

Abstract
Introduction
Co-administration of a diuretic or calcium channel blocker with an ACE inhibitor are both preferred combinations in patients with hypertensive chronic kidney disease (CKD). According to the available evidence, it is still unknown which combination plays a more active role in renal protection. We hypothesised that a combination of fosinopril and benidipine may delay the progression of CKD more effectively than a combination of fosinopril and hydrochlorothiazide (HCTZ).
Methods and analysis
This study will be a multicentred, prospective, double-blind, randomised parallel controlled trial for hypertensive CKD patients in China. Patients will be randomised to one of two treatment groups: a combination of benidipine 4–8 mg/day and fosinopril 20 mg/day; or a combination of HCTZ 12.5–25 mg/day and fosinopril 20 mg/day. Patients will be followed up for 24 months after a month's fosinopril run-in. There will be dose-titration after 1 and 2 months. The primary endpoint is changes in estimated glomerular filtration rate (eGFR) from baseline to month 24. Secondary endpoints include changes in home blood pressure (BP), ambulatory BP, proteinuria, urinary albumin/creatinine ratio, and composite renal events in 24 months. Inclusion criteria are: age 18–80 years, non-dialysis CKD patients with eGFR >30 mL/min/1.73 m2, home BP >130 mm Hg systolic or BP >80 mm Hg diastolic at the screening and randomisation, and 24 hour proteinuria <3.5 g. Principal exclusions are hypertensive crisis, transplantation, cancer, severe diabetes complications, hyperkalaemia and severe allergy. The required sample size was 511 patients for detecting a difference in the change of eGFR (one sided α=0.025, power 1-β=0.90).
Ethics and dissemination
BEAHIT (Benidipine and Hydrochlorothiazide in Fosinopril Treated Chronic Kidney Disease Patients with Hypertension) was approved by Changzheng Hospital Ethics Committee (CZ-20160504-16). The outcomes will be published in a peer-reviewed journal.
Trial registration number
Keywords: glomerular filtration rate
Strengths and limitations of this study.
This is a multicentred, prospective, double-blind, randomised, parallel controlled trial involving chronic kidney disease (CKD) patients with diabetes and non-diabetes.
Outcomes may help future guidelines regarding antihypertensive combinations in CKD.
A limitation may be the relatively short follow-up time.
Loss to follow-up, especially non-responders within follow-up, is possible.
Introduction
Patients with chronic kidney dysfunction or injury which has affected their health over a 3 month period are diagnosed with chronic kidney disease (CKD). China has a high overall prevalence of CKD of 10.8% and the number of CKD patients is estimated to be approximately 119.5 million.1 The prevalence, awareness and treatment of hypertension (HTN) in non-dialysis CKD patients has been reported at 67.3%, 85.8%, and 81.0%, respectively.1 However, of the hypertensive CKD patients, only 14.1% had controlled blood pressure (BP) to <130/80 mm Hg. Racial and geographical differences play important roles in HTN of Chinese CKD patients. With successive CKD stages, the risk of uncontrolled HTN increased, and uncontrolled HTN was associated with renal disease progression. Managing BP effectively in CKD patients is still a large unmet medical need in China.
Abnormal activation of the renin–angiotensin–aldosterone system (RAAS) in CKD plays an important role in the pathogenesis of HTN. Renin–angiotensin system inhibitors (RASI), including ACE inhibitors (ACEIs) and angiotensin II type 1 receptor blockers (ARBs), have been confirmed as having apparent renoprotective effects in patients with CKD. The guidelines recommend that ACEI or ARB is the preferred antihypertensive agent in CKD patients with HTN or albuminuria. However, RASI monotherapy does not usually achieve adequate BP control. Patients with CKD usually need combination antihypertensive therapy to achieve the recommended BP goal of <130/80 mm Hg.2 Co-administration of diuretics and calcium-channel blockers (CCBs) with ACEIs or ARBs are the most common combinations. Dihydropyridine-type CCB plus RASI is a preferred combination in clinical practice with mild adverse effects.3 Thiazide or thiazide-like diuretic plus RASI is another widely used combination, providing the synergistic function of antihypertensive action while offsetting mutual adverse effects (such as hyperkalemia and impaired glucose tolerance).
