

Abstract
Objective:
This aim of the study is to investigate whether there are possible plasma urotensin-II (U-II) and neurokinin B (NKB) level changes in patients with acute myocardial infarction (AMI) or not and plasma urotensin-II (U-II) and neurokinin B (NKB) level changes in patients with acute myocardial infarction (AMI) and stable coronary artery disease (CAD) and to evaluate whether there is any relationship between these changes and the pathogenesis of these diseases.
Methods:
This is a prospective case-control study. Three groups were formed from randomly admitted patients with AMI, stable CAD, and controls. Biochemical parameters and U-II and NKB levels were measured. Patients with congestive heart failure, chronic hepatic and renal failure, severe cardiac valve disease, and severe pulmonary hypertension were excluded from the study. The normality of the data was evaluated using the Kolmogorov-Smirnov test. We compared the three groups with one-way ANOVA and Tukey test (Kruskal-Wallis test and Mann-Whitney U test).
Results:
Compared with controls (n=31) and CAD patients (n=32), AMI patients (n=32) had lower U-II and NKB levels. In cases of stable CAD, U-II and NKB levels were similar. A positive correlation was found between U-II and NKB (r=0.720; p=0.000). U-II and NKB were poorly correlated with left ventricle ejection fraction but not with C-reactive protein.
Conclusion:
We found that U-II and NKB levels were lower in patients with AMİ in than those with CAD or the control group. According to our findings, the decreased U-II and NKB levels were related to complicated atherosclerotic events.
Keywords: urotensin II, neurokinin B, acute myocardial infarction
Introduction
Acute myocardial infarction (AMI) is one of the most common causes of morbidity and mortality. Atherosclerosis results in plaque formation in coronary arteries. The formation of thrombosis after a tear of the atherosclerotic plaque is an important, well-known mechanism in AMI pathogenesis (1). Diabetes mellitus (DM), hypertension, hyperlipidemia, cigarette smoking, and positive family history are among the most important risk factors for coronary artery disease (CAD) and AMI. However, these factors can not explain all cases with cardiovascular events. Among the patients who had experienced a coronary event, 20% had none of the traditional risk factors, and 50% of the patients had only one risk factor (2). Therefore, clinical studies are focusing on new risk factors.
Urotensin-II (U-II) is a cyclic undecapeptide that is obtained from the caudal neurosecretory system of fish (3). U-II is secreted mainly from the heart, brain, endothelium, and vascular smooth muscle cells and is found in plasma circulation. It is the most potent vasoconstrictor known in mammals and can contribute to cardiovascular disease through participating in hypertension and atherosclerosis processes (4, 5).
Neurokinin B (NKB) is classified as a neurotransmitter and is found in immune cells and discrete neurons. It plays a role in the functions of vascular reactivity, smooth muscle contraction, immune system activation, neurogenic inflammation, and pain transmission (6-9). NKB acts via binding to one of the neurokinin receptors, NK3. Experimental animal studies on rats demonstrated that NKB causes contraction in mesenteric and portal veins and an increase in heart rate through NK3 receptor activation (10-12). However, there are conflicting studies on the relationship between U-II and cardiovascular disease. There are no studies questioning the relationship between cardiovascular diseases and NKB in the literature.
For these reasons, this study aims to detect U-II and NKB levels in individuals with AMI and stable CAD and in normal individuals and to determine the relationship of U-II and NKB levels with these situations.
Methods
A total of 95 individuals involving 32 AMI patients, 32 stable CAD patients, and 31 individuals with normal coronary arteries (control) who were admitted to our outpatient clinic and angiography unit were enrolled to the study.
Our current study is a prospective randomized case-control study. The sample size was not calculated in the ethical board, because no relevant report was found in the literature. However, the power analysis was done according to results of the comparison of AMI and CAD groups in plasma urotensin II levels, which is one of the main measurement parameters of the study. The power of the study was 82.6%, with a reliability of 95%.
All individuals signed informed consent forms for participation in the study. The diagnosis of AMI was made with an increase in the cardiac biological markers [preferably cardiac troponin (cTr)] with at least one value higher than the 99% of the reference level and having at least one of the following signs. These signs were symptoms of ischemia, new or accepted new ST-segment-T wave (ST-T) changes or newly formed left bundle branch block (LBBB), pathological Q waves on ECG, newly occurring tissue loss or regional wall motion dysfunction in living myocardium, and thrombosis within the coronary artery on angiography (13).
