

Aging increases angiotensin-converting enzyme (ACE)-independent production of cardiac angiotensin II (Ang II), a response that is driven by chymase in an exercise-reversible manner. These findings highlight chymase, in addition to ACE, as an important therapeutic target in the treatment and prevention of Ang II-induced deterioration of cardiac function in the elderly.
Keywords: aging, chymase, angiotensin-converting enzyme, angiotensin II, exercise
Abstract
Age-dependent alteration of the renin-angiotensin system (RAS) and generation of angiotensin II (Ang II) are well documented. By contrast, RAS-independent generation of Ang II in aging and its responses to exercise have not been explored. To this end, we examined the effects of chymase, a secretory serine protease, on the angiotensin-converting enzyme (ACE)-independent conversion of Ang I to Ang II. We hypothesized that age-dependent alteration of cardiac Ang II formation is chymase dependent in nature and is prevented by exercise training. Experiments were conducted on hearts isolated from young (3 mo), aged sedentary (24 mo), and aged rats chronically exercised on a treadmill. In the presence of low Ang I levels and downregulation of ACE expression/activity, cardiac Ang II levels were significantly higher in aged than young rats, suggesting an ACE-independent response. Aged hearts also displayed significantly increased chymase expression and activity, as well as upregulation of tryptase, a biological marker of mast cells, confirming a mast cell-sourced increase in chymase. Coincidently, cardiac superoxide produced from NADPH oxidase (Nox) was significantly enhanced in aged rats and was normalized by exercise. Conversely, a significant reduction in cardiac expression of ACE2 followed by lower Ang 1-7 levels and downregulation of the Mas receptor (binding protein of Ang 1-7) in aged rats were completely reversed by exercise. In conclusion, local formation of Ang II is increased in aged hearts, and chymase is primarily responsible for this increase. Chronic exercise is able to normalize the age-dependent alterations via compromising chymase/Ang II/angiotensin type 1 receptor/Nox actions while promoting ACE2/Ang 1-7/MasR signaling.
NEW & NOTEWORTHY Aging increases angiotensin-converting enzyme (ACE)-independent production of cardiac angiotensin II (Ang II), a response that is driven by chymase in an exercise-reversible manner. These findings highlight chymase, in addition to ACE, as an important therapeutic target in the treatment and prevention of Ang II-induced deterioration of cardiac function in the elderly.
Listen to this article's corresponding podcast @ http://ajpheart.podbean.com/e/renin-angiotensin-system-signaling-in-aged-and-age-exercised-rats/.
angiotensin ii (Ang II) is the major biologically active molecule of the renin-angiotensin system (RAS), which is involved in the homeostasis of blood pressure through its effects on water and electrolyte balance, as well as peripheral vascular resistance (46). Ang II is produced from the decapeptide angiotensin I (Ang I) via cleavage of two COOH-terminal amino acids by angiotensin-converting enzyme (ACE). When bound to the angiotensin type 1 receptor (AT1R) on the vasculature, Ang II initiates vasoconstriction, whereas it elicits nitric oxide (NO)-mediated vasodilation via binding to the endothelium-located AT2R (18). Alternatively, cleavage of Ang II by ACE2 produces the heptapeptide angiotensin 1-7 (Ang 1-7), which, when bound to the Mas receptor (MasR) (48), initiates downstream vasodilator activity that can counteract the hypertensive effects of AT1R signaling. Ang 1-7 therefore possesses cardioprotective properties (9) through effects that can prevent and/or reverse heart failure and blunt hypertensive cardiac remodeling (3, 38, 43). In addition to the systemic RAS, local RAS has been found in many tissues, including the heart, which functions both independently and in correlation with systemic RAS components (1, 9, 44). Importantly in the heart, there is an alternate pathway for Ang II synthesis by the endopeptidase chymase, a serine protease that is predominantly found in granulocytes called mast cells located within the interstitium. Mast cells contain granules that are packaged with cytokines and proteases (including chymase) that are released during the inflammatory process via a classically mediated ligand-dependent pathway (9, 13, 55). Chymase acts in much the same manner as ACE but with a 20-fold higher catalytic activity for the conversion of Ang I to Ang II (2). In this context, chymase is believed to serve as a major Ang II-forming enzyme in the human heart (21, 22) and is reported to be involved in many pathological processes, such as hypertension, atherosclerosis, vascular proliferation, development of cardiomyopathies, myocardial infarction, and heart failure, as well as cardiac fibrosis (20, 21, 25, 31, 54, 61).
Ultimately, the impact of altered RAS signaling in the cardiovascular system extends beyond just the control of vascular tone and actually significantly contributes to the pathogenesis of a variety of cardiovascular diseases, including aging-induced cardiac dysfunction (14). During the process of aging, chronic elevation of Ang II evokes generation of reactive oxygen species (ROS) via binding with AT1R to activate NADPH oxidase (Nox) (11, 60). This Ang II-AT1R–Nox signaling cascade may, in turn, trigger downstream stress- or inflammatory-related signaling to further produce ROS (57). Alternatively, oxidative stress per se can serve as a trigger to stimulate the release of chymase from mast cells, via a nonligand-mediated signaling pathway (32), leading to the formation of a positive feedback loop during the process of Ang II stimulation of oxidative stress and vice versa. Interestingly, regular exercise has been reported to reduce age-related increases in tissue oxidative stress and inflammation (4) and has been demonstrated to be cardioprotective in both pathological and physiological conditions (26, 52, 53). One of the underlying mechanisms involved in the protective actions of exercise is via the potentiation of the ACE2/Ang1-7/MasR pathway that can balance or even outweigh actions of the ACE/Ang II/AT1R/ROS signaling in modifying acute and chronic inflammation, fibrogenesis, and cellular proliferation (14, 26).
The pathological significance of changes in systemic RAS is well established (24); however, the mechanisms responsible for aging-dependent changes in cardiac Ang II signaling, which are specifically driven by chymase, are much less understood. One study found an ~20% increase in total mast cell number with greater than a fourfold increase in activated mast cells in aged lymphatic tissue (10), findings that are important because chymase is packaged in and released from mast cells. To date, neither the roles of chymase in age-dependent cardiac formation of Ang II nor changes in chymase activity in response to exercise have been reported in the literature. Given that chymase is an important Ang II synthase in the heart, along with the lack of studies focused on the pathological significance of chymase-sourced Ang-II during cardiac aging, we hypothesized that chymase is a key contributor to the age-dependent increase in cardiac formation of Ang II, a response that we posited can be prevented by exercise training via compromising chymase actions and recruiting ACE2/Ang1-7/MasR signaling. We specifically focused on the association between cardiac aging and chymase action by determining chymase-dependent formation of Ang II in myocardium of young and aged rats.
