recognizing the polyubiquitin chain (Baboshina and Haas, 1996; Husnjak et al., 2008; van Nocker et al., 1996).
In neurons, the proteasome has widespread roles as will be explained later. Although there have not been extensive studies of individual subunits of the proteasome, at least one ATPase subunit, Rpt6, is known to have a role in activity-dependent growth of dendritic spines and the function of Rpt6 is regulated by NMDA receptor (NMDAR)- and CaMKII- mediated phosphorylation (Hamilton et al., 2012).
3. The UPP and long-term synaptic plasticity
Ubiquitin was familiar to researchers as a marker for brain pathology such as neurofibrillary tangles in Alzheimer’s disease and Lewy bodies in Parkinson’s disease (Mori et al. 1987; Lowe et al. 1988) but no physiological or pathological role for ubiquitin in the nervous system was found until the 1990s. The first discovery of degradation by the UPP of a substrate critical for synaptic plasticity in the nervous system was that of regulatory (R) subunits of cAMP-dependent protein kinase (PKA) (Hegde et al., 1993). Since then, several substrates of the ubiquitin-proteasome pathway in the nervous system have been identified (Hegde, 2010a).
3.1. Degradation of R subunits of PKA and proteolytic removal of a CREB repressor
Initial discovery on the role of the UPP in synaptic plasticity came during studies on persistent activation of PKA. Investigations on the biochemical mechanism of long-term facilitation (LTF) (Greenberg et al., 1987) in Aplysia indicated that PKA was persistently activated in the absence of elevated cAMP. LTF underlies behavioral sensitization of defensive reflexes in Aplysia which is a simple form of memory (Abrams, 1985). How is PKA activated in the absence of sustained increase in cAMP? It was found that the R subunits of PKA were decreased without any change in the catalytic (C) subunit during induction of LTF. Because there was no change in mRNA for either the R subunit or the C subunit, the inference was that quantity of R subunits was reduced probably through proteolysis. What is the mechanism of R subunit degradation? Hegde et al (1993) found through a series of biochemical experiments that R subunits were substrates for ubiquitin conjugation and degradation by the proteasome. In addition, it was found that in response to LTF-causing stimuli (such as five pulses of the neurotransmitter 5-HT on to sensory neurons), a UCH (Aplysia ubiquitin C-terminal hydrolase, Ap-uch) that interacts with the proteasome was induced (Fig. 2). Electrophysiological experiments showed that Ap-uch was critical for induction of LTF (Hegde et al., 1997). Subsequently Chain et al. demonstrated that at sensory-motor neuron synapses, injection of lactacystin, a specific proteasome inhibitor blocked induction of LTF (Chain et al., 1999). Since R subunits inhibit the activity of C subunits of PKA, the results suggested that the ubiquitin-proteasome pathway operates to remove inhibitory constraints on formation of long-term memory.
Fig. 2.
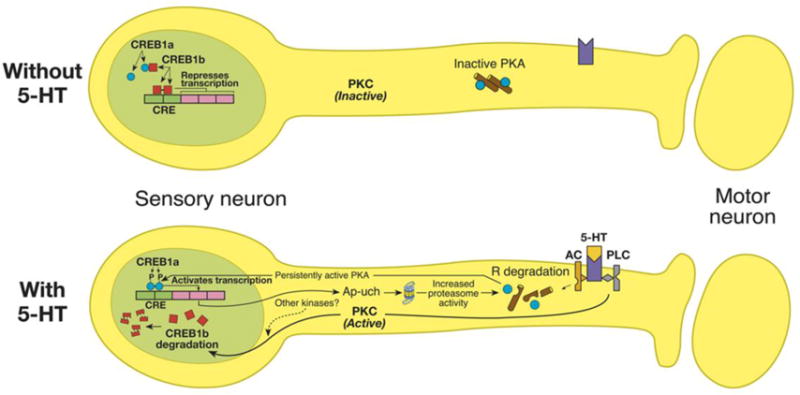
Role of the ubiquitin–proteasome pathway in long-term facilitation in Aplysia. When sensory neurons are stimulated with the neurotransmitter serotonin (5-HT), which induces long-term facilitation, R subunits of protein kinase A (PKA) are ubiquitinated and degraded by the proteasome, making the kinase persistently active. The catalytic subunit of PKA (blue circle) translocates to the nucleus and phosphorylates cAMP response element binding protein 1a (CREB1a), the activator form of CREB. Concomitantly, the repressor form of CREB, CREB1b, is degraded by the ubiquitin–proteasome pathway. Protein kinase C (PKC), which is also activated by 5-HT-mediated signaling, stimulates ubiquitin conjugation to CREB1b and subsequent degradation. AC, adenylyl cyclase; PLC, phospholipase C.
Work on the Aplysia model provided further evidence that the UPP might have a role in degrading proteins that normally inhibit long-term synaptic plasticity. Experimental protocols that induce LTF in Aplysia, cause ubiquitination and degradation of a CREB repressor called CREB1b (Upadhya et al. 2004) (Fig. 2). These observations appear to hold true in vertebrates as well. The mammalian orthologue of CREB1b, ATF4 is degraded by the UPP during induction of long-term synaptic plasticity in the murine hippocampus (Dong et al., 2008).
3.2. Modulation and essential function of a DUB in long-term synaptic plasticity
Subsequent to the finding on ubiquitin-proteasome-mediated degradation of R subunits of PKA, crucial role in LTF for a neuronal specific Ap-uch was discovered. Ap-uch is the homolog of human UCH-L1 and is induced by stimuli that produce LTF but not stimuli that lead to short-term facilitation. Injection of antibodies or antisense oligonucleotides specific to Ap-uch into sensory neurons synapsing onto motor neurons in culture blocked induction of LTF (Hegde et al., 1997). Investigation on biochemical functions of Ap-uch indicated that Ap-uch is capable of cleaving small attachments to linearly attached ubiquitin molecules such as ubiquitin–ubiquitin–cysteine but not large attachments like GST in substrates like ubiquitin–GST. Interestingly, additional biochemical analyses showed that Ap-uch associates with the proteasome. The association of Ap-uch increases the rate of degradation by the proteasome. For example, addition of recombinant Ap-uch to in vitro degradation systems showed that there was approximately a two-fold increase in degradation of R subunit of PKA. Since persistent activation of PKA has been shown to be critical for induction of LTF and R subunits of PKA were found to be substrates for the ubiquitin-proteasome pathway, the experiments on Ap-uch provided some molecular explanation for the role of regulated proteolysis in LTF (Hegde et al., 1997). Computational modeling has provided support for the idea that persistently active PKA induces Ap-uch which in turn provides a positive feedback loop for increasing PKA activity through enhancement of R subunit degradation (Song et al., 2006).
How does Ap-uch enhance the rate of degradation by the proteasome? Using recombinant ubiquitin with its Lysine-48 mutated to Arg that cannot support Lys-48 type of polyubiquitin linkage to protein substrate, it was shown that Ap-uch stimulates the release of ubiquitin from substrates in the presence of the proteasome (Hegde et al., 1997). Ubiquitin with Arg-48 can form single or multiple monoubiquitin linkages on the substrate. Therefore, it can be inferred that Ap-uch perhaps cleaves the first ubiquitin in the polyubiquitin chain attached to the peptide remnant of the substrate. Such a function of Ap-uch has to occur after the DUBs that are tightly associated with the proteasome finish bulk of the polyubiquitin chain disassembly as the unfolding of the substrate and its degradation progresses. Function of UCHs in synaptic plasticity appears to be evolutionarily conserved. It was found that the mammalian counterpart of Ap-uch, Uch-L1 is required for normal synaptic plasticity and memory. The same study also showed a link between UCH-L1 and R subunit degradation in the mouse hippocampus (Gong et al., 2006).
Other studies have expanded the role of Ap-uch and the proteasome in Aplysia to LTD. In Aplysia, sensory-motor neuron synapses undergo transcription-dependent LTD in response to treatment with the neuropeptide Phe-Met-Arg-Phe-NH2 (FMRFa). Application of the proteasome inhibitor lactacystin blocked FMRFa-induced LTD. Also, FMRFa was found to upregulate Ap-uch mRNA (Fioravante et al., 2008). Thus Ap-uch could have a role in LTD in Aplysia perhaps through its action on a different set of downstream targets compared to those affected by Ap-uch during LTF. Subsequent studies indicate that proteasome has a role in mammalian LTD as well. In rat hippocampal neurons, NMDAR-dependent LTD was shown to be independent of proteasome-mediated degradation whereas mGluR-dependent LTD was limited by ubiquitination and proteasome-mediated degradation (Citri et al., 2009). These results, however, contradict earlier studies showing that proteasome inhibition reduced the magnitude of LTD dependent on NMDARs (Colledge et al., 2003) or mGluRs (Hou et al., 2006). The role of the UPP in LTD may be more complex than previously thought. Recently, Sajikumar and colleagues showed that in rat hippocampus, proteasome activity is necessary for protein synthesis-independent early-LTD and inhibition of the proteasome converts early-LTD into protein synthesis-dependent late LTD (Li et al., 2015). In addition, work from another group showed that precise coordination between protein synthesis and proteasome-mediated degradation is essential in regulating induction of mGluR-dependent LTD (Klein et al., 2015).
3.3. The roles of the UPP in histone modification underlying synaptic plasticity
Recent studies indicate that, in addition to modulating transcription factors, the UPP has other roles in regulating transcription. For example, a novel role of the proteasome in modulation of epigenetic histone modifications was described. This study demonstrated that trimethylation of histone 3 at lysine 4 (H3K4me3), acetylation of histone H3 at lysines 9 and 14 (H3K9/14ac), and monoubiquitination of histone H2B at lysine 120 (H2BK120ub) are enhanced immediately after cLTP induction and their enhancement is blocked by β-lactone pretreatment (Bach et al., 2015).
H3K4me3 and H3K9/14ac are transcription-favoring epigenetic modifications previously shown to be important for learning and memory in rodents (Day and Sweatt, 2011; Jarome and Lubin, 2013; Zovkic et al., 2013). The Bach et al study reported that these modifications were dynamic. They showed that both H3K4me3 and H3K9/14ac were upregulated soon after cLTP induction and returned to baseline after 30 min (Bach et al., 2015). The experiments reported earlier described histone modifications that lasted hours or days after synaptic stimulation or behavioral training (Gupta-Agarwal et al., 2012; Levenson et al., 2004). It has also been shown, however, that histone modifications can occur rapidly, in minutes (Buro et al., 2010; Lopez-Atalaya et al., 2013; Riffo-Campos et al., 2015). Some researchers have postulated that lasting cellular changes in synaptic plasticity can be triggered by a transient histone modification signal (Levenson and Sweatt, 2005). Evidence from the Aplysia model indicates that transient acetylation of histone H3 is critical during long-term synaptic plasticity (Guan et al., 2002). Therefore, short-lived proteasome-dependent histone acetylation and methylation may be sufficient to trigger long-lasting upregulation of plasticity-related genes.