In recent years, clinical studies focusing on the addition of a CCB or diuretic to RASI have led to inconsistent results. The GUARD trial found that the reduction of albuminuria in the hydrochlorothiazide (HCTZ) plus benazepril group was greater than in the amlodipine plus benazepril group in hypertensive patients with diabetic nephropathy.4 Hayashi et al5 found the addition of diuretics or CCB to ARB was equally effective for the control of HTN in CKD, while diuretics may be better than CCB in reducing proteinuria (PROTECT study). However, Ishimitsu et al6 found losartan plus nifedipine showed a superior antihypertensive and renoprotective effect in CKD patients compared to losartan plus HCTZ, but nifedipine was not associated with lower urinary albumin excretion compared to HCTZ. The ACCOMPLISH study7 concluded that hypertensive patients treated with benazepril plus amlodipine had a lower risk of progression to CKD than those treated with benazepril plus HCTZ.
Until now, no large-scale studies have compared the effect of initial treatment with two different combinations of antihypertensive drugs in CKD patients on the progression of kidney disease, especially in subsets of diabetes, non-diabetes, microalbuminuria and macroalbuminuria. Moreover, CKD patients have elevated levels of cardiovascular disease risk and endothelial dysfunction markers compared with healthy individuals (such as serum aldosterone (SA), Von Willebrand factor (vWF) and E-selectin (ES)). These markers are elevated early in the disease and are associated with decline in renal function. Based on this knowledge, we aim to investigate changes of the markers in response to antihypertensive therapy.
Recent studies found that CCBs varied in their effects on glomerular arterioles, reflecting the results of differential blockage of T-channel receptors (on the afferent and efferent arteriole) versus L-channel receptors (predominantly on the afferent arteriole).8 Previous studies found benidipine had more favourable renoprotective effects when compared with other CCBs.8 T-channel blockade leads to a reduction in intra-glomerular pressure, and accordingly may lead to a fall in the concentration of urine albumin. This study will use benidipine (L/T-type CCB), which is different from ACCOMPLISH study.7
BEAHIT (Benidipine and Hydrochlorothiazide in Fosinopril Treated Chronic Kidney Disease Patients with Hypertension) will test whether the superiority of benidipine in protecting renal function exists over 2 years' treatment. This trial is large enough for estimates such as proteinuria, BP and other related clinical parameters. In consideration of the controversy between diuretic and CCB in renal protection, we aim to conduct a large-scale study to compare benidipine with HCTZ in fosinopril treated hypertensive CKD patients. This protocol was approved by the Changzheng Hospital Ethics Committee and registered in clinicaltrials.gov (NCT02646397). All the items complied with the SPIRIT 2013 guideline.9
Primary objectives
To determine whether the combination of fosinopril and benidipine may delay the progression of CKD more effectively than the combination of fosinopril and HCTZ.
Secondary objectives
To demonstrate whether the combination of fosinopril and benidipine may improve proteinuria, HTN or risk of renal events more effectively compared with the combination of fosinopril and HCTZ; and to determine the effects on endothelial dysfunction markers and safety in the two treatment groups.
Methods and analysis
Study design
This study will be a multicentred, prospective, two arm, double-blind, randomised, parallel controlled trial for hypertensive CKD patients in China. Patients will be randomised to either the benidipine+fosinopril group or the HCTZ+fosinopril group. Patients will receive treatment for 24 months after a month's fosinopril run-in. This is a superiority trial with a 1:1 allocation ratio. The design of the trial is outlined in figure 1. The composition and responsibilities of the principal investigator, steering committee (SC), trial management committee (TMC) and data monitoring committee (DMC) overseeing the trial are listed in the online supplement 1.
Figure 1.

Outline of the study design. ACEI, angiotensin-converting enzyme inhibitor; CCB, calcium channel blocker; HCTZ, hydrochlorothiazide.
bmjopen-2016-013672supp.pdf (15.5KB, pdf)
Date and version identifier
Issue date: 11 December 2016, protocol amendment number: 06.
Revision chronology: 29 December 2015, original. 2 January 2016 amendment 01: primary amendment was the procedure of drug titration. 18 March 2016 amendment 02: endpoints of endothelial function markers added at the protocol. 3 May 2016 amendment 03: the sample size estimation was optimised. 21 June 2016 amendment 04: optimising of subjects' consent. 6 November 2016 amendment 05: secondary endpoints: cardiovascular events were deleted due to the limited follow-up; the office BP was replaced with the home BP. Make β-blocker as concomitant medications more specific: carvedilol, metoprolol, nebivolol or propranolol. 11 December 2016, amendment 06: inclusion criteria changes: proteinuria <1.5 g/24 hours changed to non-nephrotic proteinuria (<3.5 g/24 hours); estimated glomerular filtration rate (eGFR) 30–60 mL/min/1.73 m2 changed to >30 mL/min/1.73 m2.