The individuals without any plaque, wall irregularities, ectasis, and slow flow during angiography were considered as having normal coronary arteries. The cases with at least one of the angiographic findings above were considered as having coronary artery disease.
The stable CAD patient group was formed out of the patients who had a diagnosis of CAD with previous angiography performed in our unit. The clinical and demographic data of the groups are given in Table 1. The study had started after taking local ethical approval corresponding with the Helsinki Declaration, Patients’ Rights Regulation, and ethical rules. The participants had been informed about the study, and they were involved in the study after their consent had been taken. The patients with congestive heart failure (LVEF<45%), chronic hepatic failure, chronic renal failure, severe cardiac valve disease, and severe pulmonary hypertension were excluded from the study. In addition to the blood samples for routine biochemical tests and complete blood count parameters, separate blood samples were taken and stored under -80 degrees to detect serum U-II and NKB after we received consent from the patients. The samples were taken during angiography in AMI patients. The samples of the rest of the individuals were taken after outpatient clinic control visits.
Table 1.
Clinical, demographic, and laboratory data of the groups
| Variable | AMI (n=32) | CAD (n=32) | Control (n=31) |
|---|---|---|---|
| Age, years | 58.5±11.8 | 62±9.2 | 56.5±10 |
| Gender, male (%) | 22 (68%) | 23 (71%) | 21 (68%) |
| Hypertension (%) | 19 (59%) | 20 (62%) | 18 (58%) |
| DM (%) | 10 (31%) | 11 (34%) | 9 (29%) |
| BMI, kg/m2 | 25.4±2.7 | 23.8±4.1 | 24.4±2.5 |
| Hyperlipidemia | 21 (65%) | 22 (68%) | 20 (64%) |
| Total-C, mg/dL | 208 (12-319) | 210 (122-353) | 207 (151-316) |
| LDL-C, mg/dL | 123±38.7 | 127±38 | 129±44 |
| HDL-C, mg/dL | 39±9 | 42±10 | 44±9 |
| TG, mg/dL | 227 (75-635) | 162 (78-570) | 202±97 |
| Fasting glucose, mg/dL | 142 (75-295) | 134 (74-390) | 123 (74-259) |
| EF | 55 (30-60) | 57 (30-65) | 58 (30-68) |
AMI - acute myocardial infarction; BMI - body mass index; CAD - coronary artery disease; DM - diabetes mellitus; EF - ejection fraction; HDL-C - high-density lipoprotein cholesterol; LDL-C - low-density lipoprotein cholesterol; TG - triglyceride; Total-C - total cholesterol
Biochemical analysis
Venous blood samples of the patients were centrifuged. From the serum obtained, fasting blood sugar (FBS), total cholesterol (total chol), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), triglyceride (TG), and C-reactive protein levels were measured in a biochemistry autoanalyzer [Cobas C501 Roche (Japan)] device.
The samples taken for U-II and NKB were put into ETDA tubes and centrifuged immediately for 10 minutes in 5000 rpm and preserved in a refrigerator at -80 degrees until analysis time. U-II and NKB levels were measured with tests of the Human Urotensin II ELISA Kit (Hangzhou East Biopharm Co. Ltd, Wuhan, China) and Human Neurokinin B ELISA Kit (Hangzhou East Biopharm Co. Ltd, Wuhan, China) according to the instructions of the producer companies. Test results were given as pg/mL. The intra- and intervariability values for U-II were 3.1% and 4.2%, respectively. The detection range was 15.6 pg/mL-1000 pg/mL.
The intra- and intervariability values for NKB were 3.6% and 4.7%, respectively. The detection range was 13.4 pg/mL-1000 pg/mL. The normal values of U-II and NKB in healthy individuals were not given by the producer companies of the kits.
Transthoracic echocardiography
Echocardiography was performed for all of the patients. Transthoracic echocardiographic examinations were performed by an experienced cardiologist with an Esaote My Lab 50 (3.5 mHz, Florence, Italy) device. Two-dimensional M-mode measurements were calculated. Left ventricle (LV) dimensions were measured in the parasternal long axis according to the recommendations of the American Echocardiography Foundation (14). From the parasternal axis, the aorta, left atrium, LV systolic and diastolic diameters, interventricular septum (IVS), and posterior wall thicknesses (PWT) were measured while the cases were placed in the left lateral decubitus position. In addition, left ventricular ejection fractions (LVEFs) were measured with modified Simpson technique.