The Bach et al study also investigated the role of the transcription-favoring H2BK120ub in synaptic plasticity. This investigation showed that H2BK120ub levels oscillate after the induction of cLTP: an increase in histone H2B monoubiquitination was observed immediately after cLTP induction and at 30 min after cLTP induction, but not at 15 min (Bach et al., 2015). This finding is consistent with previous studies of histone H2B monoubiquitination in yeast transcriptional regulation, where multiple rounds of histone ubiquitination and deubiquitination are required for transcription initiation and elongation, respectively (Minsky et al., 2008; Weake and Workman, 2008; Wyce et al., 2007). Histone H2B mono-ubiquitination has also been described as a precursor to other histone modifications (Gonzalez et al., 2002; Lee et al., 2007). The dynamic nature of global histone modifications in cLTP suggests that the role of histone modifications in synaptic plasticity may be more complex than previously believed.
4. The UPP, short-term plasticity and synaptic transmission: presynaptic and postsynaptic roles
Proteolysis by the UPP regulates key proteins at the synaptic terminals (presynaptic) as well as in the postsynaptic compartment. The UPP has been shown to control synaptic transmission as well as short-term synaptic plasticity.
4.1. Presynaptic roles of the UPP
The UPP, in addition to its role in regulating molecules such as PKA which are critical for long-term synaptic plasticity, also has a function in acute modulation of proteins which affects synaptic transmission and short-term synaptic plasticity. For example, a protein Dunc-13, which is critical in priming the synaptic vesicles, is ubiquitinated and degraded by the proteasome in Drosophila neuromuscular synapse. Administration of proteasome inhibitors and the dominantnegative mutation in a core subunit (β6) of the Drosophila proteasome both lead to an increase in the quantity of Dunc-13 protein in presynaptic terminals. In addition, application of the proteasome inhibitors lactacystin and epoxomycin cause an increase in the excitatory junctional current suggesting that stabilization of Dunc-13 and the resultant increase in the net Dunc-13 quantity leads to increased synaptic transmission (Speese et al., 2003).
The UPP seems to have a wider role in controlling short-term synaptic plasticity and has been found to regulate the amounts of other presynaptic proteins. For example, synaptic vesicle proteins such as syntaxin 1 and RIM1α are degraded by the UPP. Syntaxin 1 is a presynaptic protein that has a role in synaptic vesicle exocytosis. Evidence for ubiquitin-proteasomemediated degradation of syntaxin 1 was obtained through identification of a ubiquitin ligase called staring (syntaxin 1-interacting RING finger protein) using the yeast two-hybrid system (Chin et al., 2002). Co-expression of staring with syntaxin 1 in HeLa cells increases the degradation of syntaxin 1 which can be inhibited by the proteasome inhibitor MG132. The physiological effect of ubiquitin-proteasome-mediated degradation of syntaxin 1 remains to be determined. RIM1α functions to form a presynaptic scaffold that links synaptic vesicle with fusion machinery. A ubiquitin ligase named SCRAPPER (an acronym whose derivation is not clearly defined) has been shown to regulate the amount of RIM1α (Rab3-interacting molecule 1α). Studies using miniature postsynaptic current (mEPSC) measurements established that SCRAPPER regulates synaptic transmission. It was also found that in mice lacking SCRAPPER short-term synaptic plasticity was impaired (Yao et al., 2007).
The proteasome has been shown to function in recycling of synaptic vesicles in hippocampal neurons in primary culture. Proteasome inhibition causes an increase in the size of the recycling pool of vesicles. Blockade of neuronal activity significantly reduces the effect of proteasome inhibition, decreasing vesicle numbers. Inhibition of the proteasome, however, does not increase transmitter release probability. Therefore, it seems that in vertebrate neurons, the proteasome functions to maintain vesicle homeostasis (Willeumier et al., 2006). Results from later experiments add another layer to the complexity of UPP function in neurons. In cultured mammalian hippocampal neurons, proteasome inhibitors increase mEPSC frequency without any effect on the amplitude indicating a presynaptic role for the UPP. Although expected, stabilization of the presynaptic proteins (RIM1 or Munc13) was not observed (Rinetti and Schweizer, 2010). A different study, however, found a decrease in Rim 1 and Munc 13 during persistent presynaptic silencing induced by depolarization (Jiang et al. 2010). The results from these two sets of investigations seem to be at odds with each other even though both used postnatal rat hippocampal neurons in culture and antibodies against Rim 1 and Munc 13 from the same commercial sources. Perhaps the discrepancy was due to the fact that Jiang et al study measured Rim 1 and Munc 13 after K+-induced depolarization whereas Rinetti and Schweizer study tested Rim 1 and Munc 13 levels in relation to changes in mEPSCs and spontaneous EPSCs. Therefore, it is likely that degradation of Rim 1 and Munc 13 is triggered by neuronal depolarization rather than baseline activity.
4.2. Postsynaptic roles of the UPP
Several studies have indicated that the UPP modulates neurotransmitter receptors, structural proteins and regulatory molecules in the postsynaptic compartment. Regulation of the neurotransmitter receptors mainly occurs through ubiquitination that marks proteins for endocytosis which is mainly mediated by attachment of a single ubiquitin (monoubiquitination) or a Lys-63-linked polyubiquitin chain. The ubiquitinated protein that is endocytosed may be recycled back to the plasma membrane if the ubiquitin is removed by DUBs or targeted to the lysosome via the multivesicular body. Some membrane proteins, upon endocytosis are degraded by the proteasome instead of being routed to the lysosome for degradation.
Earlier investigations on Caenorhabditis elegans showed a role for ubiquitin in endocytosis of GLR-1 type of glutamate receptor (Burbea et al., 2002). In mammalian hippocampal neurons, treatment with the proteasome inhibitor MG132 blocks agonist-induced endocytosis of AMPA-type glutamate receptors (Patrick et al., 2003). In addition, NMDA-induced AMPA receptor internalization is prevented by application of the proteasome inhibitor. Later studies showed that AMPA receptor endocytosis and In support of this idea, a postsynaptic density protein PSD-95 was shown to be regulated by ubiquitin-proteasome-mediated degradation (Colledge et al., 2003). PSD-95 is a major component of the postsynaptic scaffold which through interaction with another protein called stargazin provides a docking site for AMPA receptors (Schnell et al., 2002). Proteolytic removal of PSD-95 leads to AMPA receptor internalization and mutations that block PSD-95 ubiquitination block NMDA-induced AMPA receptor endocytosis (Colledge et al., 2003). Furthermore, application of the proteasome inhibitor MG132 to hippocampal slices reduces the magnitude of hippocampal long-term depression (LTD) (Colledge et al., 2003). Because the transient, protein-synthesis independent LTD (Sajikumar and Frey, 2003) requires a net reduction in synaptic AMPA receptors (Malenka and Bear, 2004), these data further support a role for the proteasome in decreasing AMPA receptor amount at synaptic sites. The signal for regulating AMPA receptor internalization and degradation has been investigated in the last few years. Studies showed activity-dependent ubiquitination of the GluA1 subunit in hippocampal neurons. This ubiquitination was mediated by ubiquitin ligase Nedd4–1(neural-precursor cell-expressed developmentally downregulated gene 4–1) (Schwarz et al., 2010). A recent investigation demonstrated that ubiquitination of GluA1 and GluA2 was critical in directing the internalized AMPA receptors to late endosomes and then into lysosomes for degradation (Widagdo et al., 2015). Given that monoubiquitination on one or multiple sites in a protein generally directs the substrates to late endosomes and lysosome, and polyubiquitination directs substrates to the proteasome, how AMPA receptors are degraded may have consequences on synaptic plasticity.
A role for the proteasome in short-term synaptic plasticity has also been obtained through experiments on LTP. A type of LTP called early phase LTP (E-LTP), which is independent of protein synthesis, is enhanced by pre-incubation of hippocampal slices with the proteasome inhibitor β-lactone (Dong et al. 2008). Although the mechanisms by which E-LTP is enhanced by proteasome inhibition have not been elucidated, it is likely that AMPA receptor stabilization and consequent increase in AMPA receptor number at postsynaptic sites might contribute to the increase in E-LTP.
It is likely that the UPP has a broad role in regulating neurotransmitter receptors. NMDARs are retrotranslocated and degraded by the UPP in an activity-dependent fashion. Ubiquitination of the NR1 subunit of NMDARs by an F-box protein called Fbx2 is critical for this process (Kato et al., 2005) suggesting that an SCF-type ligase targets the NR1 subunits for ubiquitination. Subsequent studies showed that another NMDAR subunit NR2B is targeted for ubiquitination by an E3 ligase called Mind bomb-2 in a phosphorylation-dependent manner (Jurd et al., 2008). Endocytosis of other neurotransmitter receptors might be regulated by ubiquitination. Glycine receptor has been shown to be internalized upon ubiquitination (Buttner et al., 2001). A protein Plic-1, which is associated with GABAA receptors, indirectly controls removal of GABAA through endocytosis (Bedford et al., 2001). It was shown that proteasome inhibitors prevent degradation of internalized GABAA receptors. Later studies showed that GABAA receptor ubiquitination is controlled by neuronal activity. Chronic blockade of neuronal activity by tetrodotoxin increases the level of GABAA receptor ubiquitination and increase in neuronal activity decreases GABAA receptor ubiquitination and improves insertion of these receptors into the plasma membrane (Saliba et al., 2007). GABAA receptors are heteropentameric proteins typically consisting of two α subunits, two β subunits and one γ subunit. In the brain, the β subunits of the GABAA receptors are either β2 or β3 (Rudolph and Mohler, 2006). The site of ubiquitination is the β3 subunit of the receptor. Activity blockade reduces the insertion of β3-containing GABAA wild type receptor but not of the receptor containing mutant β3 that cannot be ubiquitinated (Saliba et al., 2007).
The UPP also degrades several other proteins in addition to PSD-95 in the postsynaptic density including several structural proteins. For example, Shank, GKAP and AKAP79/150 are degraded through the ubiquitin-proteasome pathway. Unlike for the degradation of PSD-95, physiological relevance of proteolytic removal of Shank, GKAP and AKAP79/150 is not clear because the studies were correlative and a direct link between ubiquitin-proteasome-mediated degradation of the PSD proteins and structural remodeling was not established (Ehlers, 2003).
There is also evidence that the UPP controls a protein that regulates spine shape. SPAR controls dendritic spine shape by reorganizing the actin cytoskeleton. During activity-dependent remodeling of synapses, SPAR was shown to be degraded by the ubiquitin-proteasome pathway. Degradation of SPAR is stimulated by serum inducible kinase (SNK). Activity induces SNK mRNA in the cell body and the induced SNK is targeted to the dendritic spines. Because of the time required for SNK mRNA to travel to the spines, the conjecture is that SPAR may function to oppose synaptic remodeling after elevated activity (Pak and Sheng, 2003).
5. Local proteolysis and synaptic plasticity: Roles for spatial control of proteasome-mediated degradation
I previously proposed a role for local ubiquitin-proteasome-mediated degradation in synaptic plasticity (Hegde, 2004). Many studies carried out since then provide support to this idea. Others working in this field are embracing the idea of local degradation as well (Segref and Hoppe, 2009). It appears that local, regulated degradation of substrate proteins plays an important role in synaptic plasticity as well as many other aspects of the nervous system such as development and fine-tuning of synaptic connections. Spatially restricted degradation can achieve synapse-specific effects. Cell-wide degradation would have consequences on all synapses made by a given neuron (Hegde, 2004).