Protocol amendments
Modifications of the protocol, which could impact on the conduct of the study and may affect patient safety or benefit to the patient, including changes of study design, study objectives, patient population, study procedures, sample sizes, or significant administrative changes, will require an amendment to this protocol. Amendments from 01 to 06 have been approved by SC, TMC, DMC and the ethics committee before implementation. Administrative changes of the protocol belong to minor clarifications or corrections which have no significant effect on the conduct of the trial. The administrative changes will be agreed on by SC, TMC and DMC, and will be documented in a memorandum.
Study population
The trial will be open to most CKD patients with HTN screened from eight hospitals in China. The BEAHIT programme made changes to the definition of HTN introduced by the KDIGO guideline of 2012:10 office BP consistently >130 mm Hg systolic or >80 mm Hg diastolic.
Inclusion criteria: (1) Age from 18 to 80 years. (2) Patients diagnosed with non-dialysis CKD with eGFR ≥30 mL/min/1.73 m2 at the screening and randomisation calculated according to the CKD-EPI formula.11 (3) Home BP >130 mm Hg systolic or BP >80 mm Hg diastolic at the screening and randomisation. Patients could not receive more than two antihypertensive medications before the screening period, and should discontinue the prior antihypertensive medications after the screening period. (4) 24 hour proteinuria <3.5 g at the screening and randomisation period. (5) Patients who signed the informed consent form.
Exclusion criteria: Serum creatinine (SCr) >3 mg/dL or kidney transplantation. Medical history in the past 6 months: (1) hypertensive crisis (office BP >180 mm Hg systolic or BP >110 mm Hg diastolic); (2) refractory HTN; (3) patients diagnosed as having severe cardiac arrhythmia (severe extra-beats, supraventricular tachycardias, ventricular arrhythmias, or bradyarrhythmias), heart failure, New York Heart Association (NYHA) functional class >III, angina, stroke, left ventricular hypertrophy or myocardial infarction; (4) diagnosis of cancer or severe sepsis; (5) haematological system disorders: myelodysplastic syndrome, granulocytopenia, hypereosinophilic syndrome, polycythaemia, thrombocytopenia; (6) restrictive pericarditis; (7) systemic lupus erythematous; (8) severe diabetes complications such as diabetic ketoacidosis, hyperosmolar coma, retinopathy, amputation; (9) diagnosis of hyperkalemia (>5.5 mmol/L); (10) renal artery stenosis or vascular embolism disease; (11) patient is currently pregnant or lactational; (12) aspartate aminotransferase/alanine aminotransferase (AST/ALT) >three times the upper limit of standard value; (13) any severe allergy to CCB, diuretic or ACEI; (14) history of severe side effects from CCB, diuretic or ACEI; (15) long-term use of non-steroidal anti-inflammatory drugs; (16) use of other investigational drugs within 30 days or five half-lives of last visit, whichever is longer. Other patients judged unsuitable by the investigators.
Recruitment strategy
Potentially suitable patients will be identified from general populations and the hospitals. Patients will be recruited for the BEAHIT trial through local advertising and identification from a CKD patient database. The screening in each site will continue until the target number is achieved during the enrolment period (12 months). A medical investigator will provide patients with written informed consent forms. Nurses will record baseline variables, take urine and blood for baseline haematology and biochemistry tests, and record medical histories. Blood samples will be analysed at the local laboratory centre by the usual methods. Serum and blood for future analyses will be stored at the local centre. First, patients need to sign the informed consent forms and meet the exclusion and inclusion criteria. Then, patients will be treated with fosinopril (10 mg) for 2 weeks in order to screen and washout previous antihypertensive medications. Two weeks later, the dose will increase to 20 mg for another 2 weeks.
Randomisation
Eligible subjects will then be randomised (1:1) to one of the following two groups: a combination of fosinopril 20 mg/day plus benidipine 4 mg/day; or a combination of fosinopril 20 mg/day plus HCTZ 12.5 mg/day. Research staff responsible for recruitment will request the randomisation codes from the statistics research section (SRS) of the second military medical university. Then the SRS will send the randomisation code to the research physicians in sealed opaque envelopes. Each physician will open the envelope and assign the treatment to the relevant patient. The physician is not allowed to provide information about the treatment allocation to either the patient or the recruitment staff. The randomisation code will not be released until the patient has completed baseline measurements and has been included in this trial.