MATERIALS AND METHODS
Animals.
Young (3-mo-old) and aged (20-mo-old) male Fischer 344 rats were obtained from Charles River Laboratories. Aged rats were randomly divided into two groups of sedentary and exercised rats and housed for an additional 4 mo to yield 24-mo-old sedentary (Aged) and 24-mo-old exercised (Aged-Ex) groups. Protocols were approved by the Institutional Animal Care and Use Committee of New York Medical College and conformed to the current guidelines of the National Institutes of Health and American Physiological Society for the care and use of laboratory animals.
Exercise training.
Rats were exercised on a treadmill (model 4215; Quinton Instruments) starting at 20 mo of age. As described previously (52), a jet of air and a mild electrical tail shock stimulus were used to keep the rats running on the treadmill but caused no injuries to the rat tail. The exercise protocol was similar to that used in our previous studies (52, 53). Exercise activity on the treadmill was carried out for 5 days/week for 4 mo. The length of time on the treadmill was initially 5 min/day and was progressively increased to a maximum of 60 min/day by the end of the sixth week. The speed and angle of the treadmill were increased from 11 m/min at a 0° grade on day 1 to a maximum of 38 m/min at a 5° grade by the end of the sixth week. All parameters were subsequently kept at a maximal level until the end of the exercise period. Sedentary rats were handled daily on the treadmill without running. At the end of exercise training, rats were anesthetized with isoflurane and euthanized. Following bilateral thoracotomy, the heart was excised and immediately pulverized in liquid N2.
Measurement of angiotensin peptides.
Ang I, Ang II, and Ang 1-7 peptides in cardiac tissue were measured by ELISA (Mybiosource). On the basis of instructions provided by the manufacturer, pulverized whole hearts were homogenized in a sample buffer and centrifuged at 1,000 g for 10 min, and the supernatant fraction was collected. In a 96-well plate, 50 μl of samples or peptide standard solution was added to each well, followed by the addition of 100 μl of horseradish peroxidase-conjugate reagent. The plate was gently mixed, covered with a plastic membrane, and incubated at 37°C for 60 min. Following incubation, the plate was washed with wash buffer four times. The plate was subsequently blotted dry, and 50 μl of each Chromogen Solution A and Chromogen Solution B were added to each well. The plate was gently mixed and incubated for an additional 15 min at 37°C, and 50 μl of stop solution was then immediately added. The absorbance was read at 450 nm using a SpectraMax 96-well plate spectrophotometer (Molecular Devices). The amount of angiotensin peptides was calculated by comparing absorbance readings of samples to those of the standard curve. Values were normalized to milligrams of tissue protein.
Enzymatic activity.
Nox activity was measured using an established lucigenin detection analysis (28). Liquid N2-pulverized hearts were homogenized in phosphate buffer containing 20 mM monophasic potassium phosphate (KPO4), 1 mM EDTA, and protease inhibitor cocktail. Samples were centrifuged at 1,000 g, the supernatant was aspirated, and protein content was determined using the Bradford method (Bio-Rad). The reaction was conducted in a final volume of 1 ml, which contained reaction assay buffer (50 mM KPO4, 150 mM sucrose, and 1 mM EDTA, pH 7.0), 5 μM lucigenin, and 100 μM NADPH. The reaction was initiated by the addition of 50 μl of homogenate (~200 μg protein) that was preincubated with or without 100 μM apocynin (inhibitor for Nox). The chemiluminescence generated by the reaction was recorded for 2 min by scintillation counter (LS6000IC, Beckman Instruments). Nox activity was calculated by subtracting the value of apocynin-treated fraction from total values (without apocynin), and reported as counts/μg protein per minute after background subtraction.
Activities of ACE and ACE2 were determined using specific fluorometric substrates Abz-FRK(Dnp)-P and Mca-APK(Dnp) (Enzo Life Sciences), respectively (19). Hearts were homogenized in 0.1 M Tris·HCl buffer containing 50 mM NaCl (pH 7.0) for ACE and 0.2 M Tris·HCl containing 200 mM NaCl (pH 7.5) for ACE2. Samples were centrifuged at 1,000 g for 10 min, and the supernatant fraction was collected for analysis. Activity assays were conducted in the same buffers with an additional 10 μM ZnCl2. The appropriate fluorogenic substrate (10 μM) was added to ~40-μg protein samples in a final reaction volume of 100 μl. The hydrolytic rate of either fluorogenic substrate was determined continuously for 40 min by incrementally reading fluorescence at 320-nm excitation and 420-nm emission. In separate experiments, the enzymatic specificity was validated by testing the reaction in the presence of 10−6 mol/l captopril (ACE inhibitor) or DX600 (ACE2 inhibitor). Protein concentration of samples was measured by the Bradford method (Bio-Rad) and used to normalize detected tissue fluorescence. The maximum rate of reaction (Vmax) was defined as changes in relative fluorescence units (RFU) per unit time (minute). The enzyme activity was expressed as Vmax per microgram of protein.
Chymase activity was determined by the rate of cleavage of the chromogenic substrate Suc-AAPF-pNA (Enzo Life Sciences) (39). Specifically, hearts were homogenized in the assay buffer containing 125 mM NaCl2, 10 μM ZnCl2, and 25 mM HEPES (pH 7.4) with the addition of 0.01% Triton X-100 (22). After homogenization, samples were centrifuged at 1,000 g for 10 min, the supernatant fraction was collected, and protein content was determined using the Bradford method. Approximately 50 μg of protein were preincubated with or without 100 μM chymastatin (chymase inhibitor) in assay buffer for 5 min, followed by incubation with 4 mM Suc-AAPF-pNA at 37°C in a total reaction volume of 250 μl. Spectrophotometric determination of absorbance at 405 nm was started immediately after adding Suc-AAPF-pNA and continued for 60 min. Chymase activity was calculated by subtracting the value of chymastatin-treated fraction from total values (without chymastatin) and presented as Vmax per milligram of protein.
Western blot analysis.
