

Abstract
Three decades ago a revolutionary idea was born that ascribed to dysfunctional endothelia some manifestations of diabetes, the Steno hypothesis, so named after the Steno Diabetes Center, Gentofte, in Denmark. Here I briefly outline the accomplishments accrued in the past 15 years to buttress this hypothesis. Those include development of novel technological platforms to examine microcirculatory beds, deeper understanding of patterns of microvascular derangement in diabetes, pathophysiology of nitric oxide synthesis and availability, nitrosative and oxidative stress in diabetes, premature senescence of endothelial cells and the role of sirtuin 1 and lysosomal dysfunction in this process, and the state of endothelial glycocalyx and endothelial progenitor cells in diabetes. These pathophysiological findings may yield some therapeutic benefits.
Keywords: premature cell senescence, sirtuin 1, nitric oxide, endothelial glycocalyx, lysosomal dysfunction
thirty years ago, delivering a Claude Bernard lecture awarded by the European Association for the Study of Diabetes, T. Deckert (14) formulated a hypothesis named after the institution where he worked, Steno Diabetes Center, Steno hypothesis. The essence of this hypothesis, revolutionary for that time, was the fact that it recognized the important role played by the global dysfunction of the vascular endothelium in the development of albuminuria in type 1 diabetes mellitus. Ten years later, his compatriot, C. Mogensen, was giving a Claude Bernard lecture where he confirmed the role of vascular dysfunction in development of microalbuminuria and hypertension developing in the course of type 1 and type 2 diabetes (2, 34). A year later, at the meeting of the Hypertension Council of the American Heart Association, I gave a lecture summarizing new developments of the Steno hypothesis, in particular, focusing on our studies of nitric oxide (NO) in diabetes and metabolic syndrome and explaining endothelial dysfunction by the uncoupling of endothelial nitric oxide synthase (eNOS) (20). I must say that the field has advanced at a steady pace since then. Therefore, in summarizing advances made in the intervening 15 years, plenty of material had to be sacrificed to fit the strict page limitation. The present review of the subject is based on my recent presentation at the American Society of Nephrology 2015 meeting and focuses on the Steno hypothesis and recent discoveries in the field that advance deeper understanding of the pathogenesis, manifestations, and sequelae of endothelial cell dysfunction in diabetes mellitus. It is by no means exhaustive, rather it emphasizes a few selected pathways leading to microvasculopathy, such as eNOS uncoupling and NO deficiency, oxidative and nitrosative stress, lysosomal membrane permeabilization and sirtuin 1 (SIRT1) deficiency, premature senescence of endothelial and endothelial progenitor cells, and disintegration of endothelial glycocalyx (Fig. 1).
Fig. 1.

The evolution of the Steno hypothesis from its original (A) to our interpretation of the subject in 2001 (B) and my present-day view on endothelial dysfunction in diabetes mellitus (C). A: summary of the original Steno hypothesis that, most importantly, considered albuminuria as a manifestation of widespread microvascular damage. It was hypothesized that this defect is a result of impaired synthesis of what we would today consider endothelial glycocalyx. B: schematic view of hyperglycemia-induced transient and cumulative effects on vascular wall leading to impaired endothelial nitric oxide synthase (eNOS) activity and nitric oxide (NO) synthesis that in turn affect matrix synthesis and degradation, angiogenesis, and vascular permeability (from Ref. 20 with permission). C: several pathways activated in vascular endothelial cells exposed to hyperglycemia and advanced glycosylation end products (AGEs). Notably, all of these alterations lead to endothelial cell dysfunction manifesting in microalbuminuria. PAI-1, plasminogen activator inhibitor-1; ECM, extracellular matrix; PAR-2, protease-activated receptor-2.
Technical advances in imaging techniques.
The progress in deciphering the functions of microvasculature in diabetes to a significant degree can be attributed to developments in the field of intravital imaging. Many known as well as the advent of new approaches to examining microvasculature proved to be indispensable in advancing our knowledge of microvasculature. During these years confocal and multiphoton microscopy approaches have become routine, and their application for intravital (video)microscopy has been broadening. Laser Doppler flow probes and imagers have advanced, as have microscopy devices based on novel platforms, as exemplified by the orthogonal polarization spectral imaging (a method for imaging small blood vessels), side stream dark field imaging, optical coherence tomography, endomicroscopy (a technique for obtaining histology-like images from inside the human body in real time, an “optical biopsy”), thermography, and laser speckle imaging (based on the speckled interference pattern modified by moving red blood cells); all of the above techniques have been detailed elsewhere (reviewed in Ref. 59). Development and refinement of microcomputed tomography allowed for the three-dimensional reconstruction of the microvasculature (29). These techniques have a sufficient resolution to detect minute aberrations of microvasculature in diabetes.
Patterns of microvascular derangement in diabetes.
Indeed, studies of microvasculature are paramount to understanding diabetic vessels because the pathology of macrovascular complications is very much similar to that of accelerated atherosclerosis without diabetes, whereas microvascular lesions, in the opinion of Shah and Brownlee, represent diabetes mellitus signature, a trio of retinal-, renal-, and nerve-associated microvessels (51). Indeed, dysfunctional endothelium lining microvascular beds is responsible, at least in part, for such manifestation of diabetes, as microalbuminuria of early diabetic nephropathy, peripheral neuropathy, retinopathy, or impaired wound healing. In accord with the Steno hypothesis, Weil et al. (57), using transmission electron microscopy, detected ultrastructural changes in glomerular endothelial cells from patients with type 2 diabetes but with normoalbuminuria. Moreover, podocyte foot processes in these Pima Indian patients were unperturbed, but endothelial fenestrae significantly diminished. The similar observations of ultrastructural alterations of glomerular endothelial cells were reported in type 1 diabetics before establishment of microalbuminuria (54).