Statistical analysis
Statistical analysis was performed using SPSS software (Version 18.0, SPSS Inc., Chicago, IL, USA). If continuous variables were normal, they were described as mean±standard deviation [p>0.05 in Kolmogorov-Smirnov test or Shapiro-Wilks test (n<30)], and if the continuous variables were not normal, they were described as median (minimum-maximum). The continuous variables were compared by the use of one-way ANOVA or Kruskal-Wallis test, depending on parametric or non-parametric values, respectively. Since analysis of variance was significant, comparisons were applied using the post hoc analysis Tukey test and MannWhitney U test. The categorical variables between the groups were analyzed by using the chi-square test or Fisher’s exact test.
Results
Demographic, anthropometric, and biochemical parameters
The clinical, demographic, laboratory, and echocardiographic features of the individuals participating in the study are shown in Table 1. The mean age of the participants was 58.5±11.8 years in the AMI group, 62±9.2 years in the CAD group, and 56.5±10 years in the control group. In all three groups, there was no statistically significant age difference (p>0.05), and there were no significant differences in terms of cardiovascular disease risk factors, including hypertension, DM, and hyperlipidemia (p>0.05). Additionally, there were no significant differences in total cholesterol, LDL-cholesterol, HDL-cholesterol, triglyceride, and FBS levels between the groups (p>0.05). The C-reactive protein (CRP) levels were significantly higher in the AMI group (9 pg/mL) compared to the CAD (3.8 pg/mL) and control (3.9 pg/mL) groups. (p=0.003 and p=0.003, respectively). There was no difference between the CAD and control groups’ CRP levels (p=0.912) (Table 2). Serum mean U-II levels were significantly lower in the AMI group [74 (53-181 pg/mL)] compared to the CAD [94 (54-324) pg/mL] and control [95 (48-196) pg/mL] groups (p=0.014 and p=0.028, respectively). There was no significant difference in U-II levels between the CAD [94 (54-324) pg/mL] and control [95 (48-196) pg/mL] groups (p=0.407) (Table 2).
Table 2.
Plasma CRP, U-II, and NKB levels in the three groups
| Variable | AMI (n=32) | CAD (n=32) | Control (n=31) |
|---|---|---|---|
| CRP mg/L | 9 (1-25)a | 3.8 (0-10) | 3.9 (0-12) |
| Urotensin-II, pg/mL | 74 (53-181)b | 94 (54-324) | 95 (48-196) |
| Neurokinin-B, pg/mL | 77 (19-192)c,d | 125 (59-282) | 96 (52-194) |
AMI - acute myocardial infarction; CAD - coronary artery disease; CRP-C-reactive protein
P<0.005 between AMI and CAD and between AMI and control groups
P<0.05 between AMI and CAD and between AMI and control groups
P<0.001 between AMI and CAD groups
P<0.05 between AMI and control groups
Serum mean NKB levels were significantly lower in the AMI group [77 (49-192) pg/mL] compared to the CAD [125(59-282) pg/mL] and control [96 (52-194) pg/mL] groups (p<0.001 and p=0.025, respectively). There was no significant difference between the CAD [125 (59-282) pg/mL] and control [96 (52-194) pg/mL] groups in NKB levels (p=0.070) (Table 2).
Although we detected a positive correlation between serum U-II and NKB levels (r=0.720; p<0.001) (Fig. 1), there was no positive correlation of serum U-II and NKB values with CRP levels. Neither U-II nor NKB values correlated with age, body mass index, or CRP levels, though they had a weak correlation with left ventricular ejection fraction (LVEF) (Fig. 2 and 3).
Figure 1.