The randomly assigned codes will be generated by the SRS through a dynamic allocation method after stratification by gender and the presence/absence of diabetes according to the data at the first screening. The randomisation lists will remain with the SRS for the duration of the study without influence from the principal investigators, the TMC or the physicians.
Blinding
Trial patients and outcome assessors will be blinded to the allocation. Trial patients also will be blinded to the study hypothesis. Outcome assessors will attend an assessment training programme. Employees outside this research team will enter data into the computer on separate datasheets so that the outcome assessors can analyse data without knowing the allocation. Trial medication will be provided in identical containers with blank labels for each of the two assignments at the Changzheng Hospital Pharmacy. The time of the final unblinding of all trial patients will be after the creation of the locked dataset.
Emergency unblinding should happen only in exceptional circumstances when knowing the actual intervention is absolutely necessary for further treatment of the patient. The DMC provides 24 hour telephone unblinding services. Physicians will be encouraged to maintain the blinding as long as possible. Actual allocation should not be open to the patient or other study investigators after the emergency unblinding.
Trial treatments
Initial treatments for the two groups are as described above. After 1 month, patients who do not fulfil the home BP target in each group will be force-titrated to twice the starting dose, namely a combination of fosinopril 20 mg/day plus benidipine 8 mg/day or a combination of fosinopril 20 mg/day plus HCTZ 25 mg/day (figure 1). Add-on antihypertensive agents (ie, β-blockers, α-blockers or clonidine) will be allowed at month 2 in patients who still have not reached the target home BP after titration. A β-blocker is suggested as the third agent in both groups. According to the KDIGO guideline of 2012,10 bisoprolol or atenolol which could accumulate in CKD patients is not recommended. Carvedilol, metoprolol or propranolol are suitable for CKD patients. Nebivolol is also adaptive in patients with an eGFR >30 mL/min/1.73 m2. If the BP reaches <90 mm Hg systolic and <60 mm Hg diastolic or the patients have had symptoms/signs of hypotension at any visit, the investigators will decrease either the benidipine or HCTZ to half of the former dose, maintaining the constant fosinopril dose.
Study procedure
The test items and procedure are shown in table 1. The tests should be performed at each visit (V0−V2±2 days, V3−V12±1 weeks). After randomisation, included patients will need to receive follow-up over the study period. Safety parameters, the interim endpoints and adverse events (AEs) will be recorded at each visit. At each visit, blood and urine will be collected for determination of eGFR, 24 hour urine protein, albumin/creatinine ratio (ACR), biochemical analysis and other related clinical parameters. BP will be measured in triplicate at visits by the same doctor. If the study medication is withdrawn by the investigators for any reason, the patient should finish the last follow-up according to V12 as soon as possible. If a patient needs to start therapy for a specific clinical indication or emergency such as an infection, the investigator could prescribe additional therapy on top of the study drug.
Table 1.
The items and procedure of the study
| V0 | V1 | V2 | V3 | V4 | V5 | V6 | V7 | V8 | V9 | V10 | V11 | V12/early termination | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Items | (−4 weeks) | (−2 weeks) | (0 months) | (1 months) | (2 months) | (3 months) | (6 months) | (9 months) | (12 months) | (15 months) | (18 months) | (21 months) | (24 months) |
| Informed consent | X | ||||||||||||
| Inclusion/exclusion criteria | X | X | |||||||||||
| Demographic data | X | ||||||||||||
| Medical history | X | ||||||||||||
| Randomisation | X | ||||||||||||
| Safety evaluation | |||||||||||||
| Physical examination | X | X | X | X | X | X | X | X | X | X | X | X | X |
| Vital signs | X | X | X | X | X | X | X | X | X | X | X | X | X |
| CKD classification | X | X | X | X | X | X | X | X | X | X | |||
| Electrocardiography | X | X | X | X | X | X | X | X | X | X | |||
| Adverse events | X | X | X | X | X | X | X | X | X | X | X | X | X |
| Drug combination | X | X | X | X | X | X | X | X | X | X | X | X | X |
| Laboratory tests | |||||||||||||
| Blood cellular evaluation | X | X | X | X | X | X | X | X | X | X | X | X | |
| Blood biochemical analysis | X | X | X | X | X | X | X | X | X | X | X | X | |
| Serum electrolytes | X | X | X | X | X | X | X | X | X | X | X | X | |
| Routine urine test | X | X | X | X | X | X | X | X | X | X | X | X | |
| 24 hour urine protein | X | X | X | X | X | X | X | X | X | X | X | X | |
| Urinary albumin /creatinine ratio | X | X | X | X | X | X | X | X | X | X | X | X | |
| Therapeutic effect evaluation | |||||||||||||
| eGFR | X | X | X | X | X | X | X | X | X | X | X | X | |
| Blood pressure (BP)* | X | X | X | X | X | X | X | X | X | X | X | X | X |
| Proteinuria | X | X | X | X | X | X | X | X | X | X | X | X | |
| Fosinopril dispensed | X | X | X | X | X | X | X | X | X | X | X | X | |
| Benidipine and hydrochlorothiazide dispensed | X | X | X | X | X | X | X | X | X | X | |||
| Drugs collected | X | X | X | X | X | X | X | X | X | X | X | X | |
*The first BP measurement will be taken before patients receive antihypertensive treatment. The mean BP of three measurements should be recorded.
CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate.
Tolerability
AEs will be recorded on the electronic case database for each visit. Investigators consider that a 2-week drug holiday is acceptable at any point, which may allow patients to remain in the study without early withdrawal.
Prohibited concomitant medications
The combined use of fosinopril with potassium-sparing drugs or potassium supplements could increase the risk of hyperkalemia. The combination of fosinopril and sirolimus or everolimus could increase the risk of angioedema. Benidipine should be avoided with digitalis, cimetidine and rifampicin. Alcohol, narcotics or barbiturates together with HCTZ is discouraged to prevent an additive effect. HCTZ is not permitted to be taken with lithium, corticosteroids and pain drugs.
Adherence
There will be pill counts at each study visit to assess medication adherence, and unused pills will be counted and recorded. Brief discussions about the reasons for missed doses will be undertaken, and a simple strategy such as linking tablets to breakfast or other activities will be provided.
Endpoints
The primary endpoint is the change of eGFR in the fosinopril plus benidipine group compared with the fosinopril plus HCTZ group from baseline to month 24.
Secondary endpoints from baseline to month 24 include: (1) values and changes in home BP in the two groups at each time point; (2) values and changes in ambulatory BP (daytime and nighttime average BP); (3) composite renal events in 2 years: 30% reduction in eGFR, doubling of SCr concentration, end-stage renal disease (ESRD, eGFR <15 mL/min/1.73 m²) or chronic dialysis;12 (4) changes in 24 hour proteinuria and urinary ACR; (5) SA, vWF and ES; (6) safety data collection, including AEs as well as abnormal laboratory tests.
Adverse event monitoring
An AE is defined as any new untoward medical occurrence or worsening of a pre-existing medical condition in an enrolled patient, which does not necessarily have a causal relationship with the treatment. The causal relationship to study drug is determined by the physician. All AEs will be collected and recorded after the patient has provided consent and is included in the trial. An AE which meets the criteria for a serious adverse event (SAE) between study enrolment and hospital discharge will be reported to the DMC and TMC within 24 hours. SAE is any untoward medical occurrence at any dose, including: death; life-threatening conditions; patient requires inpatient hospitalisation or prolongation of existing hospitalisation; persistent or significant disability/incapacity; important medical event; overdose, cancer or potential drug induced liver injury. An SAE report should be completed for any event where doubt exists regarding its status of seriousness. All SAEs should be followed to resolution or stabilisation. A non-SAE is an AE not classified as serious. Non-serious AEs should be followed to resolution or stabilisation, or reported as SAEs if they become serious. Follow-up is also required for non-serious AEs that cause interruption or discontinuation of study drug.
Drop-out criteria
Patients who meet the following dropout criteria and discontinue or withdraw the treatment should finish the last follow-up according to V12 as soon as possible.
(1) Patients who want to withdraw the treatment. (2) Patients progress into ESRD or the investigators ascertain the patients need renal replacement therapy. (3) After enrolment, the investigators ascertain the patients are unsuitable for the treatment or the patients do not comply with the clinical scheme. (4) Investigators ascertain the patients appear to have a severe drug allergy. (5) Investigators ascertain the CKD patients with accompanying glomerulonephritis need to use a high dose of immunosuppressive agent. (6) Investigators ascertain the patients suffer from acute kidney injury or SCr ascends too fast. (7) If the BP is >130/80 mm Hg after taking >3 antihypertensive drugs for more than a month, the involvement of the patient is terminated early. (8) Proteinuria ≥3.5 g/day. (9) Patients who are required to start prednisone therapy. (10) Patients lost to follow-up. (11) Other situations according to the judgment of the investigators. When patients withdraw from the study, there will be no replacements.