How might local protein degradation be achieved in neurons? A simple way would be to restrict the substrate to a subcellular location. For example, proteins whose expression is largely restricted to the synapses could be locally degraded because all the requisite components of the UPP are present at the synapse. Also substrates can be made vulnerable (or resistant) to ubiquitination by phosphorylation or glycosylation which can be locally controlled in neurons. For example, NR2B subunit of NMDAR is phosphorylated by Fyn tyrosine kinase and made susceptible for ubiquitination by the E3 ligase Mind Bomb which is localized to the apical dendrites (Jurd et al., 2008). A transcription factor critical for LTF in Aplysia called C/EBP is made resistant to ubiquitin-proteasome-mediated degradation upon phosphorylation by MAP kinase (Yamamoto et al., 1999). Other posttranslational modifications of substrates add another level to the regulation of substrate ubiquitination. For example, attachment of O-linked N-acetylglucosamine (O-GlcNAc) is known regulate phosphorylation of substrates and their consequent ubiquitination (Guinez et al., 2008; Ruan et al., 2013).
Activation (or inactivation) of ubiquitin ligases by phosphorylation or other posttranslational modifications can be locally controlled as well. Phosphorylation can either positively or negatively regulate E3 ligase activity (Im and Chung, 2015; Stacey et al., 2012). Linkage to O-GlcNAc of an E3 has been reported to occur which can potentially modify ligase activity (Zaro et al., 2011). Moreover, specific E3 ligases can also be sequestered to specific cellular compartments. The removal of attached ubiquitin by DUBs has been found to be locally regulated as well. Experimental evidence has been obtained for local regulation of E3s as well as that of DUBs, mainly from work on neuronal development in Drosophila (Hegde, 2010b). Accumulating evidence indicates that proteasome activity is also differentially regulated in different neuronal compartments, which is the main focus of discussion here.
5.1. Local roles of the proteasome in synaptic plasticity
Modulation of the proteasome adds another level to regulation of proteolysis by the UPP. Even though it was not previously appreciated, the data from the last several years indicate that proteasome is not homogenous throughout the neuron. Hints for local functions of the proteasome initially came from work on LTF in Aplysia. Later studies on hippocampal late phase of long-term potentiation (L-LTP) provided strong evidence for local roles of the proteasome in long-term synaptic plasticity.
5.1.1. Local roles of the proteasome in LTF in Aplysia
The impetus for closely looking at the regulation of the neuronal proteasome came from conflicting results obtained with proteasome inhibitors on LTF in Aplysia. Originally, it was found that proteasome inhibitors block induction of LTF (Chain et al., 1999). Later studies on LTF, however, showed that bath application of the active form of lactacystin, clasto-lactacystin β-lactone, to sensory-motor neuron synapses resulted in enhanced LTF and an increase in neurite outgrowth in isolated sensory neuron (Zhao et al., 2003). The increase in neurite elongation is consistent with results obtained in PC12 and Neuro2A cells in which lactacystin induces neurite outgrowth (Fenteany et al., 1994). Both sets of results can be reconciled if one postulates that proteasome has different roles in different cellular compartments (Hegde, 2004). In the same neuron, the proteasome is likely to carry out different tasks in different subcellular compartments resulting in different physiological consequences at different loci. Therefore, blocking different roles of the proteasome during induction of memory would lead to distinct and even opposite effects on synaptic strength. For example, the proteasome is known to degrade transcription repressors. Degradation of transcription repressors should allow transcription activators to induce gene expression which in turn leads to development of LTF. If the proteasome is inhibited only in the nucleus before the repressors are degraded, gene expression and hence induction of LTF should be blocked. Degradation of the CREB repressor CREB1b by the UPP in response to LTF-inducing protocols (Fig. 2) (Upadhya et al., 2004) supports this idea. On the other hand, if the degradation of proteins needed at the synapse for developing LTF is inhibited by the proteasome, LTF should be enhanced. As previously proposed, the main purpose of transcription during induction of LTF or other forms of long-term memory is to provide mRNAs for synthesis of ‘rapidly turning over proteins’ needed for memory formation (Hegde, 2004). If the degradation of these proteins is prevented, then long-term memory formation becomes independent of transcription. In support of this idea, Zhao et al found that proteasome inhibitor-induced synaptic strengthening depends on translation but not transcription (Zhao et al., 2003).
The biochemical experiments on the proteasome also support differential function of the proteasome in different neuronal compartments. The results of these experiments showed that both in Aplysia nervous system and mouse brain, proteasome activity in the synaptic terminals is significantly higher than that of the nuclear proteasome. Moreover, the proteasome activity in the two compartments is differentially regulated by protein kinases relevant to synaptic plasticity such as PKA, PKC, and MAP kinase (Upadhya et al., 2006). Later, others found that CaMKII can stimulate proteasome activity in cultured hippocampal neurons (Djakovic et al., 2009).
5.1.2. Local regulation of the proteasome in L-LTP in murine hippocampus
As discussed above, differential activity of the proteasome in Aplysia might explain conflicting results obtained in different studies. Does differential proteasomal activity affect synaptic plasticity differentially in vertebrates? It has been found that the proteasome has differential roles during induction and maintenance phases of hippocampal late phase of long-term potentiation (L-LTP) (Dong et al., 2008).
Evidence for functional significance of local roles of the proteasome came from studies on hippocampal late phase LTP (L-LTP). Investigations by Dong et al showed that the proteasome inhibitor application to hippocampal slices prior to induction of L-LTP caused an increase in the magnitude of the early, induction phase but an inhibition of the late, maintenance phase (Dong et al., 2008). What is the basis of these differential effects of the proteasome on phases of L-LTP? The enhancement of the early, induction phase (This early part of L-LTP is referred to as Ep-L-LTP for convenience) by the proteasome inhibitor β-lactone is blocked by prior application of the translation inhibitor anisomycin but not by a transcription inhibitor actinomycin D. The increase in Ep-L-LTP caused by β-lactone is also prevented by prior application of rapamycin which blocks signaling that controls translation of a subset of mRNAs (Gingras et al., 2001). Moreover, Ep-L-LTP is augmented in dendrites isolated from the cell body by means of a surgical cut. These lines of evidence suggest that proteasome inhibition enhances Ep-L-LTP by stabilizing proteins locally translated from pre-existing mRNAs (Dong et al., 2008) (Fig. 3 top).
Fig. 3.
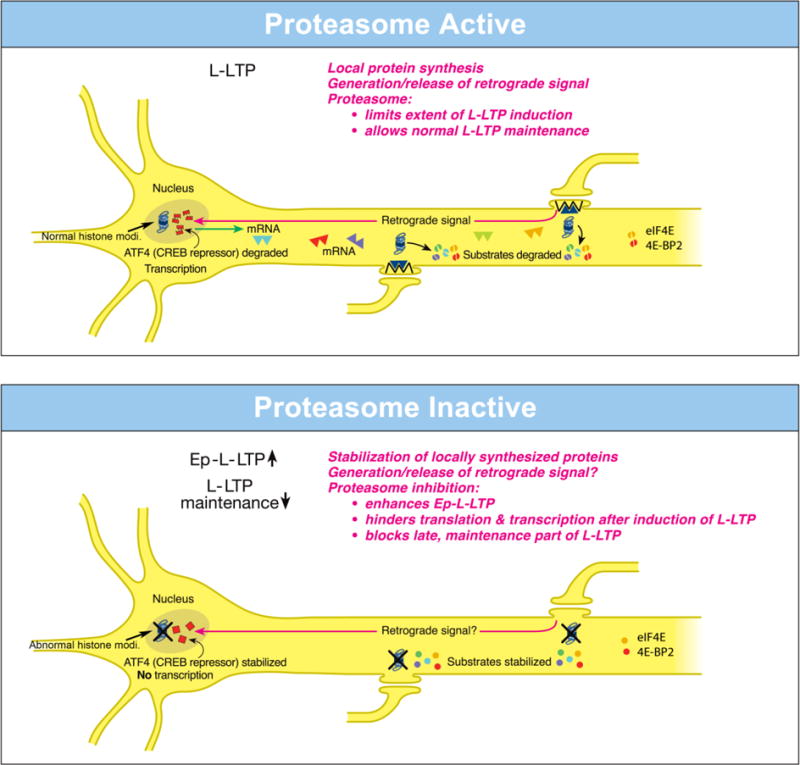
Dissimilar local roles of the proteasome in dendrites and in the nucleus during L-LTP.
(Top)-Proteasome Active: The proteasome in dendrites is highly active, translational activators such as eIF4E are degraded (broken green spheres) and protein substrates that positively regulate L-LTP are degraded (broken spheres). Therefore extent of L-LTP is limited and only normal L-LTP ensues. A retrograde signal is likely transmitted to the nucleus. Proteasome aids transcription of genes by degrading the CREB repressor ATF4 (broken squares in the nucleus) thus allowing for normal L-LTP maintenance. The proteasome also enables histone modifications (Normal histone modi.). Transcribed mRNAs (triangles) travel to activated synapses.
(Bottom)-Proteasome Inactive: When the proteasome is inhibited (indicated by X marks on the proteasome), translational activators are stabilized (intact green spheres) leading to increased protein synthesis in dendrites. Also the newly synthesized proteins in dendrites are stabilized (intact spheres) and L-LTP-inducing stimulation protocols dramatically increase (upward arrow) the early part of L-LTP (Ep-L-LTP). Proteasome inhibition obstructs CREB-mediated transcription by preventing the degradation of transcription repressor ATF4 (intact squares in the nucleus). Proteasome inhibition could inhibit the generation of the retrograde signal as well. Therefore, L-LTP is not maintained but decays (downward arrow). Proteasome inhibition also perturbs histone modifications (Abnormal histone modi.). In addition, proteasome inhibition causes failure of sustained translation because of stabilization of translation repressors such as 4E-BP (intact red spheres) which accumulate after induction of L-LTP thus contributing to blockade of L-LTP maintenance [Modified from (Hegde et al., 2014)].
How does proteasome inhibition block maintenance of L-LTP? The proteasome inhibitor β-lactone blocks maintenance of L-LTP only if applied prior to induction of L-LTP but not if applied 2 h after induction of L-LTP. Previous studies by others have established that the critical time window for transcription required for maintenance of L-LTP is 2 h (Nguyen et al., 1994). These results suggest that proteasome inhibition blocks maintenance of L-LTP by inhibiting transcription. Additional molecular evidence supports this notion. Application of β-lactone to hippocampal slices significantly reduced induction of BDNF (Brain-derived neurotrophic factor) mRNA by chemically induced LTP (cLTP) or L-LTP induced by a theta-burst protocol (Dong et al., 2008). BDNF is a CREB-inducible gene linked to maintenance of L-LTP (Barco et al., 2005).