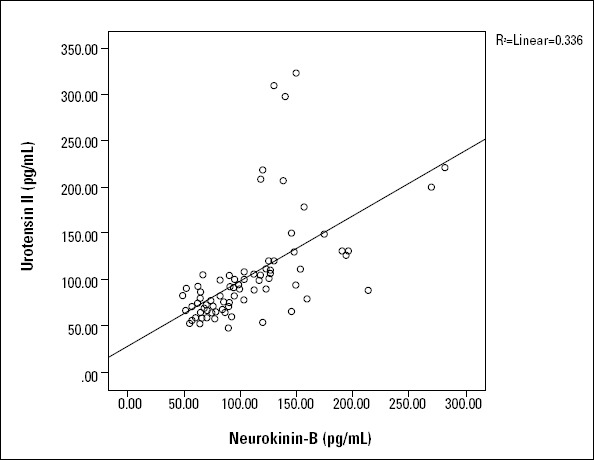
The correlation between urotensin II and neurokinin B (r=0.720; p=0.000)
Figure 2.
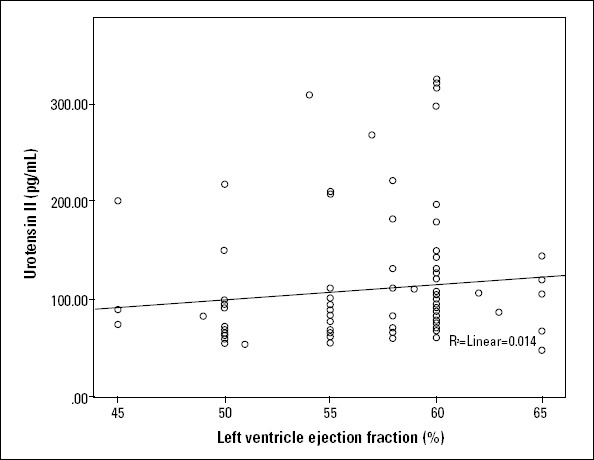
A weak correlation between urotensin II and LVEF (r=0.262; p=0.013)
LVEF - left ventricular ejection fraction
Figure 3.
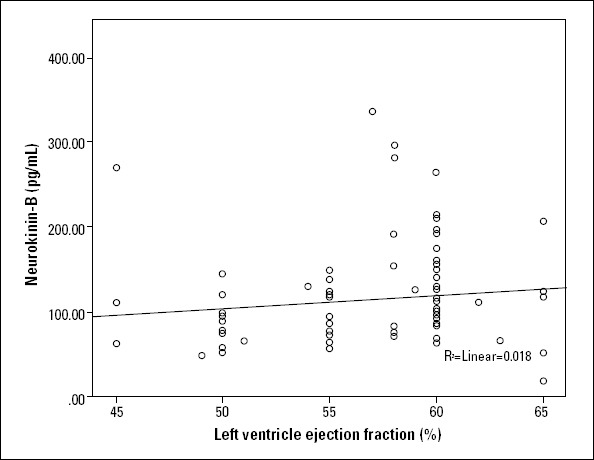
The correlation between neurokinin B and LVEF (r=209; p=0.05)
LVEF - left ventricular ejection fraction
Discussion
This is the first study that researches the relationship between NKB and cardiovascular diseases. The effect of U-II and NKB on vascular structures and cardiovascular events is still a subject of debate. In this study, we found that there was a significant decrease in serum U-II and NKB levels in AMI patients compared to stable CAD patients and individuals with normal coronary arteries. In addition, we discovered that in all three groups, there was a positive correlation between U-II and NKB.
The increased secretion of U-II in atherosclerotic carotid arteries and aortic plaques in the aorta has been proven. It is postulated that this secretion results in the proliferation of vascular smooth muscles and accelerates the formation of atherosclerotic plaque (15, 16). In addition, it has been determined that U-II causes coronary vasoconstriction, fibrosis, and hypertrophy in cardiomyocytes. Therefore, we believe that U-II might play a role in the pathogenesis of atherosclerosis (17). Animal studies have shown that the stimulation of U-II receptors of endothelial cells results in the secretion of nitric oxide and prostaglandins (18). However, the effect of these vasodilators may be neutralized by the direct vasoconstrictor effect of U-II on smooth muscle cells. Therefore, it is still unclear whether U-II has a cardioprotective or cardiodepressive function.