Recent study of the causes of increased microvascular permeability in diabetes associated with the metabolic syndrome focused on cathepsin S (27). Authors demonstrated that cathepsin S secreted by the invading macrophages activates protease-activated receptor-2 on endothelial cells, resulting in the increase in microvascular permeability manifesting as albuminuria. In accord with this, administration of inhibitors of either cathepsin S or protease-activated receptor-2 prevented ultrastructural and functional endothelial abnormalities and attenuated albuminuria and glomerulosclerosis.
Vascular endothelial growth factors (VEGF) exert multiple effects on endothelial cells, such as an increase in permeability, prosurvival signaling, and proliferative and angiogenic programs; therefore, a great deal of studies examined expression of VEGF in diabetes. These studies yielded highly controversial results: from overexpression to suppression and from beneficial to adverse effects of VEGF neutralizing antibodies (reviewed in Ref. 31). Perhaps, it could be explained by the fact that at the early stages of diabetic nephropathy VEGF is produced in excess, thus neutralizing antibodies may improve renal dysfunction and reduce albuminuria (13, 52). With further disease progression VEGF expression decreases in human diabetic nephropathy (30) and is accompanied by microvascular rarefaction. In our studies, Gealekman et al. (19) observed only a modest impairment of VEGF but a profound reduction in Flk expression in kidneys with overt microvascular rarefaction of Zucker diabetic fat (ZDF) compared with Zucker lean rats at 22 wk of age. We attempted to dissect the role of hyperglycemia in angiogenesis (25) by studying “sandwich” cocultures of endothelial cells with either glomerular or proximal tubular epithelial cells in the absence of serum: the only survival and angiogenic factor, VEGF, was secreted by the epithelial cells. When ambient d-glucose (but not l-glucose) level was increased to 30 mM, VEGF was found to be induced in both glomerular and tubular epithelial cells. Under these 30 mM d-glucose conditions, angiogenesis, which was normally well supported by both types of epithelia, became dissociated from VEGF abundance, even despite the fact that mRNAs encoding VEGF receptors were elevated. To understand this paradoxical lack of correlation between VEGF and angiogenesis at elevated glucose levels, it is necessary to consider effects of high d-glucose on eNOS and its function and role in angiogenesis, as detailed below.
Hyperglycemia and NO.
An acute effect of high d-glucose level on the availability of NO was studied by Brodsky et al. (4) who demonstrated using an amperometric electrochemical detection with an NO-selective porphyrinic microelectrode that NO-generating responses of endothelial cells to bradykinin or a calcium ionophore were diminished by elevated glucose levels. The similar effect of glucose was observed when we used purified bovine eNOS or even a solution of NO, thus suggesting that this effect could be the result of NO scavenging. This was, in fact, confirmed by mass spectrometry studies of NO and glucose solutions by demonstrating the formation of a covalent unitary addition of NO to glucose. We speculated that in a patient with diabetes, every hyperglycemic episode is associated with NO scavenging, sharp reduction in NO bioavailability, and impairment of NO-dependent endothelial functions, such as deterrence of leukocyte infiltration, platelet aggregation, imbalance in vasoreactive regulators, and reduced response to angiogenic cues (20). Notably, diabetic mice lacking eNOS are characterized by a more severe diabetic nephropathy (24, 60). In addition, studies of endothelial cell motility and angiogenic proficiency demonstrated that NO is necessary for scalar motions (we termed it “podokinesis”) and acquisition of vectorial movement when guidance cues are present (40, 41). Additional relevant effects of NO include activation of plasminogen activators and matrix metalloproteinases, inhibition of interstitial collagen synthesis, and downregulation of TGF-β and plasminogen activator inhibitor-1. These effects, together with eNOS uncoupling resulting from oxidative stress (see below), can explain in part the sequelae of chronic decline in NO bioavailability.
Nitrosative and oxidative stress of diabetes as a trigger for endothelial cell dysfunction.
As mentioned above, plasminogen activator inhibitor-1, a well-established marker of endothelial dysfunction, was detected in our differential display screens of endothelial cells cultured in the presence of nonenzymatically advanced glycosylated (AGE) collagen I compared with the native one (10). In the same model, Chen et al. (11) demonstrated induction of premature senescence of endothelial cells by AGEs. This process could be attenuated, both in vitro and in vivo, by administration of the peroxynitrite scavenger ebselen. Analysis of the mitochondrial generation of superoxide and NO revealed uncoupling of eNOS, normally docked to the outer mitochondrial membrane, and a surge in superoxide/hydrogen peroxide production (5) (Figs. 2 and 3).
Fig. 2.

Hyperglycemic switch from mitochondrial NO to superoxide production. A: human umbilical vein endothelial cells (HUVEC) respond to the calcium ionophore (A-23187) with an increase in mitochondrial NO production, as detected using diacetate (4-amino-5-methylamino-2′,7′-difluorofuorescein (DAF) fluorescence (after selective loading of DAF in mitochondria). This process is inhibited by elevated ambient d-glucose (but not l-glucose) level. B: pretreatment of HUVEC with a cell-permeable superoxide dismutase (SOD) mimetic, manganese (III) tetrakis (4-benzoic acid)porphyrin chloride (Mn-TBAP), protects mitochondrial NO production against impairment by high d-glucose. C: eNOS expression in mitochondrial fractions (mt) or whole cell lysates (cell) is not affected by high levels of ambient d-glucose. Cytochrome c oxidase (COX) was used as a mitochondrial marker. D: NO production by mitochondria isolated from the livers of Zucker diabetic fat (ZDF) or Zucker lean (ZL) rats. Note that NO generation by mitochondria of ZDF rats is impaired under basal and stimulated conditions (*P < 0.05).
Fig. 3.