What is the mechanism of transcription blockade caused by inhibition of the proteasome? One possibility is that normally the UPP aids the degradation of transcription repressors. Hence proteasome inhibition would result in accumulation of these repressors thus blocking transcription. In support of this idea, it was found that a CREB repressor ATF4 is degraded by the UPP during cLTP and β-lactone application to hippocampal slices prevents degradation of ATF4. Furthermore, ATF4-ubiquitin conjugates accumulate during cLTP when proteasome is inhibited (Dong et al., 2008) (Fig. 3 bottom).
These studies have also revealed the changing role of the proteasome even in dendrites through progression of L-LTP. Application of β-lactone to isolated dendrites also blocks maintenance of the dendritic L-LTP (Dong et al., 2008). Under these conditions, there is no supply of newly transcribed mRNA from the cell body. Thus blockade of transcription by proteasome inhibition does not explain this phenomenon. The most likely possibility is that proteasome inhibition leads to a slow accumulation of translation repressors in dendrites. Buildup of translation repressors would also occur in the cell body which would hinder translation of newly transcribed mRNAs. Thus late stages of translation in both dendrites and the cell body would be blocked by stabilization of translation repressors by proteasome inhibition. In support of this idea, confocal microscopy experiments at various time points after L-LTP induction showed that proteasome inhibition causes accumulation of translational activators eukaryotic initiation factors 4E (eIF4E) and eukaryotic elongation factor 1A (eEF1A) early during L-LTP (Dong et al., 2014). Translational repressors such as polyadenylate-binding protein interacting protein 2 (Paip2) and eukaryotic initiation factor 4E-binding protein 2 (4E-BP2) buildup at later stages of L-LTP in response to proteasome inhibition (Dong et al., 2014). Other negative regulators of translational repressors such as Mov10 might be stabilized by proteasome inhibition as well. For example, in cultured hippocampal neurons Mov10, which inhibits translation of key plasticity-related mRNAs such as that of CaMKIIα, is degraded by the proteasome in an NMDA- and activity- dependent manner (Banerjee et al., 2009). The requirement for coordination between protein synthesis and proteasome-mediated degradation has also been reported for induction of mGluR-dependent LTD (Klein et al., 2015).
Other studies have investigated the effect of proteasome inhibition on LTP. Although these studies reported reduction in magnitude of LTP by proteasome inhibitors, they failed to discover differential dendritic and nuclear functions of the proteasome in LTP perhaps because one study used MG-132 (Karpova et al., 2006) which is not a highly specific proteasome inhibitor (Chain et al., 1999; Tang and Leppla, 1999) and the other used proteasome inhibitors lactacystin and epoxomycin at nanomolar concentration (Fonseca et al., 2006) which is significantly lower than the effective concentration (micromolar) required to block proteasome activity.
5.1.3. Evidence for local roles of the proteasome in other model systems
Evidence from other studies using cultured rat hippocampal neurons showed dynamic local regulation of the proteasome at the dendrites. It was found that proteasome is redistributed from dendritic shafts to synaptic spines in an NMDAR-dependent manner. How does the redistribution of the proteasome occur? The experiments showed that activity only modestly increased the entry of the proteasome into dendritic shafts but significantly reduced their exit. Furthermore, the results suggested that the proteasome was sequestered persistently in the spines through association with cytoskeleton (Bingol and Schuman, 2006). Subsequent studies showed that a protein called NAC1, which is induced by psychostimulants, modulates the recruitment of the proteasome into the dendritic spines (Shen et al., 2007). Since the bulk of the evidence in this study is for the catalytic 20S core of the proteasome, it remains to be seen whether the recruitment of the full proteasome complex (26S) that degrades polyubiquitinated proteins is also regulated by NAC1. Another study has suggested that CaMKIIα subunit acts as a scaffold for the proteasome (Bingol et al., 2010). It is not clear how or if the functions of NAC1 and CaMKIIα relate to each other in sequestering the proteasome.
Proteasome might function to locally regulate other processes required for synaptic plasticity such as translation of mRNA. For example, fragile X mental retardation protein (FMRP), which is thought to regulate translation of a subset of mRNAs in dendrites, is regulated by the proteasome. Furthermore, regulation of FMRP by the proteasome appears to be critical for metabotropic glutamate receptor-dependent LTD (Hou et al., 2006).
6. The UPP and memory
Considering the role of the UPP in synaptic plasticity, it can be expected that the UPP would have a role in memory. Accordingly, experimental evidence has been accumulating in support for roles of the UPP in various stages memory including induction, consolidation and reconsolidation. Initial results were obtained from experiments on the rat hippocampus. Lopez-Salon and co-workers demonstrated that bilateral infusion of lactacystin to the CA1 region of the rat hippocampus caused total retrograde amnesia for a one-trial avoidance learning. They also showed that total ubiquitination increases in the hippocampus 4 h after the training (Lopez-Salon et al., 2001). These results are consistent with the idea that a decrease in some critical inhibitory proteins during long-term memory formation (Abel et al., 1998) is mediated by the ubiquitin-proteasome pathway.
Later studies on vertebrates suggest that the UPP may have much broader and more complex roles than just degrading the inhibitory constraints on long-term synaptic plasticity and memory such as R subunits and the CREB repressor. For example, infusion of the proteasome inhibitor β-lactone into the CA1 region of the hippocampus prevents extinction of contextual fear memory (Lee et al., 2008). Also, using infusion of lactacystin into the CA3 region of the hippocampus it was shown that protein degradation is important for consolidation as well as reconsolidation of spatial memory (Artinian et al., 2008). The mechanistic details as to how the UPP contributes to the extinction of fear memory or reconsolidation of spatial memory are not clearly understood.
In the past few years, there has been a resurgence of interest in the connection between the UPP and memory. Several studies have examined the role of the UPP in memory. Helmstetter and colleagues showed that infusion of a specific proteasome inhibitor β-lactone into amygdala of rats blocks fear memory. These investigators also showed a global increase K-48-linked polyubiquitination of proteins in response to NMDAR stimulation suggesting that protein degradation is required (Jarome et al., 2011). These researchers, in a different study, demonstrated the importance of protein degradation by the UPP in the prefrontal cortex in development of trace fear memory (Reis et al., 2013). Subsequently they also found that during development of long-term fear memory, Serine-120 phosphorylation of an ATPase subunit of the proteasome namely Rpt-1 occurs. They also found increase in 20S proteasome activity (Jarome et al., 2013). Given that Rpt1 is a subunit in the 19S RC, it is not clear how Rpt-1 phosphorylation leads to enhancement of 20S proteasome activity. One possibility is allosteric regulation of the 20S by the 19S RC although this is yet to be tested. Earlier, others had established that Rpt-1 phosphorylation decreases the amplitude of miniature postsynaptic currents in cultured rat hippocampal neurons and suggested that Rpt-1 phosphorylation may be important for homeostatic synaptic plasticity (Djakovic et al., 2012).
There have been other studies on the role of the UPP both in vertebrates and invertebrates. For example, in the conditioned taste aversion model, long-term memory is impaired by infusion of the proteasome inhibitor lactacystin into amygdala and the insular cortex (Rodriguez-Ortiz et al., 2011). Others infused protein synthesis inhibitors anisomycin and proteasome inhibitor β-lactone into the CA1 region of the hippocampus of rats and tested for memory using novel object recognition task. This study showed that the proteasome inhibitor did not have an effect on memory consolidation and reconsolidation but was able to reverse the impairment of these processes caused by inhibition of protein synthesis (Furini et al., 2015). Other investigations have implicated a role for a deubiquitinating enzyme USP14 (Jarome et al., 2014) and the ubiquitin ligase APC (Kuczera et al., 2011) in memory in vertebrates. A study carried out on honey bees showed that drugs that inhibit the proteasome enhance memory formation (Felsenberg et al., 2014) which is an opposite result compared to that obtained in vertebrate model systems. This may be a case of the drugs affecting proteasome in one part of the circuitry more than the others (i.e. local effects). Given the complex nature of the role of the UPP in synaptic plasticity and memory, only deep mechanistic investigations can resolve such discrepancies.
How might the UPP regulate memory consolidation? One possibility is that the UPP regulates gene expression critical for memory. For example, ubiquitin-proteasome-mediated degradation of transcription repressors might facilitate initiation of transcription. For example, gene expression pathway mediated by CREB requires the removal of repressors. In Aplysia a CREB repressor called CREB1b is degraded by the UPP during LTF (Upadhya et al., 2004) as described elsewhere in this review. A second CREB repressor called CREB2 is an important negative regulator of LTF (Bartsch et al., 1995). The mammalian counterpart of CREB2 called ATF4 is polyubiquitinated and is degraded by the proteasome during chemically induced long-lasting LTP in the mouse hippocampus (Dong et al., 2008). A repressor of another transcriptional pathway mediated by NF-κB called IκB is degraded by the proteasome (Yaron et al., 1998). Although transcription by the NF-κB pathway has been implicated in some forms of memory (Meffert et al., 2003), its contribution to memory-forming gene expression relative to the CREB pathway remains unclear.
The UPP might have other roles in memory such as reconsolidation. It has been argued that protein degradation by the UPP is necessary for making memories labile before the reconsolidation process occurs (Sol Fustinana et al., 2014). Although this argument makes intuitive sense, much remains to be learned about how proteolysis relates to the lability of memory. Progress in this regard is beginning to be made. A recent study showed that phosphorylation of Rpt6 by the proteasome by CaMKII is critical for memory destabilization after retrieval (Jarome et al., 2016). To advance towards a thorough mechanistic understanding of the role of the UPP in memory, additional studies pursuing specific substrates and integrating molecular, electrophysiological and behavioral approaches in the same experimental model system would be necessary.
7. The UPP and diseases of the synapse
Apart from the numerous roles of in normal synaptic function, the UPP has also been linked to synaptic malfunction observed in many diseases and disorders of the brain. Deficiencies in the UPP are believed to play some role in development of Alzheimer’s disease (AD) (Gentier and van Leeuwen, 2015; Upadhya and Hegde, 2007), Parkinson’s disease (PD) (Atkin and Paulson, 2014) and Huntington’s disease (HD) (Ortega and Lucas, 2014). Because of its role in synaptic plasticity the UPP may also play a role in synaptic defects underlying cognitive impairment observed in these diseases.
Deficits in synaptic plasticity and its association with the UPP is better understood in AD compared to PD and HD. Cognitive defects observed early in AD likely occur because of synaptic failure (Selkoe 2002). In mouse models of AD, deficits in LTP and memory are known to occur and have been shown to correlate well with accumulation of Aβ (Hsiao et al. 1996). Ubiquitin immunoreactivity is found in plaques and tangles of AD brains. Blockade of the UPP in the neurons of AD brains might be responsible for accumulation of ubiquitinated proteins (Upadhya and Hegde, 2005; 2007). Although how the UPP connects to AD pathology and cognitive impairment is not understood, some hints regarding the role of the UPP in AD have been discovered. For instance, application of oligomeric Aβ inhibits LTP which can be rescued by treatment with exogenous Uch-L1 (mammalian homolog of Ap-uch). In AD model mice carrying amyloid precursor protein and presenilin1 transgenes, deficits in LTP and memory can also be rescued by treatment with exogenous Uch-L1(Gong et al., 2006).