Isolated hearts were pulverized in liquid N2. Equal amounts of total protein (50 µg) extracted were loaded onto and separated by 10% SDS-PAGE gel and transferred to a PVDF membrane. The membrane was probed with specific primary antibodies for ACE (1:200 dilution), ACE2 (1:500), chymase (1:200), mast cell tryptase (1:1,000), and MasR (1:500) purchased from Abcam, AT1R (1:1,000 sc-1173) and AT2R (1:500) obtained from Santa Cruz Biotechnology, and GAPDH (1: 2,500) from Millipore, followed by appropriate secondary antibodies conjugated with horseradish peroxidase (1:2,000–1:10,000). Specific bands were visualized with a chemiluminescence kit (Thermo Scientific) and normalized to GAPDH. The X-ray film was scanned into a computer, and band densitometry was digitalized with UN-SCAN-IT software (Silk Scientific).
Calculation and statistical analysis.
Data are expressed as means ± SE, and n refers to the number of rats. Tests of data normality were conducted, following which one-way ANOVA or the Kruskal Wallis test was performed where appropriate. Statistical significance was accepted at a level of P < 0.05.
RESULTS
Aging induced imbalance between Ang II and Ang 1-7 axes.
Cardiac angiotensin peptide contents are depicted in Fig. 1. Cardiac tissue levels of Ang I were significantly lower in aged than in young control animals (Fig. 1A), findings that are consistent with most previous studies (16). Alternatively, Ang II in hearts of aged animals was significantly increased compared with that in young controls (Fig. 1B), whereas the levels of the cardioprotective peptide, Ang 1-7, were markedly reduced (Fig. 1C). The age-dependent changes in angiotensin peptides were normalized by exercise activity (Aged-Ex).
Fig. 1.

Cardiac levels of angiotensin I (Ang I) (A), Ang II (B), and Ang 1-7 (C) in young and aged rats and aged rats with exercise training (Aged-Ex) (n = 5–6 in each group). *Significant difference from young group. #Significant difference from aged group.
Aging-dependent alterations/reductions of ACE and ACE2 in an exercise-reversible manner.
Expression of ACE and its activity are summarized in Fig. 2. Interestingly, ACE expression (Fig. 2A) and activity (Fig. 2B) were reduced significantly in aged compared with young rats, implying that the age-induced increase in Ang II (shown in Fig. 1B) appears to be independent of ACE. Moreover, exercise training prevents the age-related reductions in both ACE expression and activity. In separate experiments, ACE activity was tested in the presence of captopril (10−5 mol/l; specific inhibitor of ACE) to validate the specificity of the enzyme. As shown in Fig. 2C, cardiac ACE activity in the three groups of rats was eliminated by captopril, confirming the specificity of the response.
Fig. 2.

Protein expression of angiotensin-converting enzyme (ACE) (3 blots) (A) and ACE activity (n = 6 in each group) (B) in cardiac tissue of young and aged rats and aged rats with exercise training (Aged-Ex) are shown. C: ACE activity in the 3 groups, represented as changes in relative fluorescence units (RFU) over time (minute) in the presence (+) and absence (−) of captopril (10−5 M), a specific inhibitor of ACE (n = 6 in each group). *Significant difference from young group. #Significant difference from aged group.
ACE2 expression and activity are summarized in Fig. 3. As shown in Fig. 3A, the age-associated downregulation of cardiac ACE2 was, not only normalized, but also upregulated by exercise training. In a similar fashion, the pattern of decreased ACE2 activity was completely reversed by exercise training to levels observed in control animals (Fig. 3B). The specificity of ACE2 was also validated by experiments, in which DX600 (10−5 mol/l; specific inhibitor of ACE2) abolished the enzyme activity in all groups of rats (Fig. 3C).
Fig. 3.

Protein expression of ACE2 (3 blots) (A) and ACE2 activity (n = 5) (B) in cardiac tissue of young and aged rats and aged rats with exercise training (Aged-Ex) are shown. C: ACE2 activity measured in the presence (+) and absence (−) of DX600 (10−5 M), a specific inhibitor of ACE2 (n = 6 in each group). *Significant difference from young group. #Significant difference from aged group.
Age-induced increase in chymase expression/activity paralleled with changes in mast cell content.
To clarify the nature of the ACE-independent increase in cardiac Ang II in aged rats (Figs. 1 and 2), cardiac chymase expression and activity were evaluated and are presented in Fig. 4. Contrary to the downregulation of ACE expression and attenuation of ACE activity (Fig. 2, A and B), aged rats display an upregulation of chymase expression (Fig. 4A), accompanied by a significant increase in chymase activity (Fig. 4B). To identify the source of chymase in the cardiac tissue, mast cell tryptase expression was used as an indirect determinant of mast cell content, based on the fact that the tryptase is highly specific for mast cells and its release is proportional to the activation of mast cells (45). As shown in Fig. 4C, changes in tryptase expression, characterized as an age-dependent upregulation, were paralleled with those of chymase expression/activity (Fig. 4, A and B), confirming the specific correlation between the two enzymes. Both increments in chymase and tryptase were presented in an exercise-reversible manner, even though their protein expression did not fully recover to levels observed in young rats (Fig. 4A). Thus the age-dependent increase in cardiac Ang II appears to be a chymase-driven response (Fig. 4).
Fig. 4.

Chymase protein expression (3 blots) (A) and activity (B) (n = 5–6) and tryptase protein expression (2 blots) (C) in cardiac tissue of young and aged rats and aged rats with exercise training (Aged-Ex) are shown. *Significant difference from young group. #Significant difference from aged group.
Reciprocal potentiation between oxidative stress and chymase-Ang II.
To evaluate roles of superoxide in the age-induced altered Ang II signaling, Nox activity was assessed. In correlation with changes in chymase, tryptase, and Ang II, Nox activity was significantly elevated in aged compared with young hearts, a response that was also present in an exercise-reversible manner (Fig. 5), revealing the specific linkage among them.
Fig. 5.