Hyperglycemia enhances mitochondrial reactive oxygen species (ROS) production. A: representative images of dichlorodihydrofluorescein diacetate (DDF) colocalizing with MitoTracker-labeled mitochondria in HUVEC. B: HUVEC respond to the calcium ionophore with a minor “hump” in mitochondrial hydrogen peroxide production, as detected using DDF fluorescence (after selective loading of DDF in mitochondria). This process is dramatically enhanced by elevated ambient d-glucose (but not l-glucose) level. C: treatment of HUVEC cultured in 30 mM d-glucose for 24 h with Mn-TBAP abolishes the A-23187-induced increase in ROS production. D: ROS production by mitochondria isolated from livers of ZDF and ZL rats. Note that both the baseline and stimulated ROS production are enhanced in ZDF rats (*P < 0.05).
Endothelial cell dysfunction was detectable in ZDF rats at the age of 22 wk when diabetes becomes rampant. Notably, these metabolic changes were correctable with a mitochondrial-targeted superoxide dismutase mimetic. Furthermore, ZDF rats exhibited signs of peroxynitrite formation, as judged by 3-nitrotyrosine modification of proteins in renal microvasculature, and nephropathy could be prevented in these rats by treatment with a scavenger of peroxynitrite, ebselen (8).
This series of studies produced ex juvantibus evidence that 1) nitroso-oxydative stress contributes to endothelial dysfunction associated with diabetes and 2) endothelial functions can be partially restored by reducing this stress, in our case with the aid of ebselen.
AGEs and activation of receptor for advanced glycation end product-inducing premature senescence of endothelial cells.
AGEs are keepers of diabetic memory recording cumulative prevalence and duration of hyperglycemic episodes. The chemistry and biology of nonenzymatically glycosylated proteins have been exhaustively reviewed (Ref. 42 and references therein). We have previously demonstrated that a nonenzymatically glycosylated long-lived protein collagen I (GC) increased the proportion of senescent endothelial cells threefold compared with the native collagen I (NC) (Fig. 4). When a silencing RNA construct targeting receptor for AGEs, receptor for advanced glycation end product (RAGE), was added to the culture medium and cells were coincubated with GC or NC, the proportion of senescent endothelial cells was drastically reduced. These findings (J. Chen, unpublished observations) argue in favor of the key role played by AGE signaling via RAGE in inducing premature senescence of endothelial cells. Other diabetes-induced increased protein modifications, as for instance those accomplished by O-linked β-N-acetylglucosamine, have been reviewed elsewhere (51).
Fig. 4.
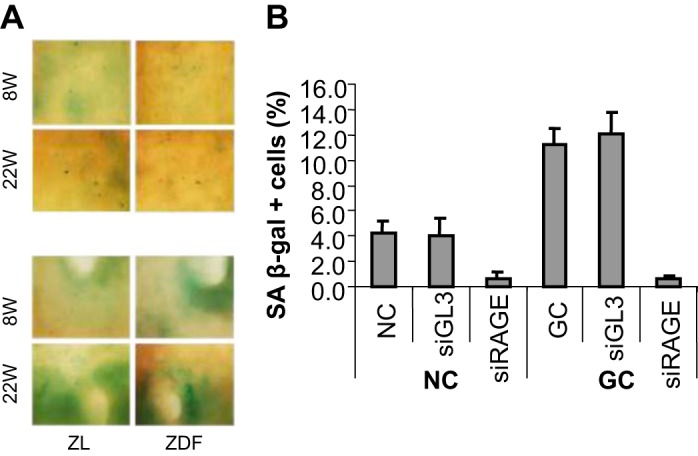
AGE-induced senescence of endothelial cells is receptor for advanced glycation end product (RAGE) dependent. A: representative images of aortic endothelium (top, plain of the aorta; bottom, orifices of intercostal arteries) of ZL and ZDF rats at ages of 8 and 22 wk. B: HUVEC were cultured for 5 days in the presence of native collagen I (NC) or glycosylated long-lived protein collagen I (GC) (100 μM each), and expression of senescence-associated β-galactosidase (SA-β-gal) was quantified. Parallel cultures were exposed to siRAGE or Luciferase GL3 siRNA used as a negative control. siRAGE, but not siGL3, dramatically reduced the proportion of SA-β-gal-positive cells (J. Chen and M. S. Goligorsky, unpublished observations).
In summarizing this group of studies, it appears that the AGE-RAGE pathway of endothelial cell activation is critically involved in pathogenesis of endothelial dysfunction, and controlling this pathway by either AGE breakers (see below) or by silencing RAGE signaling may ameliorate endothelial dysfunction.
Role of the SIRT1 system in promoting and maintaining premature cell senescence.
SIRT1, an NAD-dependent deacetylase, is one of the key regulators of cell longevity. In our studies (J. Chen, unpublished observations) we detected the reduction of SIRT1 expression in the whole kidney lysates of db/db mice (compared with dbm controls) at the age of 30 wk but not at the young age of 12 wk when hyperglycemia is just appearing (Fig. 5A).
Fig. 5.

Expression of sirtuin 1 (SIRT1) in diabetes. A: SIRT1 expression in kidney lysates obtained from db/db and dbm mice at ages 12 (time of a shift from normoglycemia to hyperglycemia) and 30 (time of persistent hyperglycemia) wk. Note that at 30 wk the expression of Sirt1 is reduced. The panel on the bottom summarizes these Western blotting studies. *P < 0.05 (unpublished observations). B: left, results of Western blot analysis of Sirt1 expression after its overexpression (SIRT1), inhibition (shRNA), and control transfection (Mock). Right, results of Sirt1 overexpression or suppression on apoptosis and senescence in cells subjected to GC or NC. *P < 0.05. Reprinted with permission from Ref. 9.