Evidence for directly connecting the UPP to pathogenesis of AD came from the observation that the brains of some AD patients contained an aberrant form of ubiquitin that has 20 additional amino acids at its C-terminus (UBB+1) (van Leeuwen et al., 1998). Postnatal expression of UBB+1 in neurons of transgenic mice showed proteasome dysfunction and deficits in contextual memory (Fischer et al., 2009). Given that proteasome inhibition throughout the neuron blocks maintenance of L-LTP because it hinders transcription and sustained translation (Dong et al., 2008), it is interesting to speculate that the memory deficits in the UBB+1 mice result from impaired synaptic plasticity owing to neuron-wide proteasome dysfunction.
Another type link between the UPP and neurodegeneration has also been reported. It was shown that another cellular degradative process, autophagy, utilizes an enzymatic pathway similar to ubiquitin conjugation which attaches ubiquitin-like proteins such as Atg12 to some proteins that regulate the autophagic process (Nakatogawa et al., 2009). In autophagy, a double membrane vesicle (called the autophagosome) engulfs parts of the cytoplasm or organelles and delivers it to the lysosome. Ubiquitination has also been found to be linked to autophagy. In SH-SY5Y cells, Lys-63-linked polyubiquitination promotes inclusion bodies which are cleared by autophagy (Tan et al., 2008). In addition, it was observed that parkin, which is an E3 ligase, promotes Lys-63-linked polyubiquitin chain attachment to misfolded proteins. Lys-63-polyubiquitin chain seems to serve as a signal to couple the misfolded proteins to dynein motor complex through histone deacetylase 6 (which serves as an adaptor) and thus aiding in sequestration of misfolded proteins into specialized inclusion bodies called aggresomes which are cleared by autophagy (Olzmann and Chin, 2008). A subsequent study found that autophagy promotes synapse development in Drosophila (Shen and Ganetzky, 2009). The UPP and autophagy might work in concert to remove aggregated proteins observed in many neurodegenerative disease (Ciechanover and Kwon, 2015). It remains to be seen whether autophagy is connected to synaptic malfunction observed in neurodegenerative diseases.
8. Other proteolytic pathways that play a role in synaptic plasticity and memory
In addition to the UPP, two other cellular proteolytic mechanisms are known to play a role in synaptic plasticity and memory: the lysosomal pathway and autophagy. It must be noted that both of these degradation mechanisms do have connection to the UPP.
8.1. Roles of the lysosome in synaptic plasticity and memory
A major way in which the lysosome participates in synaptic plasticity and memory is through control of the number of neurotransmitter receptors on the plasma membrane. As described elsewhere in this article, the receptors that undergo endocytosis through ubiquitin signaling can either be recycled back to the plasma membrane or routed to the lysosome for degradation. Therefore, any effect on neurotransmitter receptors such as glutamate receptors or GABA receptors would affect synaptic strength and plasticity. A study carried out several years ago showed that in cultured rat hippocampal neurons, kainate receptors (KRs) undergo endocytosis in response to stimulation by kainate (Martin and Henley, 2004). The internalization of KRs in response to kainate stimulation is Ca2+- and PKC- dependent and the internalized KRs are targeted to lysosomes for degradation. Activation of NMDARs, however, results in Ca2+- and PKA- dependent endocytosis of KRs and recycling of the KRs back to the plasma membrane (Martin and Henley, 2004).
There have been some mechanistic studies using stimulation of cultured rat neurons NMDA and other reagents. It was found that stimulation of NMDARs caused dephosphorylation of a DUB called Usp8. This leads to recycling of the AMPARs that have been endocytosed as a result of ubiquitination by the ligase Nedd4–1. The same study also showed that increased synaptic activity through prolonged bicuculline treatment reduced Usp8 levels and caused an increase in the recruitment of Nedd4–1 at synapses and consequent enhancement in ubiquitination of AMPARs and trafficking of the endocytosed receptors to the lysosome for degradation (Scudder et al., 2014).
The number of inhibitory neurotransmitter receptors such as GABAA receptors at synaptic sites is controlled by lysosomal degradation as well. This was demonstrated using behavioral adaptation in the worm C. elegans to acute exposure of GABAA agonist muscimol (Davis et al., 2010). Initially the worms are paralyzed because of hyperpolarization of postsynaptic cells. After several hours of exposure to muscimol the worms adapt. Using electrophysiological recordings and visualization through immunofluorescence it was shown that during adaptation GABAA receptors are selectively removed from synaptic sites and are routed to the lysosome for degradation. The role of lysosome in GABAA receptor degradation was established based on the fact that the mutant worms with a defect in lysosomal function have elevated levels of synaptic GABAA receptors (Davis et al., 2010).
The role of the lysosome in memory has not been investigated much. There is a study in Drosophila, however, showing a link between lysosomal degradation and long-term olfactory memory. A genetic screen to identify new genes with a role in memory revealed that a gene called debra is linked to long-term memory (Kottler et al., 2011). Previous results had established that debra protein functions to induce polyubiquitination of a protein called Ci and directs it to the lysosome for degradation (Dai et al., 2003). The exact role of debra in lysosomal trafficking of Ci is not clear because debra in neither a ligase not does it have any homology to any other enzymes of the UPP. It has been suggested that Ci ubiquitination is mediated by Slimb, a protein containing F-box/WD40 repeats. The vertebrate homolog of Slimb is βTrCP which is part of an SCF ligase. Dai et al suggested that debra possibly binds to the ligase complex that ubiquitinates Ci (Dai et al., 2003). Even though these results provide a hint for possible function of debra in the Drosophila mushroom bodies in mediating long-term memory, much remains to be learned regarding the molecular mechanisms by which debra contributes to long-term memory.
8.2 Roles of autophagy in synaptic plasticity and memory
There are three main types of autophagy: macroautophagy, microautophagy and chaperone-mediated autophagy. Macroautophagy is an elaborate process comprising the formation of a double-membraned structure called the autophagosome which can fuse with the lysosome. In microautophgy, the lysosome directly takes up cytosolic components through invagination of its membrane. Chaperone-mediated autophagy denotes translocation of unfolded proteins across the lysosomal membrane with the aid of chaperones such as heat shock proteins. The focus in this article is on macroautophagy (henceforth referred to as “autophagy”) which requires the action of many proteins some of which are functionally akin to the enzymes of the UPP. The molecules that regulate autophagy (Atg proteins) were originally discovered in yeast (Mizushima et al., 2011). Since then, orthologues of Atg proteins have been discovered in other organisms.
Although there have not been many systematic investigations on the roles of autophagy in synaptic plasticity, some studies indicate a role for autophagy in phenomena such as LTP and LTD. Investigations on streptozotocin-induced diabetes in mice showed that LTP impairment in diabetic mice was exacerbated in the presence of an inhibitor of autophagy called 3-methyl adenine (Li et al., 2016). In the context of chemically induced LTD, it has been shown that neuronal stimulation induces autophagy (Shehata et al., 2012). When chemical LTD is induced by low doses of NMDA, a marker of autophagosome formation called light chain protein 3-II (LC 3-II) increases along with the number of auophagosomes. The increase in LC 3-II is coincides with dephosphorylation of Akt and mammalian target of rapamycin and degradation of GluR1, an AMPA receptor subunit. RNA interference-mediated knockdown of ATG7, an enzyme which is like E1 of the UPP and is critical for formation of auophagosomes, blocks autophagy induced by chemical LTD (Shehata et al., 2012).
There have been some studies on relating autophagy to memory. A CNS-specific knockout of the Wdr45 gene (one of the orthologues of yeast Atg18), whose protein product has an essential function in autophagosome formation, leads to memory impairment as judged by performance in Morris water maze and contextual fear conditioning. Mutations in the human WDR45 cause a type of neurodegeneration called β-propeller associated neurodegeneration which is characterized by cognitive impairment (Haack et al., 2012; Hayflick et al., 2013; Saitsu et al., 2013). Therefore, it is likely that autophagy has a role in memory formation in humans as well. In the study on diabetic mice, it was found that spatial memory was not impaired but spatial reversal memory was and the memory impairment was exacerbated by inhibition of autophagy (Li et al., 2016). Even though these results are suggestive of the link between autophagy and memory, the caveat is that the studies were conducted in the context of diabetes. Definitive conclusion on the role of autophagy in memory will have to await CNS-specific and conditional suppression of molecules critical for autophagy.
The connection between autophagy and memory was observed in another model organism, Drosophila. It was found that spermidine ameliorates age-related memory impairment and this beneficial effect on memory requires autophagy (Gupta et al., 2013). Spermidine is a naturally occurring polyamine with pleiotropic effects on cells. Previous research showed that spermidine promotes longevity in yeast cells and one of the mediators of spermidine’s effect is autophagy (Eisenberg et al., 2009). Although these results suggest a role for autophagy in memory and the mechanisms are likely to be evolutionarily conserved, it would be necessary to test the effects of compounds such as spermidine in vertebrate model systems of memory.
9. Looking ahead
Despite numerous investigation into the roles of ubiquitin-proteasome-mediated proteolysis in synaptic plasticity and memory, there are many open questions. An important area of for future investigation is likely to be elucidation of mechanisms that determine the spatial and temporal regulation of proteolysis in the nervous system. Although many substrates in the nervous system have been identified, roles for many more are likely to be revealed. Function of the UPP in wiring and fine-tuning the nervous system has not been investigated much and needs to be explored. Studies on the roles of the UPP in memory would benefit from genetic manipulations such as gene editing. Connections between the UPP and neurodegenerative diseases will also likely to be a fruitful area of research because of the potential for therapeutic development.
HIGLIGHTS.
Proteolysis plays a role in changing synaptic strength.
Ubiquitin-proteasome pathway has a major role in synaptic plasticity and memory.
Local regulation of proteolysis is critical for synaptic plasticity.
Protein degradation in neurons also occurs through the lysosome and autophagy.
Dysregulation of proteolysis could lead to diseases and disorders of the brain.