An increase in plasma U-II levels was shown in cardiac failure, CAD, ischemic cardiomyopathy, hypertension, DM, and chronic renal failure (19-21). There are conflicting data in the literature regarding U-II levels in acute coronary syndrome or AMI. In some studies, an increase in plasma U-II levels in acute coronary syndrome (ACS) was observed. In contrast, other studies indicated a decrease in U-II levels (22, 23). Joyal et al. (24) carried out a study on 54 acute coronary syndrome (ACS) patients, 51 stable coronary artery patients, and 29 healthy volunteers, reporting lower U-II levels in comparison with the stable CAD and control groups. Our results are in accordance with this. In addition, Joyal et al. (24) found a negative relationship between plasma U-II levels and systemic arterial pressure in both the ACS and stable KAH groups. The hypertension ratios in our study were similar, and no correlation was determined between hypertension and U-II and NKB levels. In contrast to many studies, they also measured significantly lower U-II levels in patients with low LVEF. Similarly, our study revealed low levels of U-II in the AMI group, but we found a weak positive correlation with LVEF. In another study, lower U-II levels were detected in high-risk ACS patients (22). However, Khan et al. (23) found higher U-II levels in ACS patients than in patients without CAD. They also showed a positive correlation between increased U-II levels and adverse clinical events. In general, high U-II levels were found in hypertensive patients (25). No correlation was found between U-II levels and troponin levels (23). Therefore, the hypothesis of U-II acting as a marker for myocardial necrosis was disproven. It was postulated that the increase in U-II levels in myocardial necrosis could result from the impairment of left ventricular function. This hypothesis was supported by the positive correlation between U-II and LVEF and the negative correlation between U-II and N-terminal pro-brain natriuretic peptide (NT-proBNP) (22). We observed a weak correlation between U-II and LVEF
In our study, we detected significantly low NKB levels in patients with myocardial infarction compared to patients having stable CAD and individuals with normal coronary arteries. There is no study researching the relationship between cardiovascular diseases and NKB in the literature. NKB is a neurotransmitter with various roles, such as vascular reactivity, smooth muscle contraction, and vascular inflammation. A previous study has claimed that substance P which, like NKB, is from the tachykinin family and increases mast cell-dependent atherosclerotic plaque destabilization. NKB might also have a similar effect (26).
In vitro studies suggest that the activation of NKB could have a role in the pathogenesis of hypertension and preeclampsia through increasing the secretion of some cytokines, involving interleukin 1 (IL-1), IL-2, IL-6, and tumor necrosis factor (TNF-a), causing endothelial cell destruction, vascular smooth muscle dysfunction, and endothelin secretion (27, 28). A number of studies suggest that oversecretion of NKB by the placenta can decrease vasodilatation and increase vasoconstriction, resulting in the development of preeclampsia. Moreover, a positive correlation between NKB and preeclampsia was found (29, 30). NKB and U-II were studied together in preeclampsia patients, and high levels of these markers were considered to relate to the development of high blood pressure. Liu et al. (27) found high levels of U-II and NKB in patients with preeclampsia compared to healthy individuals, and they revealed a positive correlation between U-II and NKB. In our study, we found lower NKB levels in the AMI group compared to the CAD and control groups. We also found a positive correlation between U-II and NKB.
There are two possible reasons for the low U-II and NKB levels in the AMI group. The first is increased sympathetic activation, though studies researching blood norepinephrine, epinephrine, and cortisol levels are necessary to prove it. The second is a secondary immune response against myocardial degradation products. There is no detailed study in the literature explaining these mechanisms.
The producer companies did not give the normal ranges of either kit for healthy people. In the literature, U-II and NKB were studied with different kits from different companies. Therefore, the values in our study were different from the values of other studies. The normalization of the values with internationally accepted methods is necessary.
Study limitations
The most important limiting factor of our study is the low number of cases. If we included patients with systolic cardiac failure, the value of our study would be higher. Because many existing studies have examined U-II and cardiac failure, we chose to study a group with normal systolic function. In addition, this study is not a follow-up study but rather a cross-sectional one. Thus, we did not monitor the prognosis of the myocardial infarction group.
Conclusion
Our study has importance, because it is the first study evaluating U-II and NKB in AMI and CAD, revealing a positive correlation between them. Our study revealed significantly low levels of U-II and NKB in AMI patients. However, prospective randomized studies on U-II and NKB levels in AMI including large populations are necessary.
Footnotes
Conflict of interest: None declared.
Peer-review: Partially peer-reviewed.