We then inquired whether it is affected by GC and in the course of diabetes. GC was responsible for accelerated apoptosis and premature senescence of cultured endothelial progenitor cells (Fig. 5B). Moreover, by overexpressing SIRT1, it was possible to attenuate apoptosis of GC-exposed endothelial progenitor cells, whereas further downregulation of SIRT1 using shRNA targeting SIRT1 resulted in deterioration of GC-induced apoptosis and premature senescence. Combined with the benefits of SIRT1-activating pharmaceuticals (Ref. 23 and see below), data show a role that this deacetylase plays in endothelial cell biology, especially during stress.
Lysosomal dysfunction, cell senescence, and degradation of SIRT1.
Patschan et al. (44–46) discovered that exposure to GC results in lysosomal permeabilization, as detected using prolonged exposure to a fluorescently labeled dextran, which accumulates in lysosomes, and the lysosomal membrane marker lysosome-associated protein-1 (LAMP-1). As shown in Fig. 6, under control conditions both markers are colocalized. Chloroquine, a known lysosomal membrane-permeabilizing agent, resulted in dissociation of dextran and LAMP-1 fluorescence. NC did not affect the normal colocalization of markers, whereas GC, at two different dilutions, dissociated the localization of lysosomal markers. These findings are consistent with GC-induced permeabilization of the lysosomal membrane.
Fig. 6.

Lysosomal permeabilization by AGEs. A: schematic of the experimental protocol: colocalization of dextran and lysosome-associated protein-1 in control and their dissociation in a permeabilized state. B: glycated collagen induces lysosomal permeabilization in HUVEC. Studies were conducted using dextran-Texas red loading in a lysosomal compartment (followed by immunodetection of LAMP-1 and labeling of nuclei) under control conditions, following application of chloroquine (positive control for permeabilization of the lysosomal membrane) and native and glycated collagen at two different concentrations. A significant and comparable permeabilization of the lysosomal membrane is detected after application of either chloroquine or glycated collagen I.
In such a case, it is conceivable that GC is capable of the liberation of cathepsins from lysosomes. Indeed, Chen et al. (9) have shown that it is the case by detecting fluorescence of cathepsin B substrate. Furthermore, in collaboration with B. Turk at Ljubljana University, we were able to demonstrate in vitro that three different cathepsins cleaved SIRT1. It is possible that this mechanism is in part responsible for the reduction in SIRT1 expression, as shown above. On a broader scale, the studies point to the impairment of autophagy, accumulation of misfolded proteins, and dysfunctional organelles normally sequestered through the process of mitophagy and pexophagy.
Endothelial glycocalyx in diabetes.
As mentioned above, impaired anionic charge barrier was considered by Deckert to be a trigger for widespread vasculopathy and albuminuria. Endothelial glycocalyx, a surface layer of a well-balanced and intertwined mixture of glycoproteins, proteoglycans, and glycosaminoglycans creating a scaffold, is responsible for endothelial permeability, antithrombogenic properties, control of leukocyte trafficking, mechanosensing, and maintenance of electronegativity of the vascular luminal surface. It is disintegrated by oxidative stress, hence leading to impairment of the above functions (38, 48, 49, 58). Deckert and colleagues (15) observed that the fractional incorporation of [3H]glucosamine into heparan sulfate was significantly reduced in fibroblasts isolated from diabetes patients with albuminuria (but not those without or control healthy subjects). Most recently, upregulation of endothelin-1 in diabetes was found to induce heparanase in podocytes, which in turn led to impaired glomerular endothelial glycocalyx (18). In a series of studies of endothelial glycocalyx in diabetic rodents and patients with type 1 and type 2 diabetes mellitus, different investigators have demonstrated the loss of its integrity (26, 28, 36, 39). Myrup et al. (36) reported on benefits of low-dose heparin in reducing albuminuria. Furthermore, a synthetic analog of heparan sulfate, sulodexide, was able to significantly improve endothelial functions and reduce albuminuria (6). Evidently, the Steno hypothesis finds strong support in these studies.
Endothelial progenitor cells in diabetes.
Multiple investigators have demonstrated quantitative and/or qualitative defect in endothelial progenitor cells in experimental animals and patients with diabetes (16, 17, 26, 53). Chen et al. (12) have shown that in db/db mice endothelial progenitor cells are numerically deficient and show increased proportion of apoptotic cells compared with control dbm counterparts. However, the number and the viability of endothelial progenitors in db/db mice could be improved by treating animals in vivo or cells ex vivo with the peroxynitrite scavenger ebselen (Fig. 7). When these db/db mice received an adaptive transfer of bone marrow-derived cells obtained from dbm mice or when bone marrow-derived cells from db/db mice were treated ex vivo with ebselen before transplantation, this resulted in improved macrovasculopathy (as judged by the acetylcholine-induced relaxation of aortic rings) and reduced albuminuria (Fig. 8).
Fig. 7.

Analysis of bone marrow-derived endothelial progenitors. A-C: fluorescence-activated cell sorting (FACS) analysis (A and C) and direct cell culture of bone marrow-derived cells (B) indicate an increase in the proportion of apoptotic progenitors and a decrease in the number of progenitors detected through lectin+/LDL+ cells or Sca1+/Flk1+ cells in db/db mice. Pretreatment of db/db mice in vivo or db/db mice-derived bone marrow cells ex vivo with the organoselenic antioxidant and peroxinitrite scavenger ebselen results in improved viability and restored numbers of bone marrow-derived progenitors. P < 0.05 compared with db/m (*) and with TxCM (#). Reprinted with permission from Ref. 12.
Fig. 8.