Acknowledgments
The research in the author’s laboratory was supported by a grant from National Institute of Neurological Disease and Stroke (NINDS) (NS066583) and startup funds from Georgia College and State University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abel T, Martin KC, Bartsch D, Kandel ER. Memory suppressor genes: inhibitory constraints on the storage of long-term memory. Science. 1998;279:338–341. doi: 10.1126/science.279.5349.338. [DOI] [PubMed] [Google Scholar]
- Abrams TW. Activity-dependent presynaptic facilitation: an associative mechanism in Aplysia. Cell Mol Neurobiol. 1985;5:123–145. doi: 10.1007/BF00711089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artinian J, McGauran AM, De J X, Mouledous L, Frances B, Roullet P. Protein degradation, as with protein synthesis, is required during not only long-term spatial memory consolidation but also reconsolidation. European Journal of Neuroscience. 2008;27:3009–3019. doi: 10.1111/j.1460-9568.2008.06262.x. [DOI] [PubMed] [Google Scholar]
- Atkin G, Paulson H. Ubiquitin pathways in neurodegenerative disease. Front Mol Neurosci. 2014;7:63. doi: 10.3389/fnmol.2014.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baboshina OV, Haas AL. Novel multiubiquitin chain linkages catalyzed by the conjugating enzymes E2EPF and RAD6 are recognized by 26 S proteasome subunit 5. Journal of Biological Chemistry. 1996;271:2823–2831. doi: 10.1074/jbc.271.5.2823. [DOI] [PubMed] [Google Scholar]
- Bach SV, Tacon PR, Morgan JW, Hegde AN. Proteasome regulates transcription-favoring histone methylation, acetylation and ubiquitination in long-term synaptic plasticity. Neuroscience Letters. 2015;591:59–64. doi: 10.1016/j.neulet.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Patterson S, Alarcon JM, Gromova P, Mata-Roig M, Morozov A, Kandel ER. Gene expression profiling of facilitated L-LTP in VP16-CREB mice reveals that BDNF is critical for the maintenance of LTP and its synaptic capture. Neuron. 2005;48:123–137. doi: 10.1016/j.neuron.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Bartsch D, Ghirardi M, Skehel PA, Karl KA, Herder SP, Chen M, Bailey CH, Kandel ER. Aplysia CREB2 represses long-term facilitation: relief of repression converts transient facilitation into long-term functional and structural change. Cell. 1995;83:979–992. doi: 10.1016/0092-8674(95)90213-9. [DOI] [PubMed] [Google Scholar]
- Bedford FK, Kittler JT, Muller E, Thomas P, Uren JM, Merlo D, Wisden W, Triller A, Smart TG, Moss SJ. GABA(A) receptor cell surface number and subunit stability are regulated by the ubiquitin-like protein Plic-1. Nature Neuroscience. 2001;4:908–916. doi: 10.1038/nn0901-908. [DOI] [PubMed] [Google Scholar]
- Bingol B, Schuman EM. Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature. 2006;441:1144–1148. doi: 10.1038/nature04769. [DOI] [PubMed] [Google Scholar]
- Bingol B, Wang CF, Arnott D, Cheng D, Peng J, Sheng M. Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell. 2010;140:567–578. doi: 10.1016/j.cell.2010.01.024. [DOI] [PubMed] [Google Scholar]
- Burbea M, Dreier L, Dittman JS, Grunwald ME, Kaplan JM. Ubiquitin and AP180 regulate the abundance of GLR-1 glutamate receptors at postsynaptic elements in C. elegans. Neuron. 2002;35:107–120. doi: 10.1016/s0896-6273(02)00749-3. [DOI] [PubMed] [Google Scholar]
- Buro LJ, Chipumuro E, Henriksen MA. Menin and RNF20 recruitment is associated with dynamic histone modifications that regulate signal transducer and activator of transcription 1 (STAT1)-activated transcription of the interferon regulatory factor 1 gene (IRF1) Epigenetics Chromatin. 2010;3:16. doi: 10.1186/1756-8935-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttner C, Sadtler S, Leyendecker A, Laube B, Griffon N, Betz H, Schmalzing G. Ubiquitination precedes internalization and proteolytic cleavage of plasma membrane-bound glycine receptors. Journal of Biological Chemistry. 2001;276:42978–42985. doi: 10.1074/jbc.M102121200. [DOI] [PubMed] [Google Scholar]
- Chain DG, Casadio A, Schacher S, Hegde AN, Valbrun M, Yamamoto N, Goldberg AL, Bartsch D, Kandel ER, Schwartz JH. Mechanisms for generating the autonomous cAMP-dependent protein kinase required for long-term facilitation in Aplysia. Neuron. 1999;22:147–156. doi: 10.1016/s0896-6273(00)80686-8. [DOI] [PubMed] [Google Scholar]
- Cheng Y. Toward an atomic model of the 26S proteasome. Curr Opin Struct Biol. 2009;19:203–208. doi: 10.1016/j.sbi.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin LS, Vavalle JP, Li L. Staring, a novel E3 ubiquitin-protein ligase that targets syntaxin 1 for degradation. Journal of Biological Chemistry. 2002;277:35071–35079. doi: 10.1074/jbc.M203300200. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med. 2015;47:e147. doi: 10.1038/emm.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Soler-Llavina G, Bhattacharyya S, Malenka RC. N-methyl-D-aspartate receptor- and metabotropic glutamate receptor-dependent long-term depression are differentially regulated by the ubiquitin-proteasome system. Eur J Neurosci. 2009;30:1443–1450. doi: 10.1111/j.1460-9568.2009.06950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai P, Akimaru H, Ishii S. A hedgehog-responsive region in the Drosophila wing disc is defined by debra-mediated ubiquitination and lysosomal degradation of Ci. Dev Cell. 2003;4:917–928. doi: 10.1016/s1534-5807(03)00158-8. [DOI] [PubMed] [Google Scholar]
- Davis KM, Sturt BL, Friedmann AJ, Richmond JE, Bessereau JL, Grant BD, Bamber BA. Regulated lysosomal trafficking as a mechanism for regulating GABAA receptor abundance at synapses in Caenorhabditis elegans. Mol Cell Neurosci. 2010;44:307–317. doi: 10.1016/j.mcn.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Day JJ, Sweatt JD. Epigenetic mechanisms in cognition. Neuron. 2011;70:813–829. doi: 10.1016/j.neuron.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- Djakovic SN, Marquez-Lona EM, Jakawich SK, Wright R, Chu C, Sutton MA, Patrick GN. Phosphorylation of Rpt6 regulates synaptic strength in hippocampal neurons. Journal of Neuroscience. 2012;32:5126–5131. doi: 10.1523/JNEUROSCI.4427-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djakovic SN, Schwarz LA, Barylko B, DeMartino GN, Patrick GN. Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. Journal of Biological Chemistry. 2009;284:26655–26665. doi: 10.1074/jbc.M109.021956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Bach SV, Haynes KA, Hegde AN. Proteasome modulates positive and negative translational regulators in long-term synaptic plasticity. Journal of Neuroscience. 2014;34:3171–3182. doi: 10.1523/JNEUROSCI.3291-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Upadhya SC, Ding L, Smith TK, Hegde AN. Proteasome inhibition enhances the induction and impairs the maintenance of late-phase long-term potentiation. Learning & Memory. 2008;15:335–347. doi: 10.1101/lm.984508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nature Neuroscience. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, Fussi H, Deszcz L, Hartl R, Schraml E, Criollo A, Megalou E, Weiskopf D, Laun P, Heeren G, Breitenbach M, Grubeck-Loebenstein B, Herker E, Fahrenkrog B, Frohlich KU, Sinner F, Tavernarakis N, Minois N, Kroemer G, Madeo F. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11:1305–1314. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- Felsenberg J, Dyck Y, Kloss A, Dahlmann B, Kloetzel PM, Eisenhardt D. Two inhibitors of the ubiquitin proteasome system enhance long-term memory formation upon olfactory conditioning in the honeybee (Apis mellifera) J Exp Biol. 2014;217:3441–3446. doi: 10.1242/jeb.108142. [DOI] [PubMed] [Google Scholar]
- Fenteany G, Standaert RF, Reichard GA, Corey EJ, Schreiber SL. A beta-lactone related to lactacystin induces neurite outgrowth in a neuroblastoma cell line and inhibits cell cycle progression in an osteosarcoma cell line. Proceedings of the National Academy of Sciences USA. 1994;91:3358–3362. doi: 10.1073/pnas.91.8.3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravante D, Liu RY, Byrne JH. The ubiquitin-proteasome system is necessary for long-term synaptic depression in Aplysia. Journal of Neuroscience. 2008;28:10245–10256. doi: 10.1523/JNEUROSCI.2139-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer DF, van DR, van TP, Hobo B, Verhage MC, van der Schors RC, Li KW, van MJ, Hol EM, van Leeuwen FW. Long-term proteasome dysfunction in the mouse brain by expression of aberrant ubiquitin. Neurobiol Aging. 2009;30:847–863. doi: 10.1016/j.neurobiolaging.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Fonseca R, Vabulas RM, Hartl FU, Bonhoeffer T, Nagerl UV. A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron. 2006;52:239–245. doi: 10.1016/j.neuron.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Furini CR, Myskiw Jde C, Schmidt BE, Zinn CG, Peixoto PB, Pereira LD, Izquierdo I. The relationship between protein synthesis and protein degradation in object recognition memory. Behav Brain Res. 2015;294:17–24. doi: 10.1016/j.bbr.2015.07.038. [DOI] [PubMed] [Google Scholar]
- Gentier RJ, van Leeuwen FW. Misframed ubiquitin and impaired protein quality control: an early event in Alzheimer’s disease. Front Mol Neurosci. 2015;8:47. doi: 10.3389/fnmol.2015.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001;15:807–826. doi: 10.1101/gad.887201. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- Gong B, Cao Z, Zheng P, Vitolo OV, Liu S, Staniszewski A, Moolman D, Zhang H, Shelanski M, Arancio O. Ubiquitin Hydrolase Uch-L1 Rescues beta-Amyloid-Induced Decreases in Synaptic Function and Contextual Memory. Cell. 2006;126:775–788. doi: 10.1016/j.cell.2006.06.046. [DOI] [PubMed] [Google Scholar]
- Gonzalez F, Delahodde A, Kodadek T, Johnston SA. Recruitment of a 19S proteasome subcomplex to an activated promoter. Science. 2002;296:548–550. doi: 10.1126/science.1069490. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Castellucci VF, Bayley H, Schwartz JH. A molecular mechanism for long-term sensitization in Aplysia. Nature. 1987;329:62–65. doi: 10.1038/329062a0. [DOI] [PubMed] [Google Scholar]
- Guan Z, Giustetto M, Lomvardas S, Kim JH, Miniaci MC, Schwartz JH, Thanos D, Kandel ER. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111:483–493. doi: 10.1016/s0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- Guinez C, Mir AM, Dehennaut V, Cacan R, Harduin-Lepers A, Michalski JC, Lefebvre T. Protein ubiquitination is modulated by O-GlcNAc glycosylation. Faseb j. 2008;22:2901–2911. doi: 10.1096/fj.07-102509. [DOI] [PubMed] [Google Scholar]