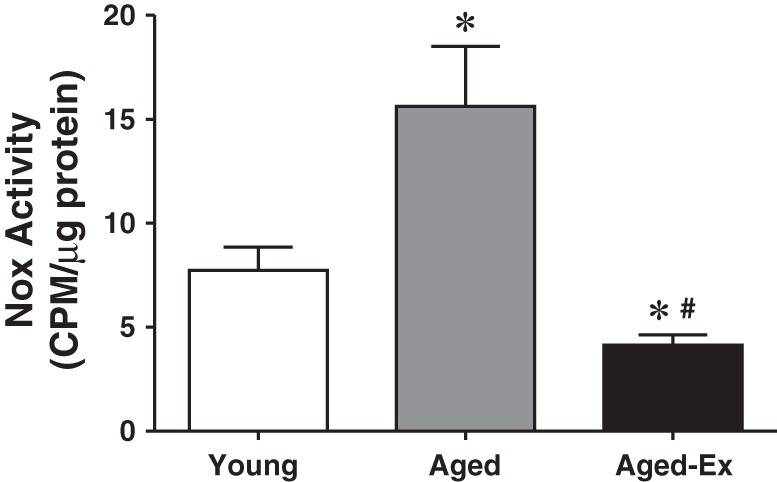
NADPH oxidase (Nox) activity (indicative of superoxide production) in cardiac tissue of young, aged, and aged rats with exercise training (Aged-Ex) (n = 5–6 for each group). *Significant difference from young group. #Significant difference from aged group.
Different responses of angiotensin receptors to aging and exercise.
Protein expressions of downstream targets of Ang II and Ang 1-7, namely AT1R, AT2R, and MasR, are presented in Fig. 6. In the presence of comparable expression of cardiac AT2R among all groups (Fig. 6B), cardiac expression of AT1R was upregulated in aged compared with that in young rats (Fig. 6A). The increased expression of AT1R was not significantly changed by exercise training, which implies an age-intensifying action of Ang II. However, the age-induced downregulation of MasR was normalized by exercise training (Fig. 6C), suggesting an exercise-favorable action on the beneficial Ang 1-7 cascade.
Fig. 6.

Protein expression of AT1R (A), AT2R (B), and MasR (C) in cardiac tissue of young and aged rats and aged rats with exercise training (Aged-Ex) (3 blots for each receptor). *Significant difference from young group. #Significant difference from aged group.
Taken together, our data unravel an ACE-independent chymase-mediated increase in cardiac Ang II, as a function of age, a response that was prevented by exercise activity via suppression of chymase-dependent Ang II production accompanied with decreases in oxidative stress and recruitment of ACE2-mediated Ang II degradation.
DISCUSSION
The salient findings of the present study are that 1) the age-dependent increase in cardiac Ang II is independent of ACE activity and is a chymase-driven response. Thus elevation of Ang II levels in aged hearts is proportional to chymase expression/activity and superoxide production and correlates inversely with ACE activity. 2) Suppression of ACE2 expression and activity contributes significantly to reduced formation of Ang 1-7 in aged hearts, which is coupled with 3) a concurrent downregulation of MasR and upregulation of AT1R, thus creating an imbalance between the chymase/Ang II/AT1R/Nox and ACE2/Ang1-7/MasR axes of the RAS. A decrease in the ACE2/Ang- (1-7) cascade diminishes its ability to counterbalance the vasoconstrictor/hypertrophic/proliferative effects of Ang II. 4) Chronic exercise of aged rats results in a decrease in chymase activity and suppression of oxidative stress, enables recovery of ACE and ACE2 activity to enhance Ang 1-7 levels, and normalizes MasR expression. As such, the overall effect of exercise reestablishes balance between the two opposite pathways (Fig. 7). Taken together, our studies clarified for the first time the pathological significance of chymase in the age-dependent alteration of cardiac Ang II signaling, an impairment that is reversible by chronic exercise training. Our results may serve as an explanation, at least in part, for the inadequate outcomes that result from long-term use of renin and ACE inhibitors compared with other antihypertensive classes (21, 37, 47). Moreover, our findings shed light on the clinical interventions that give merit to chymase as a therapeutic target, instead of the current use of ACE inhibitors alone, to reverse the adverse Ang II profile in aged populations.
Fig. 7.

Schematic illustration of changes in cardiac angiotensin signaling, as a function of aging and exercise. A: aging hearts exhibit reductions in Ang I, ACE, ACE2, and MasR, whereas upregulation of chymase and AT1R, leading to significant increase in Ang II, parallel with increases in superoxide that in turn stimulates chymase-derived Ang II and attenuation in Ang 1-7. B: exercise training primarily reverses all of the alterations via suppressing chymase and superoxide while normalizing actions of ACE, ACE2, and MasR. C: as a result, the age-dependent imbalance between the Ang II and Ang 1-7 axes is primarily corrected by exercise training. (+) indicates a stimulation; (−) indicates an inhibition.
Potentiation of chymase/Ang II/AT1R/Nox signaling, as a function of age.
The local RAS in cardiomyocytes and coronary vessels is propelled by the de novo synthesis of Ang I from renin, followed by conversion of Ang I to Ang II through the rate-limiting cleavage step by ACE (40, 44). Alternatively, an endopeptidase, chymase, has been identified in human and animal hearts and constitutes a non-ACE-related pathway for Ang II production (21, 56). In this regard, chymase is believed to play a pivotal role in Ang II formation in human hearts (55). The contribution of chymase to pathophysiological changes in the cardiovascular system has attracted considerable attention based on observations that long-term treatment of patients with ACE inhibitors is associated with a phenomenon known as “angiotensin II escape,” which is characterized as the rebound of circulating Ang II to pretreatment levels (36). Additionally, evidence exists showing that not all cardiac Ang II formation and action are sensitive to angiotensin receptor blockers (ARBs) or ACE inhibitors (6, 15, 23). Moreover, during the process of angiotensin signaling, local modifications/alterations in the synthesis of Ang II can result from changes in activity and structure of RAS enzymes; such changes can be exacerbated with age-related excess of local Ang II (30). Reports show that cardiac-derived Ang II mediates cardiomyocyte remodeling. This remodeling is responsible for age-dependent cardiac dysfunction and heart failure observed in transgenic mice that develop Ang II-mediated cardiac hypertrophy in the absence of hypertension (17). In this context, the nature of tissue-specific formation of Ang II as a function of aging formed the basis of the present study, which was aimed to extend our current understanding of age-dependent changes in cardiac RAS components and alternative formation of Ang II. Indeed, our results indicate that aged hearts exhibit a significantly high level of Ang II (Fig. 1B), a finding that is paradoxically coupled to declines in its precursor (Ang I; Fig. 1A) and its rate-limiting enzyme (ACE; Fig. 2). This negates the possibility that the response is ACE dependent and challenges a generally accepted concept that the increased level of Ang II is due exclusively to elevated ACE activity. As such, upregulation of chymase expression and greater chymase activity in aged hearts (Fig. 4) reveal a strong causal relationship between the enzyme action and formation of its product (Ang II), thus supporting chymase as the primary source for cardiac Ang II in aged rats.