Authorship contributions: Concept - D.Ç. A.; Design - A. A., Ş. A.; Supervision - A.G., N.G.; Resource - Namık Kemal University BAP; Data collection and/or processing - Ü.G., H.D.; Analysis and/or Interpretation - Ş. A., A. A.; Literature search - D.Ç. A.; Writing - D.Ç. A.; Critical review - A.G., A. A.
References
- 1.Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation. 1995;92:657–71. doi: 10.1161/01.cir.92.3.657. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 2.Adalet K. “Clinical Cardiology“. 1st. ed. “Istabul Tıp Kitapevi“; 2013. pp. 336–7. [Google Scholar]
- 3.Bern HA, Pearson D, Larson BA, Nishioka RS. Neurohormones from fish tails: the caudal neurosecretory system I. “Urophysiology“and the caudal neurosecretory system of fishes. Recent Prog Horm Res. 1985;41:533–52. doi: 10.1016/b978-0-12-571141-8.50016-0. [DOI] [PubMed] [Google Scholar]
- 4.Douglas SA, Ohlstein EH. Human urotensin-II, the most potent mammalian vasoconstrictor identified to date, as a therapeutic target for the management of cardiovascular disease. Trends Cardiovasc Med. 2000;10:229–37. doi: 10.1016/s1050-1738(00)00069-4. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 5.Maguire JJ, Kuc RE, Wiley KE, Kleinz MJ, Davenport AFP. Cellular distribution of immunoreactive urotensin-II in human tissue with evidence of increased expression in atherosclerosis and a greater constrictor response of small compared to large coronary arteries. Peptides. 2004;25:1767–74. doi: 10.1016/j.peptides.2004.01.028. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 6.Page NM. Hemokinins and endokinins. Cell Mol Life Sci. 2004;61:1652–63. doi: 10.1007/s00018-004-4035-x. [CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Page NM. New challenges in the study of the mammalian tachykinins. Peptides. 2005;26:1356–68. doi: 10.1016/j.peptides.2005.03.030. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 8.Patak EN, Pennefather JN, Story ME. Effects of tachykinin on uterine smooth muscle. Clin Exp Pharmacol Physiol. 2000;27:922–7. doi: 10.1046/j.1440-1681.2000.03362.x. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 9.Nava E, Wiklund NP, Salazar FJ. Changes in nitric oxide release in vivo in response to vasoactive substances. Br J Pharmacol. 1996;119:1211–6. doi: 10.1111/j.1476-5381.1996.tb16024.x. [CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moussaoui SM, Le Prado N, Bonici B, Faucher DC, Cuiné F, Laduron PM, et al. Distribution of neurokinin B in rat spinal cord and peripheral tissues: comparison with neurokinin A and substance P and effects of neonatal capsaicin treatment. Neuroscience. 1992;48:969–78. doi: 10.1016/0306-4522(92)90285-a. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 11.D’Orléans-Juste P, Claing A, Télémaque S, Warner TD, Regoli D. Neurokinins produce selective venoconstriction via NK-3 receptors in the rat mesenteric vascular bed. Eur J Pharmacol. 1991;204:329–34. doi: 10.1016/0014-2999(91)90860-s. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 12.Thompson GW, Hoover DB, Ardell JL, Armour JA. Canine intrinsic cardiac neurons involved in cardiac regulation possess NK1, NK2, and NK3 receptors. Am J Physiol. 1998;275:R1683–9. doi: 10.1152/ajpregu.1998.275.5.R1683. [DOI] [PubMed] [Google Scholar]
- 13.Steg PG, James SK, Atar D, Badano LP, Blömstrom-Lundqvist C, Borger MA, et al. ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force on the management of ST-segment elevation acute myocardial infarction of the European Society of Cardiology (ESC) Eur Heart J. 2012;33:2569–619. doi: 10.1093/eurheartj/ehs215. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 14.Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, et al. Recommendations for chamber quantification: a report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 2005;18:1440–63. doi: 10.1016/j.echo.2005.10.005. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 15.Bousette N, Patel L, Douglas SA, Ohlstein EH, Giaid A. Increased expression of urotensin II and its cognate receptor GPR14 in atherosclerotic lesions of the human aorta. Atherosclerosis. 