- Gupta-Agarwal S, Franklin AV, Deramus T, Wheelock M, Davis RL, McMahon LL, Lubin FD. G9a/GLP histone lysine dimethyltransferase complex activity in the hippocampus and the entorhinal cortex is required for gene activation and silencing during memory consolidation. Journal of Neuroscience. 2012;32:5440–5453. doi: 10.1523/JNEUROSCI.0147-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta VK, Scheunemann L, Eisenberg T, Mertel S, Bhukel A, Koemans TS, Kramer JM, Liu KS, Schroeder S, Stunnenberg HG, Sinner F, Magnes C, Pieber TR, Dipt S, Fiala A, Schenck A, Schwaerzel M, Madeo F, Sigrist SJ. Restoring polyamines protects from age-induced memory impairment in an autophagy-dependent manner. Nat Neurosci. 2013;16:1453–1460. doi: 10.1038/nn.3512. [DOI] [PubMed] [Google Scholar]
- Haack TB, Hogarth P, Kruer MC, Gregory A, Wieland T, Schwarzmayr T, Graf E, Sanford L, Meyer E, Kara E, Cuno SM, Harik SI, Dandu VH, Nardocci N, Zorzi G, Dunaway T, Tarnopolsky M, Skinner S, Frucht S, Hanspal E, Schrander-Stumpel C, Heron D, Mignot C, Garavaglia B, Bhatia K, Hardy J, Strom TM, Boddaert N, Houlden HH, Kurian MA, Meitinger T, Prokisch H, Hayflick SJ. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet. 2012;91:1144–1149. doi: 10.1016/j.ajhg.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton AM, Oh WC, Vega-Ramirez H, Stein IS, Hell JW, Patrick GN, Zito K. Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron. 2012;74:1023–1030. doi: 10.1016/j.neuron.2012.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick SJ, Kruer MC, Gregory A, Haack TB, Kurian MA, Houlden HH, Anderson J, Boddaert N, Sanford L, Harik SI, Dandu VH, Nardocci N, Zorzi G, Dunaway T, Tarnopolsky M, Skinner S, Holden KR, Frucht S, Hanspal E, Schrander-Stumpel C, Mignot C, Heron D, Saunders DE, Kaminska M, Lin JP, Lascelles K, Cuno SM, Meyer E, Garavaglia B, Bhatia K, de Silva R, Crisp S, Lunt P, Carey M, Hardy J, Meitinger T, Prokisch H, Hogarth P. beta-Propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain. 2013;136:1708–1717. doi: 10.1093/brain/awt095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde AN. Ubiquitin-proteasome-mediated local protein degradation and synaptic plasticity. Progress in Neuropbiology. 2004;73:311–357. doi: 10.1016/j.pneurobio.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Hegde AN. Ubiquitin-dependent protein degradation. In: Mander L, Lui H-W, editors. Comprehensive Natural Products II Chemistry and Biology. Oxford: Elsevier; 2010a. pp. 699–752. [Google Scholar]
- Hegde AN. The ubiquitin-proteasome pathway and synaptic plasticity. Learn Mem. 2010b;17:314–327. doi: 10.1101/lm.1504010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde AN, Goldberg AL, Schwartz JH. Regulatory subunits of cAMP-dependent protein kinases are degraded after conjugation to ubiquitin: a molecular mechanism underlying long-term synaptic plasticity. Proceedings of the National Academy of Sciences USA. 1993;90:7436–7440. doi: 10.1073/pnas.90.16.7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde AN, Haynes KA, Bach SV, Beckelman BC. Local ubiquitin-proteasome-mediated proteolysis and long-term synaptic plasticity. Front Mol Neurosci. 2014;7:96. doi: 10.3389/fnmol.2014.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde AN, Inokuchi K, Pei W, Casadio A, Ghirardi M, Chain DG, Martin KC, Kandel ER, Schwartz JH. Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia. Cell. 1997;89:115–126. doi: 10.1016/s0092-8674(00)80188-9. [DOI] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, Dikic I. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature. 2008;453:481–488. doi: 10.1038/nature06926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im E, Chung KC. Dyrk1A phosphorylates parkin at Ser-131 and negatively regulates its ubiquitin E3 ligase activity. J Neurochem. 2015;134:756–768. doi: 10.1111/jnc.13164. [DOI] [PubMed] [Google Scholar]
- Jarome TJ, Ferrara NC, Kwapis JL, Helmstetter FJ. CaMKII regulates proteasome phosphorylation and activity and promotes memory destabilization following retrieval. Neurobiol Learning & Memory. 2016;128:103–109. doi: 10.1016/j.nlm.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome TJ, Kwapis JL, Hallengren JJ, Wilson SM, Helmstetter FJ. The ubiquitin-specific protease 14 (USP14) is a critical regulator of long-term memory formation. Learning & Memory. 2014;21:9–13. doi: 10.1101/lm.032771.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome TJ, Kwapis JL, Ruenzel WL, Helmstetter FJ. CaMKII, but not protein kinase A, regulates Rpt6 phosphorylation and proteasome activity during the formation of long-term memories. Front Behav Neurosci. 2013;7:115. doi: 10.3389/fnbeh.2013.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome TJ, Lubin FD. Histone lysine methylation: critical regulator of memory and behavior. Rev Neurosci. 2013;24:375–387. doi: 10.1515/revneuro-2013-0008. [DOI] [PubMed] [Google Scholar]
- Jarome TJ, Werner CT, Kwapis JL, Helmstetter FJ. Activity dependent protein degradation is critical for the formation and stability of fear memory in the amygdala. PLoS One. 2011;6:e24349. doi: 10.1371/journal.pone.0024349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurd R, Thornton C, Wang J, Luong K, Phamluong K, Kharazia V, Gibb SL, Ron D. Mind bomb-2 is an E3 ligase that ubiquitinates the N-methyl-D-aspartate receptor NR2B subunit in a phosphorylation-dependent manner. J Biol Chem. 2008;283:301–310. doi: 10.1074/jbc.M705580200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova A, Mikhaylova M, Thomas U, Knopfel T, Behnisch T. Involvement of protein synthesis and degradation in long-term potentiation of Schaffer collateral CA1 synapses. Journal of Neuroscience. 2006;26:4949–4955. doi: 10.1523/JNEUROSCI.4573-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, Rouach N, Nicoll RA, Bredt DS. Activity-dependent NMDA receptor degradation mediated by retrotranslocation and ubiquitination. Proceedings of the National Academy of Sciences USA. 2005;102:5600–5605. doi: 10.1073/pnas.0501769102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein ME, Castillo PE, Jordan BA. Coordination between Translation and Degradation Regulates Inducibility of mGluR-LTD. Cell Rep. 2015;10:1459–1466. doi: 10.1016/j.celrep.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D. The emerging complexity of protein ubiquitination. Biochemical Society Transactions. 2009;37:937–953. doi: 10.1042/BST0370937. [DOI] [PubMed] [Google Scholar]
- Kottler B, Lampin-Saint-Amaux A, Comas D, Preat T, Goguel V. Debra, a protein mediating lysosomal degradation, is required for long-term memory in Drosophila. PLoS One. 2011;6:e25902. doi: 10.1371/journal.pone.0025902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczera T, Stilling RM, Hsia HE, Bahari-Javan S, Irniger S, Nasmyth K, Sananbenesi F, Fischer A. The anaphase promoting complex is required for memory function in mice. Learning & Memory. 2011;18:49–57. doi: 10.1101/lm.1998411. [DOI] [PubMed] [Google Scholar]
- Lee JS, Shukla A, Schneider J, Swanson SK, Washburn MP, Florens L, Bhaumik SR, Shilatifard A. Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell. 2007;131:1084–1096. doi: 10.1016/j.cell.2007.09.046. [DOI] [PubMed] [Google Scholar]
- Lee SH, Choi JH, Lee N, Lee HR, Kim JI, Yu NK, Choi SL, Lee SH, Kim H, Kaang BK. Synaptic protein degradation underlies destabilization of retrieved fear memory. Science. 2008;319:1253–1256. doi: 10.1126/science.1150541. [DOI] [PubMed] [Google Scholar]
- Leggett DS, Hanna J, Borodovsky A, Crosas B, Schmidt M, Baker RT, Walz T, Ploegh H, Finley D. Multiple associated proteins regulate proteasome structure and function. Molecular Cell. 2002;10:495–507. doi: 10.1016/s1097-2765(02)00638-x. [DOI] [PubMed] [Google Scholar]
- Levenson JM, O’Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. Journal of Biological Chemistry. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nat Rev Neurosci. 2005;6:108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- Li Q, Korte M, Sajikumar S. Ubiquitin-Proteasome System Inhibition Promotes Long-Term Depression and Synaptic Tagging/Capture. Cereb Cortex. 2015 doi: 10.1093/cercor/bhv084. [DOI] [PubMed] [Google Scholar]
- Li Z, Hao S, Yin H, Gao J, Yang Z. Autophagy ameliorates cognitive impairment through activation of PVT1 and apoptosis in diabetes mice. Behav Brain Res. 2016;305:265–277. doi: 10.1016/j.bbr.2016.03.023. [DOI] [PubMed] [Google Scholar]
- Lopez-Atalaya JP, Ito S, Valor LM, Benito E, Barco A. Genomic targets, and histone acetylation and gene expression profiling of neural HDAC inhibition. Nucleic Acids Res. 2013;41:8072–8084. doi: 10.1093/nar/gkt590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Salon M, Alonso M, Vianna MR, Viola H, Souza Me, Izquierdo I, Pasquini JM, Medina JH. The ubiquitin-proteasome cascade is required for mammalian long-term memory formation. European Journal of Neuroscience. 2001;14:1820–1826. doi: 10.1046/j.0953-816x.2001.01806.x. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Marques AJ, Palanimurugan R, Matias AC, Ramos PC, Dohmen RJ. Catalytic mechanism and assembly of the proteasome. Chem Rev. 2009;109:1509–1536. doi: 10.1021/cr8004857. [DOI] [PubMed] [Google Scholar]
- Martin S, Henley JM. Activity-dependent endocytic sorting of kainate receptors to recycling or degradation pathways. Embo j. 2004;23:4749–4759. doi: 10.1038/sj.emboj.7600483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-kappa B functions in synaptic signaling and behavior. Nat Neurosci. 2003;6:1072–1078. doi: 10.1038/nn1110. [DOI] [PubMed] [Google Scholar]
- Minsky N, Shema E, Field Y, Schuster M, Segal E, Oren M. Monoubiquitinated H2B is associated with the transcribed region of highly expressed genes in human cells. Nat Cell Biol. 2008;10:483–488. doi: 10.1038/ncb1712. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nature Reviews Molecular and Cellular Biology. 2009;10:458–467. doi: 10.1038/nrm2708. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265:1104–1107. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- Ohtake F, Saeki Y, Sakamoto K, Ohtake K, Nishikawa H, Tsuchiya H, Ohta T, Tanaka K, Kanno J. Ubiquitin acetylation inhibits polyubiquitin chain elongation. EMBO Reports. 2015;16:192–201. doi: 10.15252/embr.201439152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, Chin LS. Parkin-mediated K63-linked polyubiquitination: a signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy. 2008;4:85–87. doi: 10.4161/auto.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega Z, Lucas JJ. Ubiquitin-proteasome system involvement in Huntington’s disease. Front Mol Neurosci. 2014;7:77. doi: 10.3389/fnmol.2014.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak DT, Sheng M. Targeted protein degradation and synapse remodeling by an inducible protein kinase. Science. 2003;302:1368–1373. doi: 10.1126/science.1082475. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Bingol B, Weld HA, Schuman EM. Ubiquitin-mediated proteasome activity is required for agonist-induced endocytosis of GluRs. Current Biology. 2003;13:2073–2081. doi: 10.1016/j.cub.2003.10.028. [DOI] [PubMed] [Google Scholar]