Because mast cells are the predominant producers of chymase in heart tissue, the result of increases in cardiac mast cells, as indicated by upregulation of tryptase (Fig. 4C), provides molecular evidence for the mast cell-sourced increase in chymase, as a function of age. Furthermore, the augmentation of superoxide in aged hearts (Fig. 5) is able to trigger a shift of mast cells from a nonactivated to activated state, resulting in the release of more chymase (32, 33) to further generate Ang II. Specifically, the activated mast cell is defined as the cells that have previously been matured by either binding with their immunoglobin ligands or through nonligand-mediated pathways, such as by ROS, and are ready for degranulation (release of their granules) in response to the next stimulus (13, 32). Consistent with our findings, an association between aging and increases in mast cell number and activation has also been reported (10). In regards to whether ROS serves as an initiator to stimulate release of chymase or as a consequence of activation of chymase/Ang II/Nox signaling, this issue remains undetermined; however, we believe that oxidative stress and activation of chymase act as reciprocal causations during the pathogenesis of age-specific alteration of Ang II signaling (Fig. 7A).
One study has also demonstrated a switch from ACE-dependent to ACE-independent formation of Ang II during progression of metabolic syndrome, an age-dependent metabolic disorder. Accordingly, diabetic kidneys exhibit vasoconstriction in response to exogenous administration of Ang I. This response was sensitive to inhibitors of serine protease (chymase) but resistant to those of ACE. An effect directly opposite to this was observed in kidneys of control mice (41). Thus the evidence for chymase-dependent accumulation of Ang II in aged hearts, not only offers an explanation for the increase in Ang II paradoxically associated with reductions in its precursor (Ang I) and ACE, but also additionally indicates a pathological involvement of chymase in age-specific Ang II generation via a distinctly different pathway from that of the canonical ACE-dependent pathway.
Age-dependent compromise of the ACE2/Ang-1-7/MasR axis.
ACE2 acts on Ang II to produce Ang 1-7 that binds to MasR and evokes protective properties to counterbalance ACE/AT1R-dependent detrimental activity (48). Since the discovery of ACE2 over a decade ago (18), a balance between these two opposing pathways (ACE vs. ACE2) has served as an important parameter in pathophysiological evaluation of RAS actions (42). In this context, not only the Ang II/ACE/AT1R axis, but also the ACE2/Ang 1-7/MasR arm merits consideration as regulatory targets in governing the function of RAS (58). In accordance with this concept, altered actions of both pathways have been linked to a variety of cardiovascular pathologies, including age-related cardiac dysfunction. For instance, renal ACE2 gene expression was downregulated in multiple hypertensive models (12). Moreover, heterozygous ACE2-deficient animals were susceptible to heart diseases (59), and homozygous ACE2-knockout mice developed an age-dependent cardiomyopathy with an accumulation of cardiac Ang II (12). On the other hand, myocardial infarction-induced heart failure and ischemic cardiomyopathy were reported to be alleviated significantly in response to administration of Ang 1-7 (5, 35). Consistent with these reports, we also indicate an age-dependent compromise of the protective signaling, characterized as significant suppression of ACE2 expression and activity (Fig. 3), followed by a decrease in cardiac Ang 1-7 (Fig. 1C) and concomitant association with a more than twofold downregulation of MasR (Fig. 6C) in aged compared with young rats. Results similar to these are observed in type II diabetic mice (41). ACE2, which exhibits the highest catalytic efficiency of any of the RAS enzymes, has emerged as a crucial regulator of cardiac function and blood pressure, based on its ability to hydrolyze Ang II to Ang 1-7 (43). This notion of ACE2 serving as an Ang II clearance enzyme is supported by evidence that inactivation of ACE2 is correlated with a deficiency in plasma Ang II clearance following a bolus injection of Ang II (29). As such, a decline in ACE2 and ACE participates reciprocally with the increase in chymase action, in the accumulation of Ang II with concomitant attenuation of Ang 1-7 in aged hearts. With regard to the potential use of ACE inhibitors and ARBs in improvement of physical function among older adults (8), several studies have improved our basic understanding of critical pathophysiological factors that modulate the RAS to improve cardiac function. It is important to note, however, that the long-term therapeutic efficiency of ACE inhibitors to restore age-related decline in cardiovascular function is somehow weak at best (21, 37, 47). Nevertheless, favorable outcomes are observed in the elderly patients treated with ARBs (34, 49). This phenomenon can be mechanistically explained, at least in part, by our findings that show 1) the involvement of the ACE-independent Ang II pathway and 2) an upregulation of AT1 and downregulation of Mas receptors, as a function of aging.
Exercise balances the age-induced shifts between Chymase/Ang II/AT1R and ACE2/Ang-1-7/MasR.
Accumulating evidence shows that exercise favors a shift in the RAS toward the ACE2/Ang 1-7/MasR pathway in both normal and diseased hearts (7, 19, 62), skeletal muscles (27), and kidneys (50). To the best of our knowledge, however, this is the first study that indicates an age-specific chymase-dependent increase in Ang II that is reversed by exercise training. Our findings show a phenotype of exercise-induced optimal regulation of the ACE2/Ang 1-7/MasR axis that is identical to those reported previously. This phenotype is characterized by the potentiation of ACE2 activity (Fig. 3), restoration of Ang 1-7 levels (Fig. 1C), and increase in expression of MasR (Fig. 6C), leading to a reduction in Ang II (Fig. 1B) and suppression of superoxide (Fig. 5). Nonetheless, the novel finding presented in the present study is that exercise training, independent of aging, can enhance ACE activity (Fig. 2) and decrease chymase activity (Fig. 4). As such, the exercise-induced normalization of ACE (increased) and chymase (decreased) work in concert with the upregulation of ACE2 to shift the RAS pathway away from Ang II production toward production of Ang 1-7. This shift is accomplished by favoring ACE2-mediated degradation of Ang II to Ang 1-7, and Ang I to Ang 1-9, the latter of which is subsequently converted by the corrected ACE activity to further increase Ang 1-7 content in the tissue. In addition, even in the absence of significant effects on AT1R (Fig. 6A), exercise can still amplify Ang 1-7-mediated cardioprotective responses (Fig. 7) through the upregulation of downstream MasR expression (Fig. 6C).