Analysis of acetylcholine-induced vasorelaxation of aortic rings (A) and albuminuria (B) in db/db mice and after treatment with dbm bone marrow-derived cells (Txdb) or db/db bone marrow-derived cells exposed in vivo (Txdb-Ebs in vivo) and ex vivo (Txdb-Ebs ex vivo) to the organoselenic antioxidant and peroxinitrite scavenger ebselen. Note that adoptive transfer of syngeneic bone marrow-derived cells improves macro- and microvasculopathy. P < 0.05 compared with db/db (*), with db/db and Txdb (**), and with TxCM (#). Reprinted with permission from Ref. 12.
Future therapies of endothelial dysfunction in diabetes.
Well-established therapeutic interventions designed to alleviate endothelial cell dysfunction, including angiotensin-converting enzyme inhibitors and angiotensin receptor blockers, statins, and peroxisome proliferator-activated receptors, have been comprehensively reviewed elsewhere (21, 32, 33, 47, 50, 55). It is possible to derive valuable pathogenic information on a disease by analyzing which therapies are most effective, ex juvantibus. Along this line, use of AGE breakers and RAGE blockade appear to garner significant advantages (1, 37). Metformin has been strongly advocated as effective therapy of endothelial dysfunction in diabetes, since it has been shown to reduce cardiovascular risk points in type 2 diabetes, beyond its hypoglycemic effect, probably via activation of AMP-activated protein kinase and eNOS (7).
Several additional rational experimental strategies for vascular protection have recently emerged. Based on the discovery of SIRT1 and its activation by resveratrol, small-molecule SIRT1 activators have been synthesized and are currently tested (23). Sirtuin-activating compounds (STACs) exert their effect by allosteric activation of this deacetylase. Three generations of STACs include, in addition to resveratrol, quercetin and butein (first generation), SRT-1720, -1460, and -2183 (second generation), and STAC-5, -9, and -10 (third generation), all extending life span and/or health span. These compounds are presently undergoing clinical trials.
NAD+ is a cofactor necessary for activation of several sirtuins, and its bioavailability has been shown to be reduced (56). A precursor of NAD+, nicotinamide, is paving way as a therapy to correct NAD+ deficiency.
In fact, dietary restriction, which possibly induces SIRT1, acts via mammalian target of rapamycin (mTOR) signaling and NAD-dependent pathways accompanied by a shift toward oxidative metabolism (35), both representing novel venues of rejuvenation therapy. A recently described periodic diet mimicking fasting may become the preferred strategy (3).
Another venue for future therapies is based on the findings implicating mTOR activation and the resulting defect in autophagy in senescence. Application of the mTOR inhibitor rapamycin has been shown to act as a rejuvenating therapy, even when applied late in life (22). Other pharmacological modalities aimed at preventing premature cell senescence are also therapeutic targets in type 2 diabetes, including IGF-I axis, ceramides, and targeted components of senescence-associated secretory products (43).
In conclusion, the Steno hypothesis has not only survived for the past 30 years but continues to thrive, with gains in breadth and depth of understanding mechanisms of endothelial dysfunction in diabetes, and commences the harvest of novel therapeutic strategies.
GRANTS
These studies were supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-54602, D-K052783, and DK-45462 and Westchester Artificial Kidney Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author.
AUTHOR CONTRIBUTIONS
M.S.G. conception and design of research; M.S.G. interpreted results of experiments; M.S.G. drafted manuscript; M.S.G. edited and revised manuscript; M.S.G. approved final version of manuscript.
ACKNOWLEDGMENTS
I thank Dr. Jun Chen, who through the years has contributed handsomely to the studies reported herein and to many postdoctoral fellows, mentioned in the text, for their contributions to the project.
REFERENCES
- 1.Aldini G, Vistoli G, Stefek M, Chondrogianni N, Grune T, Sereikaite J, Sadowska-Bartosz I, Bartosz G. Molecular strategies to prevent, inhibit, and degrade advanced glycoxidation and advanced lipoxidation end products. Free Radic Res 47, Suppl 1: 93–137, 2013. doi: 10.3109/10715762.2013.792926. [DOI] [PubMed] [Google Scholar]
- 2.Bianchi S, Bigazzi R, Campese VM. Microalbuminuria in patient with essential hypertension. Cardiovascular and renal implications (4th ed.). In: The Kidney and Hypertension in Diabetes Mellitus, edited by Mogensen CE. Boston, MA: Kluwer Academic Publ, 1998, p. 569–584. doi: 10.1007/978-1-4757-6752-0_53. [DOI] [Google Scholar]