- Peth A, Besche HC, Goldberg AL. Ubiquitinated proteins activate the proteasome by binding to Usp14/Ubp6, which causes 20S gate opening. Molecular Cell. 2009;36:794–804. doi: 10.1016/j.molcel.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis DS, Jarome TJ, Helmstetter FJ. Memory formation for trace fear conditioning requires ubiquitin-proteasome mediated protein degradation in the prefrontal cortex. Front Behav Neurosci. 2013;7:150. doi: 10.3389/fnbeh.2013.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riffo-Campos AL, Castillo J, Tur G, Gonzalez-Figueroa P, Georgieva EI, Rodriguez JL, Lopez-Rodas G, Rodrigo MI, Franco L. Nucleosome-specific, Time-dependent Changes in Histone Modifications during Activation of the Early Growth Response 1 (Egr1) Gene. Journal of Biological Chemistry. 2015;290:197–208. doi: 10.1074/jbc.M114.579292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinetti GV, Schweizer FE. Ubiquitination acutely regulates presynaptic neurotransmitter release in mammalian neurons. Journal of Neuroscience. 2010;30:3157–3166. doi: 10.1523/JNEUROSCI.3712-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Ortiz CJ, Balderas I, Saucedo-Alquicira F, Cruz-Castaneda P, Bermudez-Rattoni F. Long-term aversive taste memory requires insular and amygdala protein degradation. Neurobiol Learning & Memory. 2011;95:311–315. doi: 10.1016/j.nlm.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Ruan HB, Nie Y, Yang X. Regulation of protein degradation by O-GlcNAcylation: crosstalk with ubiquitination. Mol Cell Proteomics. 2013;12:3489–3497. doi: 10.1074/mcp.R113.029751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Mohler H. GABA-based therapeutic approaches: GABAA receptor subtype functions. Current Opinion in Pharmacology. 2006;6:18–23. doi: 10.1016/j.coph.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Saitsu H, Nishimura T, Muramatsu K, Kodera H, Kumada S, Sugai K, Kasai-Yoshida E, Sawaura N, Nishida H, Hoshino A, Ryujin F, Yoshioka S, Nishiyama K, Kondo Y, Tsurusaki Y, Nakashima M, Miyake N, Arakawa H, Kato M, Mizushima N, Matsumoto N. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet. 2013;45:445–449. 449e441. doi: 10.1038/ng.2562. [DOI] [PubMed] [Google Scholar]
- Sajikumar S, Frey JU. Anisomycin inhibits the late maintenance of long-term depression in rat hippocampal slices in vitro. Neuroscience Letters. 2003;338:147–150. doi: 10.1016/s0304-3940(02)01400-3. [DOI] [PubMed] [Google Scholar]
- Saliba RS, Michels G, Jacob TC, Pangalos MN, Moss SJ. Activity-dependent ubiquitination of GABA(A) receptors regulates their accumulation at synaptic sites. Journal of Neuroscience. 2007;27:13341–13351. doi: 10.1523/JNEUROSCI.3277-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proceedings of the National Academy of Sciences USA. 2002;99:13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz LA, Hall BJ, Patrick GN. Activity-dependent ubiquitination of GluA1 mediates a distinct AMPA receptor endocytosis and sorting pathway. Journal of Neuroscience. 2010;30:16718–16729. doi: 10.1523/JNEUROSCI.3686-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scudder SL, Goo MS, Cartier AE, Molteni A, Schwarz LA, Wright R, Patrick GN. Synaptic strength is bidirectionally controlled by opposing activity-dependent regulation of Nedd4–1 and USP8. J Neurosci. 2014;34:16637–16649. doi: 10.1523/JNEUROSCI.2452-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segref A, Hoppe T. Think locally: control of ubiquitin-dependent protein degradation in neurons. EMBO Reports. 2009;10:44–50. doi: 10.1038/embor.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehata M, Matsumura H, Okubo-Suzuki R, Ohkawa N, Inokuchi K. Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J Neurosci. 2012;32:10413–10422. doi: 10.1523/JNEUROSCI.4533-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Korutla L, Champtiaux N, Toda S, LaLumiere R, Vallone J, Klugmann M, Blendy JA, Mackler SA, Kalivas PW. NAC1 Regulates the Recruitment of the Proteasome Complex into Dendritic Spines. Journal of Neuroscience. 2007;27:8903–8913. doi: 10.1523/JNEUROSCI.1571-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Ganetzky B. Autophagy promotes synapse development in Drosophila. J Cell Biol. 2009;187:71–79. doi: 10.1083/jcb.200907109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sol Fustinana M, de la Fuente V, Federman N, Freudenthal R, Romano A. Protein degradation by ubiquitin-proteasome system in formation and labilization of contextual conditioning memory. Learning & Memory. 2014;21:478–487. doi: 10.1101/lm.035998.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Smolen P, Av-Ron E, Baxter DA, Byrne JH. Bifurcation and singularity analysis of a molecular network for the induction of long-term memory. Biophys J. 2006;90:2309–2325. doi: 10.1529/biophysj.105.074500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speese SD, Trotta N, Rodesch CK, Aravamudan B, Broadie K. The ubiquitin proteasome system acutely regulates presynaptic protein turnover and synaptic efficacy. Current Biology. 2003;13:899–910. doi: 10.1016/s0960-9822(03)00338-5. [DOI] [PubMed] [Google Scholar]
- Stacey KB, Breen E, Jefferies CA. Tyrosine phosphorylation of the E3 ubiquitin ligase TRIM21 positively regulates interaction with IRF3 and hence TRIM21 activity. PLoS One. 2012;7:e34041. doi: 10.1371/journal.pone.0034041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney DL, Rodriguez-Mias RA, Villen J. Phosphorylation of ubiquitin at Ser65 affects its polymerization, targets, and proteome-wide turnover. EMBO Reports. 2015;16:1131–1144. doi: 10.15252/embr.201540298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, Ho MW, Troncoso J, Gygi SP, Lee MK, Dawson VL, Dawson TM, Lim KL. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet. 2008;17:431–439. doi: 10.1093/hmg/ddm320. [DOI] [PubMed] [Google Scholar]
- Tang G, Leppla SH. Proteasome activity is required for anthrax lethal toxin to kill macrophages. Infection and Immunity. 1999;67:3055–3060. doi: 10.1128/iai.67.6.3055-3060.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhya SC, Ding L, Smith TK, Hegde AN. Differential regulation of proteasome activity in the nucleus and the synaptic terminals. Neurochemistry International. 2006;48:296–305. doi: 10.1016/j.neuint.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Upadhya SC, Hegde AN. Ubiquitin-proteasome pathway components as therapeutic targets for CNS maladies. Curr Pharm Des. 2005;11:3807–3828. doi: 10.2174/138161205774580651. [DOI] [PubMed] [Google Scholar]
- Upadhya SC, Hegde AN. Role of the ubiquitin proteasome system in Alzheimer’s disease. BMC Biochem. 2007;8(Suppl 1):S12. doi: 10.1186/1471-2091-8-S1-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhya SC, Smith TK, Hegde AN. Ubiquitin-proteasome-mediated CREB repressor degradation during induction of long-term facilitation. Journal of Neurochemisty. 2004;91:210–219. doi: 10.1111/j.1471-4159.2004.02707.x. [DOI] [PubMed] [Google Scholar]
- van Leeuwen FW, de Kleijn DP, van den Hurk HH, Neubauer A, Sonnemans MA, Sluijs JA, Koycu S, Ramdjielal RD, Salehi A, Martens GJ, Grosveld FG, Peter J, Burbach H, Hol EM. Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer’s and Down patients. Science. 1998;279:242–247. doi: 10.1126/science.279.5348.242. [DOI] [PubMed] [Google Scholar]
- van Nocker S, Deveraux Q, Rechsteiner M, Vierstra RD. Arabidopsis MBP1 gene encodes a conserved ubiquitin recognition component of the 26S proteasome. Proceedings of the National Academy of Sciences USA. 1996;93:856–860. doi: 10.1073/pnas.93.2.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weake VM, Workman JL. Histone ubiquitination: triggering gene activity. Mol Cell. 2008;29:653–663. doi: 10.1016/j.molcel.2008.02.014. [DOI] [PubMed] [Google Scholar]
- Widagdo J, Chai YJ, Ridder MC, Chau YQ, Johnson RC, Sah P, Huganir RL, Anggono V. Activity-Dependent Ubiquitination of GluA1 and GluA2 Regulates AMPA Receptor Intracellular Sorting and Degradation. Cell Reports. 2015 doi: 10.1016/j.celrep.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KD. Ubiquitination and deubiquitination: targeting of proteins for degradation by the proteasome. Seminars in Cell and Developmental Biology. 2000;11:141–148. doi: 10.1006/scdb.2000.0164. [DOI] [PubMed] [Google Scholar]
- Willeumier K, Pulst SM, Schweizer FE. Proteasome inhibition triggers activity-dependent increase in the size of the recycling vesicle pool in cultured hippocampal neurons. Journal of Neuroscience. 2006;26:11333–11341. doi: 10.1523/JNEUROSCI.1684-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyce A, Xiao T, Whelan KA, Kosman C, Walter W, Eick D, Hughes TR, Krogan NJ, Strahl BD, Berger SL. H2B ubiquitylation acts as a barrier to Ctk1 nucleosomal recruitment prior to removal by Ubp8 within a SAGA-related complex. Mol Cell. 2007;27:275–288. doi: 10.1016/j.molcel.2007.01.035. [DOI] [PubMed] [Google Scholar]
- Yamamoto N, Hegde AN, Chain DG, Schwartz JH. Activation and degradation of the transcription factor C/EBP during long-term facilitation in Aplysia. J Neurochem. 1999;73:2415–2423. doi: 10.1046/j.1471-4159.1999.0732415.x. [DOI] [PubMed] [Google Scholar]
- Yao I, Takagi H, Ageta H, Kahyo T, Sato S, Hatanaka K, Fukuda Y, Chiba T, Morone N, Yuasa S, Inokuchi K, Ohtsuka T, Macgregor GR, Tanaka K, Setou M. SCRAPPER-dependent ubiquitination of active zone protein RIM1 regulates synaptic vesicle release. Cell. 2007;130:943–957. doi: 10.1016/j.cell.2007.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaron A, Hatzubai A, Davis M, Lavon I, Amit S, Manning AM, Andersen JS, Mann M, Mercurio F, Ben-Neriah Y. Identification of the receptor component of the IkappaBalpha-ubiquitin ligase. Nature. 1998;396:590–594. doi: 10.1038/25159. [DOI] [PubMed] [Google Scholar]
- Ye Y, Rape M. Building ubiquitin chains: E2 enzymes at work. Nature Reviews Molecular and Cellular Biology. 2009;10:755–764. doi: 10.1038/nrm2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaro BW, Yang YY, Hang HC, Pratt MR. Chemical reporters for fluorescent detection and identification of O-GlcNAc-modified proteins reveal glycosylation of the ubiquitin ligase NEDD4–1. Proc Natl Acad Sci U S A. 2011;108:8146–8151. doi: 10.1073/pnas.1102458108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Hegde AN, Martin KC. The ubiquitin proteasome system functions as an inhibitory constraint on synaptic strengthening. Current Biology. 2003;13:887–898. doi: 10.1016/s0960-9822(03)00332-4. [DOI] [PubMed] [Google Scholar]
- Zovkic IB, Guzman-Karlsson MC, Sweatt JD. Epigenetic regulation of memory formation and maintenance. Learning & Memory. 2013;20:61–74. doi: 10.1101/lm.026575.112. [DOI] [PMC free article] [PubMed] [Google Scholar]