2004;176:117–23. doi: 10.1016/j.atherosclerosis.2004.03.023. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 16.Tzanidis A, Hannan RD, Thomas WG, Onan D, Autelitano DJ, See F, et al. Direct actions of urotensin II on the heart: implications for cardiac fibrosis and hypertrophy. Circ Res. 2003;93:246–53. doi: 10.1161/01.RES.0000084382.64418.BC. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 17.Bousette N, Patel L, Douglas SA, Ohlstein EH, Giaid A. Increased expression of urotensin II and its cognate receptor GPR14 in atherosclerotic lesions of the human aorta. Atherosclerosis. 2004;176:117–23. doi: 10.1016/j.atherosclerosis.2004.03.023. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 18.Bottrill FE, Douglas SA, Hiley CR, White R. Human urotensin-II is an endothelium-dependent vasodilator in rat small arteries. Br J Pharmacol. 2000;130:1865–70. doi: 10.1038/sj.bjp.0703513. [CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watanabe T, Kanome T, Miyazaki A, Katagiri T. Human urotensin II as a link between hypertension and coronary artery disease. Hypertens Res. 2006;29:375–87. doi: 10.1291/hypres.29.375. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 20.Lapp H, Boerrigter G, Costello-Boerrigter LC, Jaekel K, Scheffold T, Krakau I, et al. Elevated plasma human urotensin-II-like immunoreactivity in ischemic cardiomyopathy. Int J Cardiol. 2004;94:93–7. doi: 10.1016/j.ijcard.2003.05.008. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 21.Ng LL, Loke I, O’Brien RJ, Squire IB, Davies JE. Plasma urotensin in human systolic heart failure. Circulation. 2002;106:2877–80. doi: 10.1161/01.cir.0000044388.19119.02. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 22.Babinska M, Holecki M, Prochaczek F, Owczarek A, Kokocinska D, Chudek J, et al. Is plasma urotensin II concentration an indicator of myocardial damage in patients with acute coronary syndrome? Arch Med Sci. 2012;8:449–54. doi: 10.5114/aoms.2012.29400. [CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khan SQ, Bhandari SS, Quinn P, Davies JE, Ng LL. Urotensin II is raised in acute myocardial infarction and low levels predict risk of adverse clinical outcome in humans. Int J Cardiol. 2007;117:323–8. doi: 10.1016/j.ijcard.2006.05.016. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 24.Joyal D, Huynh T, Aiyar N, Guida B, Douglas S, Giaid A. Urotensin-II levels in acute coronary syndromes. Int J Cardiol. 2006;108:31–5. doi: 10.1016/j.ijcard.2005.04.001. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 25.Cheung BM, Leung R, Man YB, Wong LY. Plasma concentration of urotensin II is raised in hypertension. J Hypertens. 2004;22:1341–4. doi: 10.1097/01.hjh.0000125452.28861.f1. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 26.Bot I, de Jager SC, Bot M, van Heiningen SH, de Groot P, Veldhuizen RW, et al. The neuropeptide substance P mediates adventitial mast cell activation and induces intraplaque hemorrhage in advanced atherosclerosis. Circ Res. 2010;106:89–92. doi: 10.1161/CIRCRESAHA.109.204875. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Li Y, Xu X, Chen X, Chen H. Neurokinin B and urotensin II levels in pre-eclampsia. J Matern Fetal Neonatal Med. 2010;23:869–73. doi: 10.3109/14767050903358355. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 28.Dianzani C, Collino M, Lombardi G, Garbarino G, Fantozzi R. Substance P increases neutrophil adhesion to human umbilical vein endothelial cells. Br J Pharmacol. 2003;139:1103–10. doi: 10.1038/sj.bjp.0705344. [CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Page NM, Woods RJ, Gardiner SM, Lomthaisong K, Gladwell RT, Butlin DJ, et al. Excessive placental secretion of neurokinin B during the third trimester causes pre-eclampsia. Nature. 2000;405:797–800. doi: 10.1038/35015579. [CrossRef] [DOI] [PubMed] [Google Scholar]
- 30.Page NM, Woods RJ, Lowry PJ. A regulatory role for neurokinin B in placental physiology and pre-eclampsia. Regul Pept. 2001;98:97–104. doi: 10.1016/s0167-0115(00)00239-1. [CrossRef] [DOI] [PubMed] [Google Scholar]