- 3.Brandhorst S, Choi IY, Wei M, Cheng CW, Sedrakyan S, Navarrete G, Dubeau L, Yap LP, Park R, Vinciguerra M, Di Biase S, Mirzaei H, Mirisola MG, Childress P, Ji L, Groshen S, Penna F, Odetti P, Perin L, Conti PS, Ikeno Y, Kennedy BK, Cohen P, Morgan TE, Dorff TB, Longo VD. A periodic diet that mimics fasting promotes multi-system regeneration, enhanced cognitive performance, and healthspan. Cell Metab 22: 86–99, 2015. doi: 10.1016/j.cmet.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brodsky SV, Morrishow AM, Dharia N, Gross SS, Goligorsky MS. Glucose scavenging of nitric oxide. Am J Physiol Renal Physiol 280: F480–F486, 2001. [DOI] [PubMed] [Google Scholar]
- 5.Brodsky SV, Gao S, Li H, Goligorsky MS. Hyperglycemic switch from mitochondrial nitric oxide to superoxide production in endothelial cells. Am J Physiol Heart Circ Physiol 283: H2130–H21, 2002. doi: 10.1152/ajpheart.00196.2002. [DOI] [PubMed] [Google Scholar]
- 6.Broekhuizen LN, Lemkes BA, Mooij HL, Meuwese MC, Verberne H, Holleman F, Schlingemann RO, Nieuwdorp M, Stroes ES, Vink H. Effect of sulodexide on endothelial glycocalyx and vascular permeability in patients with type 2 diabetes mellitus. Diabetologia 53: 2646–2655, 2010. doi: 10.1007/s00125-010-1910-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calvert JW, Gundewar S, Jha S, Greer JJ, Bestermann WH, Tian R, Lefer DJ. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes 57: 696–705, 2008. doi: 10.2337/db07-1098. [DOI] [PubMed] [Google Scholar]
- 8.Chander PN, Gealekman O, Brodsky SV, Elitok S, Tojo A, Crabtree M, Gross SS, Goligorsky MS. Nephropathy in Zucker diabetic fat rat is associated with oxidative and nitrosative stress: prevention by chronic therapy with a peroxynitrite scavenger ebselen. J Am Soc Nephrol 15: 2391–2403, 2004. doi: 10.1097/01.ASN.0000135971.88164.2C. [DOI] [PubMed] [Google Scholar]
- 9.Chen J, Xavier S, Moskowitz-Kassai E, Chen R, Lu CY, Sanduski K, Špes A, Turk B, Goligorsky MS. Cathepsin cleavage of sirtuin 1 in endothelial progenitor cells mediates stress-induced premature senescence. Am J Pathol 180: 973–983, 2012. doi: 10.1016/j.ajpath.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J, Brodsky S, Li H, Hampel DJ, Miyata T, Weinstein T, Gafter U, Norman JT, Fine LG, Goligorsky MS. Delayed branching of endothelial capillary-like cords in glycated collagen I is mediated by early induction of PAI-1. Am J Physiol Renal Physiol 281: F71–F80, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Brodsky SV, Goligorsky DM, Hampel DJ, Li H, Gross SS, Goligorsky MS. Glycated collagen I induces premature senescence-like phenotypic changes in endothelial cells. Circ Res 90: 1290–1298, 2002. doi: 10.1161/01.RES.0000022161.42655.98. [DOI] [PubMed] [Google Scholar]
- 12.Chen J, Li H, Addabbo F, Zhang F, Pelger E, Patschan D, Park HC, Kuo MC, Ni J, Gobe G, Chander PN, Nasjletti A, Goligorsky MS. Adoptive transfer of syngeneic bone marrow-derived cells in mice with obesity-induced diabetes: selenoorganic antioxidant ebselen restores stem cell competence. Am J Pathol 174: 701–711, 2009. doi: 10.2353/ajpath.2009.080606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Vriese AS, Tilton RG, Elger M, Stephan CC, Kriz W, Lameire NH. Antibodies against vascular endothelial growth factor improve early renal dysfunction in experimental diabetes. J Am Soc Nephrol 12: 993–1000, 2001. [DOI] [PubMed] [Google Scholar]
- 14.Deckert T, Feldt-Rasmussen B, Borch-Johnsen K, Jensen T, Kofoed-Enevoldsen A. Albuminuria reflects widespread vascular damage. The Steno hypothesis. Diabetologia 32: 219–226, 1989. doi: 10.1007/BF00285287. [DOI] [PubMed] [Google Scholar]
- 15.Deckert T, Horowitz IM, Kofoed-Enevoldsen A, Kjellén L, Deckert M, Lykkelund C, Burcharth F. Possible genetic defects in regulation of glycosaminoglycans in patients with diabetic nephropathy. Diabetes 40: 764–770, 1991. doi: 10.2337/diab.40.6.764. [DOI] [PubMed] [Google Scholar]
- 16.Fadini GP, Sartore S, Albiero M, Baesso I, Murphy E, Menegolo M, Grego F, Vigili de Kreutzenberg S, Tiengo A, Agostini C, Avogaro A. Number and function of endothelial progenitor cells as a marker of severity for diabetic vasculopathy. Arterioscler Thromb Vasc Biol 26: 2140–2146, 2006. doi: 10.1161/01.ATV.0000237750.44469.88. [DOI] [PubMed] [Google Scholar]
- 17.Fadini GP, Sartore S, Schiavon M, Albiero M, Baesso I, Cabrelle A, Agostini C, Avogaro A. Diabetes impairs progenitor cell mobilisation after hindlimb ischaemia-reperfusion injury in rats. Diabetologia 49: 3075–3084, 2006. doi: 10.1007/s00125-006-0401-6. [DOI] [PubMed] [Google Scholar]
- 18.Garsen M, Lenoir O, Rops AL, Dijkman HB, Willemsen B, van Kuppevelt TH, Rabelink TJ, Berden JH, Tharaux PL, van der Vlag J. Endothelin-1 induces proteinuria by heparanase-mediated disruption of the glomerular glycocalyx. J Am Soc Nephrol 27: 3545–3551, 2016. doi: 10.1681/ASN.2015091070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gealekman O, Brodsky SV, Zhang F, Chander PN, Friedli C, Nasjletti A, Goligorsky MS. Endothelial dysfunction as a modifier of angiogenic response in Zucker diabetic fat rat: amelioration with Ebselen. Kidney Int 66: 2337–2347, 2004. doi: 10.1111/j.1523-1755.2004.66035.x. [DOI] [PubMed] [Google Scholar]
- 20.Goligorsky MS, Chen J, Brodsky S. Workshop: endothelial cell dysfunction leading to diabetic nephropathy : focus on nitric oxide. Hypertension 37: 744–748, 2001. doi: 10.1161/01.HYP.37.2.744. [DOI] [PubMed] [Google Scholar]