Perspectives and alternatives.
The literature is replete with studies that highlight a residual risk of cardiovascular events in patients who are treated with RAS inhibitors, a phenomenon that has been attributed to insufficient inhibition of Ang II synthesis or incomplete blockade of intracellular-based actions of Ang II (51). Pharmacological approaches that prevent Ang II synthesis, inhibit ACE activity, and block the peptide binding to AT1R epitomize the foundation of current treatment and prevention strategies against heart diseases. However, these therapies fail to effectively inhibit the intracellular formation or activity of Ang II. Thus the broader implication of the present study, which shows an age-specific increase in chymase-dependent Ang II production, is that ACE inhibitors alone may not be sufficient for preventing cardiac functional decline in aged populations; they may require combinatorial treatment with both ACE and chymase inhibitors to optimally protect the elderly heart from Ang II-induced deteriorations. On the other hand, while this study indicates a correlation between cardiac chymase and age, it has not yet provided a functional link between the chymase-dependent formation of Ang II and changes in cardiac function or explored possible mechanisms responsible for the exercise-dependent modulation of functional changes via providing chymase inhibitors; these issues will need to be clarified in our future studies.
GRANTS
This work was supported by NIH grants R01 HL129797 and HL070653.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
G.F., J.T.P., A.H., and D.S. conceived and designed research; G.F., Y.L., S.K., and Y.A. performed experiments; G.F., A.H., and D.S. analyzed data; G.F., J.T.P., A.H., and D.S. interpreted results of experiments; G.F., A.H., and D.S. prepared figures; G.F. and A.H. drafted manuscript; G.F., J.T.P., A.H., and D.S. edited and revised manuscript; G.F., J.T.P., Y.L., S.K., Y.A., A.H., and D.S. approved final version of manuscript.
REFERENCES
- 1.Abadir PM, Walston JD, Carey RM. Subcellular characteristics of functional intracellular renin-angiotensin systems. Peptides 38: 437–445, 2012. doi: 10.1016/j.peptides.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmad S, Simmons T, Varagic J, Moniwa N, Chappell MC, Ferrario CM. Chymase-dependent generation of angiotensin II from angiotensin-(1-12) in human atrial tissue. PLoS One 6: e28501, 2011. doi: 10.1371/journal.pone.0028501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alzayadneh EM, Chappell MC. Angiotensin-(1-7) abolishes AGE-induced cellular hypertrophy and myofibroblast transformation via inhibition of ERK1/2. Cell Signal 26: 3027–3035, 2014. doi: 10.1016/j.cellsig.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asghar M, George L, Lokhandwala MF. Exercise decreases oxidative stress and inflammation and restores renal dopamine D1 receptor function in old rats. Am J Physiol Renal Physiol 293: F914–F919, 2007. doi: 10.1152/ajprenal.00272.2007. [DOI] [PubMed] [Google Scholar]
- 5.Averill DB, Ishiyama Y, Chappell MC, Ferrario CM. Cardiac angiotensin-(1-7) in ischemic cardiomyopathy. Circulation 108: 2141–2146, 2003. doi: 10.1161/01.CIR.0000092888.63239.54. [DOI] [PubMed] [Google Scholar]
- 6.Baker KM, Kumar R. Intracellular angiotensin II induces cell proliferation independent of AT1 receptor. Am J Physiol Cell Physiol 291: C995–C1001, 2006. doi: 10.1152/ajpcell.00238.2006. [DOI] [PubMed] [Google Scholar]
- 7.Barretti DL, Magalhães FC, Fernandes T, do Carmo EC, Rosa KT, Irigoyen MC, Negrão CE, Oliveira EM. Effects of aerobic exercise training on cardiac renin-angiotensin system in an obese Zucker rat strain. PLoS One 7: e46114, 2012. doi: 10.1371/journal.pone.0046114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carter CS, Onder G, Kritchevsky SB, Pahor M. Angiotensin-converting enzyme inhibition intervention in elderly persons: effects on body composition and physical performance. J Gerontol A Biol Sci Med Sci 60: 1437–1446, 2005. doi: 10.1093/gerona/60.11.1437. [DOI] [PubMed] [Google Scholar]
- 9.Chappell MC. Biochemical evaluation of the renin-angiotensin system: the good, bad, and absolute? Am J Physiol Heart Circ Physiol 310: H137–H152, 2016. doi: 10.1152/ajpheart.00618.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chatterjee V, Gashev AA. Aging-associated shifts in functional status of mast cells located by adult and aged mesenteric lymphatic vessels. Am J Physiol Heart Circ Physiol 303: H693–H702, 2012. doi: 10.1152/ajpheart.00378.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conti S, Cassis P, Benigni A. Aging and the renin-angiotensin system. Hypertension 60: 878–883, 2012. doi: 10.1161/HYPERTENSIONAHA.110.155895. [DOI] [PubMed] [Google Scholar]
- 12.Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da Costa J, Zhang L, Pei Y, Scholey J, Ferrario CM, Manoukian AS, Chappell MC, Backx PH, Yagil Y, Penninger JM. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 417: 822–828, 2002. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 13.Dahlin JS, Hallgren J. Mast cell progenitors: origin, development and migration to tissues. Mol Immunol 63: 9–17, 2015. doi: 10.1016/j.molimm.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 14.Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res 110: 1109–1124, 2012. doi: 10.1161/CIRCRESAHA.111.246140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Danser AH. Cardiac angiotensin II: does it have a function? Am J Physiol Heart Circ Physiol 299: H1304–H1306, 2010. doi: 10.1152/ajpheart.00881.2010. [DOI] [PubMed] [Google Scholar]