- 21.Goligorsky MS. Chronic kidney disease and vascular endothelium. In: Chronic Kidney Disease, edited by Kimmel P, Rosenberg M. New York, NY: Elsevier, 2015, chapt. 15, p. 170–180. [Google Scholar]
- 22.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460: 392–395, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hubbard BP, Sinclair DA. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci 35: 146–154, 2014. doi: 10.1016/j.tips.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanetsuna Y, Takahashi K, Nagata M, Gannon MA, Breyer MD, Harris RC, Takahashi T. Deficiency of endothelial nitric-oxide synthase confers susceptibility to diabetic nephropathy in nephropathy-resistant inbred mice. Am J Pathol 170: 1473–1484, 2007. doi: 10.2353/ajpath.2007.060481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim BS, Chen J, Weinstein T, Noiri E, Goligorsky MS. VEGF expression in hypoxia and hyperglycemia: reciprocal effect on branching angiogenesis in epithelial-endothelial co-cultures. J Am Soc Nephrol 13: 2027–2036, 2002. doi: 10.1097/01.ASN.0000024436.00520.D8. [DOI] [PubMed] [Google Scholar]
- 26.Kränkel N, Adams V, Linke A, Gielen S, Erbs S, Lenk K, Schuler G, Hambrecht R. Hyperglycemia reduces survival and impairs function of circulating blood-derived progenitor cells. Arterioscler Thromb Vasc Biol 25: 698–703, 2005. doi: 10.1161/01.ATV.0000156401.04325.8f. [DOI] [PubMed] [Google Scholar]
- 27.Kumar Vr S, Darisipudi MN, Steiger S, Devarapu SK, Tato M, Kukarni OP, Mulay SR, Thomasova D, Popper B, Demleitner J, Zuchtriegel G, Reichel C, Cohen CD, Lindenmeyer MT, Liapis H, Moll S, Reid E, Stitt AW, Schott B, Gruner S, Haap W, Ebeling M, Hartmann G, Anders HJ. Cathepsin S cleavage of protease-activated receptor-2 on endothelial cells promotes microvascular diabetic complications. J Am Soc Nephrol 27: 1635–1649, 2016. doi: 10.1681/ASN.2015020208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lemkes BA, Nieuwdorp M, Hoekstra JB, Holleman F. The glycocalyx and cardiovascular disease in diabetes: should we judge the endothelium by its cover? Diabetes Technol Ther 14, Suppl 1: S3–S10, 2012. doi: 10.1089/dia.2012.0011. [DOI] [PubMed] [Google Scholar]
- 29.Lerman LO, Chade AR. Angiogenesis in the kidney: a new therapeutic target? Curr Opin Nephrol Hypertens 18: 160–165, 2009. doi: 10.1097/MNH.0b013e32831ec1db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindenmeyer MT, Kretzler M, Boucherot A, Berra S, Yasuda Y, Henger A, Eichinger F, Gaiser S, Schmid H, Rastaldi MP, Schrier RW, Schlöndorff D, Cohen CD. Interstitial vascular rarefaction and reduced VEGF-A expression in human diabetic nephropathy. J Am Soc Nephrol 18: 1765–1776, 2007. doi: 10.1681/ASN.2006121304. [DOI] [PubMed] [Google Scholar]
- 31.Maezawa Y, Takemoto M, Yokote K. Cell biology of diabetic nephropathy: Roles of endothelial cells, tubulointerstitial cells and podocytes. J Diabetes Investig 6: 3–15, 2015. doi: 10.1111/jdi.12255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mervaala E, Finckenberg P, Lapatto R, Müller DN, Park JK, Dechend R, Ganten D, Vapaatalo H, Luft FC. Lipoic acid supplementation prevents angiotensin II-induced renal injury. Kidney Int 64: 501–508, 2003. doi: 10.1046/j.1523-1755.2003.00108.x. [DOI] [PubMed] [Google Scholar]
- 33.Miyata T, Suzuki N, van Ypersele de Strihou C. Diabetic nephropathy: are there new and potentially promising therapies targeting oxygen biology? Kidney Int 84: 693–702, 2013. doi: 10.1038/ki.2013.74. [DOI] [PubMed] [Google Scholar]
- 34.Mogensen CE. Microalbuminuria, blood pressure and diabetic renal disease: origin and development of ideas. Diabetologia 42: 263–285, 1999. doi: 10.1007/s001250051151. [DOI] [PubMed] [Google Scholar]
- 35.Moroz N, Carmona JJ, Anderson E, Hart AC, Sinclair DA, Blackwell TK. Dietary restriction involves NAD+ -dependent mechanisms and a shift toward oxidative metabolism. Aging Cell 13: 1075–1085, 2014. doi: 10.1111/acel.12273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Myrup B, Hansen PM, Jensen T, Kofoed-Enevoldsen A, Feldt-Rasmussen B, Gram J, Kluft C, Jespersen J, Deckert T. Effect of low-dose heparin on urinary albumin excretion in insulin-dependent diabetes mellitus. Lancet 345: 421–422, 1995. doi: 10.1016/S0140-6736(95)90403-4. [DOI] [PubMed] [Google Scholar]
- 37.Nagai R, Murray DB, Metz TO, Baynes JW. Chelation: a fundamental mechanism of action of AGE inhibitors, AGE breakers, and other inhibitors of diabetes complications. Diabetes 61: 549–559, 2012. doi: 10.2337/db11-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nieuwdorp M, Meuwese MC, Vink H, Hoekstra JB, Kastelein JJ, Stroes ES. The endothelial glycocalyx: a potential barrier between health and vascular disease. Curr Opin Lipidol 16: 507–511, 2005. doi: 10.1097/01.mol.0000181325.08926.9c. [DOI] [PubMed] [Google Scholar]