- 16.Diz DI, Lewis K. Lewis K. Dahl memorial lecture: The renin-angiotensin system and aging. Hypertension 52: 37–43, 2008. doi: 10.1161/HYPERTENSIONAHA.107.108985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Domenighetti AA, Wang Q, Egger M, Richards SM, Pedrazzini T, Delbridge LM. Angiotensin II-mediated phenotypic cardiomyoc yte remodeling leads to age-dependent cardiac dysfunction and failure. Hypertension 46: 426–432, 2005. doi: 10.1161/01.HYP.0000173069.53699.d9. [DOI] [PubMed] [Google Scholar]
- 18.Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res 87: E1–E9, 2000. doi: 10.1161/01.RES.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 19.Fernandes T, Hashimoto NY, Magalhães FC, Fernandes FB, Casarini DE, Carmona AK, Krieger JE, Phillips MI, Oliveira EM. Aerobic exercise training-induced left ventricular hypertrophy involves regulatory MicroRNAs, decreased angiotensin-converting enzyme-angiotensin ii, and synergistic regulation of angiotensin-converting enzyme 2-angiotensin (1-7). Hypertension 58: 182–189, 2011. doi: 10.1161/HYPERTENSIONAHA.110.168252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrario CM. Cardiac remodelling and RAS inhibition. Ther Adv Cardiovasc Dis 10: 162–171, 2016. doi: 10.1177/1753944716642677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrario CM, Ahmad S, Nagata S, Simington SW, Varagic J, Kon N, Dell’italia LJ. An evolving story of angiotensin-II-forming pathways in rodents and humans. Clin Sci (Lond) 126: 461–469, 2014. doi: 10.1042/CS20130400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferrario CM, Ahmad S, Varagic J, Cheng CP, Groban L, Wang H, Collawn JF, Dell Italia LJ. Intracrine angiotensin II functions originate from non-canonical pathways in the human heart. Am J Physiol Heart Circ Physiol 311: H404–H414, 2016. doi: 10.1152/ajpheart.00219.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, Diz DI, Gallagher PE. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 111: 2605–2610, 2005. doi: 10.1161/CIRCULATIONAHA.104.510461. [DOI] [PubMed] [Google Scholar]
- 24.Ferrario CM, Strawn WB. Role of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. Am J Cardiol 98: 121–128, 2006. doi: 10.1016/j.amjcard.2006.01.059. [DOI] [PubMed] [Google Scholar]
- 25.Fu L, Wei CC, Powell PC, Bradley WE, Ahmad S, Ferrario CM, Collawn JF, Dell’Italia LJ. Increased fibroblast chymase production mediates procollagen autophagic digestion in volume overload. J Mol Cell Cardiol 92: 1–9, 2016. doi: 10.1016/j.yjmcc.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gielen S, Schuler G, Adams V. Cardiovascular effects of exercise training: molecular mechanisms. Circulation 122: 1221–1238, 2010. doi: 10.1161/CIRCULATIONAHA.110.939959. [DOI] [PubMed] [Google Scholar]
- 27.Gomes-Santos IL, Fernandes T, Couto GK, Ferreira-Filho JC, Salemi VM, Fernandes FB, Casarini DE, Brum PC, Rossoni LV, de Oliveira EM, Negrao CE. Effects of exercise training on circulating and skeletal muscle renin-angiotensin system in chronic heart failure rats. PLoS One 9: e98012, 2014. doi: 10.1371/journal.pone.0098012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 74: 1141–1148, 1994. doi: 10.1161/01.RES.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 29.Gurley SB, Allred A, Le TH, Griffiths R, Mao L, Philip N, Haystead TA, Donoghue M, Breitbart RE, Acton SL, Rockman HA, Coffman TM. Altered blood pressure responses and normal cardiac phenotype in ACE2-null mice. J Clin Invest 116: 2218–2225, 2006. doi: 10.1172/JCI16980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horton RE, Yadid M, McCain ML, Sheehy SP, Pasqualini FS, Park SJ, Cho A, Campbell P, Parker KK. Angiotensin II induced cardiac dysfunction on a chip. PLoS One 11: e0146415, 2016. doi: 10.1371/journal.pone.0146415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ihara M, Urata H, Kinoshita A, Suzumiya J, Sasaguri M, Kikuchi M, Ideishi M, Arakawa K. Increased chymase-dependent angiotensin II formation in human atherosclerotic aorta. Hypertension 33: 1399–1405, 1999. doi: 10.1161/01.HYP.33.6.1399. [DOI] [PubMed] [Google Scholar]
- 32.Levick SP, Meléndez GC, Plante E, McLarty JL, Brower GL, Janicki JS. Cardiac mast cells: The centrepiece in adverse myocardial remodelling. Cardiovasc Res 89: 12–19, 2011. doi: 10.1093/cvr/cvq272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J, Lu H, Plante E, Meléndez GC, Levick SP, Janicki JS. Stem cell factor is responsible for the rapid response in mature mast cell density in the acutely stressed heart. J Mol Cell Cardiol 53: 469–474, 2012. doi: 10.1016/j.yjmcc.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li NC, Lee A, Whitmer RA, Kivipelto M, Lawler E, Kazis LE, Wolozin B. Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: Prospective cohort analysis. BMJ 340: b5465, 2010. doi: 10.1136/bmj.b5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loot AE, Roks AJ, Henning RH, Tio RA, Suurmeijer AJ, Boomsma F, van Gilst WH. Angiotensin-(1-7) attenuates the development of heart failure after myocardial infarction in rats. Circulation 105: 1548–1550, 2002. doi: 10.1161/01.CIR.0000013847.07035.B9. [DOI] [PubMed] [Google Scholar]
- 36.Lorenz JN. Chymase: The other ACE? Am J Physiol Renal Physiol 298: F35–F36, 2010. doi: 10.1152/ajprenal.00641.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mento PF, Wilkes BM. Plasma angiotensins and blood pressure during converting enzyme inhibition. Hypertension 9: III42–III48, 1987. doi: 10.1161/01.HYP.9.6_Pt_2.III42. [DOI] [PubMed] [Google Scholar]
- 38.Mercure C, Yogi A, Callera GE, Aranha AB, Bader M, Ferreira AJ, Santos RA, Walther T, Touyz RM, Reudelhuber TL. Angiotensin(1-7) blunts hypertensive cardiac remodeling by a direct effect on the heart. Circ Res 103: 1319–1326, 2008. doi: 10.1161/CIRCRESAHA.108.184911. [DOI] [PubMed] [Google Scholar]