- 39.Nieuwdorp M, Mooij HL, Kroon J, Atasever B, Spaan JA, Ince C, Holleman F, Diamant M, Heine RJ, Hoekstra JB, Kastelein JJ, Stroes ES, Vink H. Endothelial glycocalyx damage coincides with microalbuminuria in type 1 diabetes. Diabetes 55: 1127–1132, 2006. doi: 10.2337/diabetes.55.04.06.db05-1619. [DOI] [PubMed] [Google Scholar]
- 40.Noiri E, Hu Y, Bahou WF, Keese CR, Giaever I, Goligorsky MS. Permissive role of nitric oxide in endothelin-induced migration of endothelial cells. J Biol Chem 272: 1747–1752, 1997. doi: 10.1074/jbc.272.3.1747. [DOI] [PubMed] [Google Scholar]
- 41.Noiri E, Lee E, Testa J, Quigley J, Colflesh D, Keese CR, Giaever I, Goligorsky MS. Podokinesis in endothelial cell migration: role of nitric oxide. Am J Physiol Cell Physiol 274: C236–C244, 1998. [DOI] [PubMed] [Google Scholar]
- 42.Nowotny K, Jung T, Höhn A, Weber D, Grune T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 5: 194–222, 2015. doi: 10.3390/biom5010194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palmer AK, Tchkonia T, LeBrasseur NK, Chini EN, Xu M, Kirkland JL. Cellular senescence in type 2 diabetes: a therapeutic opportunity. Diabetes 64: 2289–2298, 2015. doi: 10.2337/db14-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patschan S, Chen J, Gealekman O, Krupincza K, Wang M, Shu L, Shayman JA, Goligorsky MS. Mapping mechanisms and charting the time course of premature cell senescence and apoptosis: lysosomal dysfunction and ganglioside accumulation in endothelial cells. Am J Physiol Renal Physiol 294: F100–F109, 2008. doi: 10.1152/ajprenal.00261.2007. [DOI] [PubMed] [Google Scholar]
- 45.Patschan S, Chen J, Polotskaia A, Mendelev N, Cheng J, Patschan D, Goligorsky MS. Lipid mediators of autophagy in stress-induced premature senescence of endothelial cells. Am J Physiol Heart Circ Physiol 294: H1119–H1129, 2008. doi: 10.1152/ajpheart.00713.2007. [DOI] [PubMed] [Google Scholar]
- 46.Patschan S, Goligorsky MS. Autophagy: The missing link between non-enzymatically glycated proteins inducing apoptosis and premature senescence of endothelial cells? Autophagy 4: 521–523, 2008. doi: 10.4161/auto.5904. [DOI] [PubMed] [Google Scholar]
- 47.Pergola PE, Raskin P, Toto RD, Meyer CJ, Huff JW, Grossman EB, Krauth M, Ruiz S, Audhya P, Christ-Schmidt H, Wittes J, Warnock DG; BEAM Study Investigators . Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 365: 327–336, 2011. doi: 10.1056/NEJMoa1105351. [DOI] [PubMed] [Google Scholar]
- 48.Rabelink TJ, de Zeeuw D. The glycocalyx--linking albuminuria with renal and cardiovascular disease. Nat Rev Nephrol 11: 667–676, 2015. doi: 10.1038/nrneph.2015.162. [DOI] [PubMed] [Google Scholar]
- 49.Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch 454: 345–359, 2007. doi: 10.1007/s00424-007-0212-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schulz E, Dopheide J, Schuhmacher S, Thomas SR, Chen K, Daiber A, Wenzel P, Münzel T, Keaney JF Jr. Suppression of the JNK pathway by induction of a metabolic stress response prevents vascular injury and dysfunction. Circulation 118: 1347–1357, 2008. doi: 10.1161/CIRCULATIONAHA.108.784289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shah MS, Brownlee M. Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circ Res 118: 1808–1829, 2016. doi: 10.1161/CIRCRESAHA.116.306923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sung SH, Ziyadeh FN, Wang A, Pyagay PE, Kanwar YS, Chen S. Blockade of vascular endothelial growth factor signaling ameliorates diabetic albuminuria in mice. J Am Soc Nephrol 17: 3093–3104, 2006. doi: 10.1681/ASN.2006010064. [DOI] [PubMed] [Google Scholar]
- 53.Thum T, Fraccarollo D, Schultheiss M, Froese S, Galuppo P, Widder JD, Tsikas D, Ertl G, Bauersachs J. Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes 56: 666–674, 2007. doi: 10.2337/db06-0699. [DOI] [PubMed] [Google Scholar]
- 54.Toyoda M, Najafian B, Kim Y, Caramori ML, Mauer M. Podocyte detachment and reduced glomerular capillary endothelial fenestration in human type 1 diabetic nephropathy. Diabetes 56: 2155–2160, 2007. doi: 10.2337/db07-0019. [DOI] [PubMed] [Google Scholar]
- 55.Turner JM, Bauer C, Abramowitz MK, Melamed ML, Hostetter TH. Treatment of chronic kidney disease. Kidney Int 81: 351–362, 2012. doi: 10.1038/ki.2011.380. [DOI] [PubMed] [Google Scholar]
- 56.Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science 350: 1208–1213, 2015. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- 57.Weil EJ, Lemley KV, Mason CC, Yee B, Jones LI, Blouch K, Lovato T, Richardson M, Myers BD, Nelson RG. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int 82: 1010–1017, 2012. doi: 10.1038/ki.2012.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng 9: 121–167, 2007. doi: 10.1146/annurev.bioeng.9.060906.151959. [DOI] [PubMed] [Google Scholar]
- 59.Zafrani L, Payen D, Azoulay E, Ince C. The microcirculation of the septic kidney. Semin Nephrol 35: 75–84, 2015. doi: 10.1016/j.semnephrol.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 60.Zhao HJ, Wang S, Cheng H, Zhang MZ, Takahashi T, Fogo AB, Breyer MD, Harris RC. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol 17: 2664–2669, 2006. doi: 10.1681/ASN.2006070798. [DOI] [PMC free article] [PubMed] [Google Scholar]