- 39.Nakakubo H, Morita M, Imada T, Takai S, Shiota N, Miyazaki M, Nakamura N. Functional reconstitution of an active recombinant humanFunctional reconstitution of an active recombinant human chymase from Pichia pastoris cell lysate. Yeast 16: 1387–1396, 2000. doi: 10.1002/1097-0061(200011)16:15<1387::AID-YEA634>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen Dinh Cat A, Touyz RM. A new look at the renin-angiotensin system–focusing on the vascular system. Peptides 32: 2141–2150, 2011. doi: 10.1016/j.peptides.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 41.Park S, Bivona BJ, Kobori H, Seth DM, Chappell MC, Lazartigues E, Harrison-Bernard LM. Major role for ACE-independent intrarenal ANG II formation in type II diabetes. Am J Physiol Renal Physiol 298: F37–F48, 2010. doi: 10.1152/ajprenal.00519.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patel VB, Takawale A, Ramprasath T, Das SK, Basu R, Grant MB, Hall DA, Kassiri Z, Oudit GY. Antagonism of angiotensin 1-7 prevents the therapeutic effects of recombinant human ACE2. J Mol Med (Berl) 93: 1003–1013, 2015. doi: 10.1007/s00109-015-1285-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patel VB, Zhong JC, Grant MB, Oudit GY. Role of the ACE2/angiotensin 1-7 axis of the renin-angiotensin system in heart failure. Circ Res 118: 1313–1326, 2016. doi: 10.1161/CIRCRESAHA.116.307708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev 86: 747–803, 2006. doi: 10.1152/physrev.00036.2005. [DOI] [PubMed] [Google Scholar]
- 45.Payne V, Kam PC. Mast cell tryptase: a review of its physiology and clinical significance. Anaesthesia 59: 695–703, 2004. doi: 10.1111/j.1365-2044.2004.03757.x. [DOI] [PubMed] [Google Scholar]
- 46.Peach MJ. Renin-angiotensin system: Biochemistry and mechanisms of action. Physiol Rev 57: 313–370, 1977. [DOI] [PubMed] [Google Scholar]
- 47.Rousseau MF, Konstam MA, Benedict CR, Donckier J, Galanti L, Melin J, Kinan D, Ahn S, Ketelslegers JM, Pouleur H. Progression of left ventricular dysfunction secondary to coronary artery disease, sustained neurohormonal activation and effects of ibopamine therapy during long-term therapy with angiotensin-converting enzyme inhibitor. Am J Cardiol 73: 488–493, 1994. doi: 10.1016/0002-9149(94)90680-7. [DOI] [PubMed] [Google Scholar]
- 48.Santos RA. Angiotensin-(1-7). Hypertension 63: 1138–1147, 2014. doi: 10.1161/HYPERTENSIONAHA.113.01274. [DOI] [PubMed] [Google Scholar]
- 49.Simon CB, Lee-McMullen B, Phelan D, Gilkes J, Carter CS, Buford TW. The renin-angiotensin system and prevention of age-related functional decline: where are we now? Age (Dordr) 37: 9753, 2015. doi: 10.1007/s11357-015-9753-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Somineni HK, Boivin GP, Elased KM. Daily exercise training protects against albuminuria and angiotensin converting enzyme 2 shedding in db/db diabetic mice. J Endocrinol 221: 235–251, 2014. doi: 10.1530/JOE-13-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.St John Sutton M, Pfeffer MA, Plappert T, Rouleau JL, Moye LA, Dagenais GR, Lamas GA, Klein M, Sussex B, Goldman S. Quantitative two-dimensional echocardiographic measurements are major predictors of adverse cardiovascular events after acute myocardial infarction. The protective effects of captopril. Circulation 89: 68–75, 1994. doi: 10.1161/01.CIR.89.1.68. [DOI] [PubMed] [Google Scholar]
- 52.Sun D, Huang A, Koller A, Kaley G. Decreased arteriolar sensitivity to shear stress in adult rats is reversed by chronic exercise activity. Microcirculation 9: 91–97, 2002. doi: 10.1080/713774057. [DOI] [PubMed] [Google Scholar]
- 53.Sun D, Huang A, Koller A, Kaley G. Enhanced NO-mediated dilations in skeletal muscle arterioles of chronically exercised rats. Microvasc Res 64: 491–496, 2002. doi: 10.1006/mvre.2002.2450. [DOI] [PubMed] [Google Scholar]
- 54.Takai S, Jin D. Improvement of cardiovascular remodelling by chymase inhibitor. Clin Exp Pharmacol Physiol 43: 387–393, 2016. doi: 10.1111/1440-1681.12549. [DOI] [PubMed] [Google Scholar]
- 55.Urata H, Kinoshita A, Misono KS, Bumpus FM, Husain A. Identification of a highly specific chymase as the major angiotensin II-forming enzyme in the human heart. J Biol Chem 265: 22348–22357, 1990. [PubMed] [Google Scholar]
- 56.Urata H, Nishimura H, Ganten D. Chymase-dependent angiotensin II forming systems in humans. Am J Hypertens 9: 277–284, 1996. doi: 10.1016/0895-7061(95)00349-5. [DOI] [PubMed] [Google Scholar]
- 57.Vajapey R, Rini D, Walston J, Abadir P. The impact of age-related dysregulation of the angiotensin system on mitochondrial redox balance. Front Physiol 5: 439, 2014. doi: 10.3389/fphys.2014.00439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Varagic J, Ahmad S, Nagata S, Ferrario CM. ACE2: Angiotensin II/angiotensin-(1-7) balance in cardiac and renal injury. Curr Hypertens Rep 16: 420, 2014. doi: 10.1007/s11906-014-0420-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang W, Patel VB, Parajuli N, Fan D, Basu R, Wang Z, Ramprasath T, Kassiri Z, Penninger JM, Oudit GY. Heterozygote loss of ACE2 is sufficient to increase the susceptibility to heart disease. J Mol Med (Berl) 92: 847–858, 2014. doi: 10.1007/s00109-014-1149-y. [DOI] [PubMed] [Google Scholar]
- 60.Wolin MS. Reactive oxygen species and the control of vascular function. Am J Physiol Heart Circ Physiol 296: H539–H549, 2009. doi: 10.1152/ajpheart.01167.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang M, Huang W, Bai J, Nie X, Wang W. Chymase inhibition protects diabetic rats from renal lesions. Mol Med Rep 14: 121–128, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zucker IH, Schultz HD, Patel KP, Wang H. Modulation of angiotensin II signaling following exercise training in heart failure. Am J Physiol Heart Circ Physiol 308: H781–H791, 2015. doi: 10.1152/ajpheart.00026.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]