Data handling and record keeping
Study data will be recorded through a web-developed electronic case report form (CRF) by a remote entry. CRF data will be anonymous and identified only by the assigned trial number. Any missing data, data out of pre-limits and possible duplication for each end point will be queried and internally validated by the DMC before locking the database. The DMC is independent of the sponsor and has no competing interests. An interim analysis will be performed on changes in eGFR when half of the patients have been randomised and have completed the 12-month follow-up. The interim analysis will be performed by the SRS who is blinded to the allocation. The SRS will report to the DMC, and the DMC will discuss the results with the SC to decide whether to continue with the trial.
Data analysis
Hypothesis
The primary efficacy endpoint is change in eGFR from baseline to month 24. The study is designed to test whether fosinopril plus benidipine is superior to fosinopril plus HCTZ in delaying renal disease progression. Therefore, the primary analysis will test the following hypotheses. Null hypothesis: Difference of change in eGFR >0, 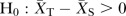 ; Alternative hypothesis: Difference of change in eGFR <0,
; Alternative hypothesis: Difference of change in eGFR <0, 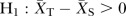 , α=0.025 (one-sided).
, α=0.025 (one-sided).
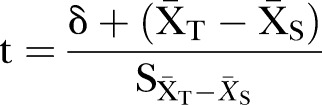 |
T is the fosinopril+benidipine group. S is the fosinopril+HCTZ group. At the α=0.025 level, if we reject H0 and conclude in favour of H1, this means fosinopril+benidipine is better than fosinopril+HCTZ in improving eGFR.
Sample size estimation
The sample size of this study was estimated according to the primary endpoint eGFR using a t-statistic under the assumption of a one-sided test of size 2.5%. Regarding the change of eGFR level, the required sample size for detecting a five difference with SD=16 between the two treatment arms (type I error, one sided α=0.025, power 1−β=0.90) was about 217 for each arm (434 in total) according to Ishimitsu et al's study.6 The formula is
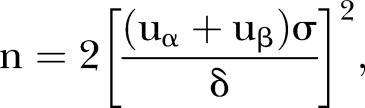 |
uα=1.96, uβ=1.282, δ=5, σ=16.
Assuming a 15% drop-out rate, at least 511 subjects will be needed.
Statistical plan
The patients in the statistical analysis will be selected as follows:
Modified intention to treat (MITT): all the patients who meet the inclusion/exclusion criteria and take the medicine and have at least one qualified record of primary endpoint evaluation post-baseline.
Per protocol (PP): all eligible patients who meet the study protocol, and complete the specified content in the case report after 24 months of follow-up.
Safety assessment patients: all the enrolled subjects who take the medicine and receive at least one safety assessment after the baseline visit.
All data will be shown as mean and SD with 95% CI. All statistical analyses will be performed using the SAS System. A two-tailed p value <0.05 will be considered significant.
Investigators will evaluate the compliance and remaining drugs of the patients at each visit of the follow-up. Drug compliance=the actual dosage of drug/the drug dosage according to the protocol. Drug compliance <80% or >120% indicates poor compliance. Patients with drug compliance <70% or >130% will exit PP analysis, but will still be included in the MITT analysis. Investigators will collect the drugs and remind patients at every visit to make sure of high treatment compliance. Researchers should deal with the situation of poor compliance and explain to the patient about the importance of time and drug dosage.
Extensive sensitivity analyses on the impact of the dropout and the robustness of results will be analysed by MITT and PP, respectively. Homogeneity analysis will be used to check the selection bias of the two groups and the baseline and demographic characteristics of the two groups.
All analysis of the endpoints will follow the MITT principle. The primary endpoint, change in eGFR in 24 months, will be analysed with the use of a linear mixed-effects model as a function of the treatment group, and month, with adjustment for treatment by month of interaction, age, sex, baseline BP, diabetes or non-diabetes, and baseline urine ACR (UACR) >300 or ≤300.
Assessments of secondary endpoints: Descriptive statistics, such as the mean, SD, maximum, minimum, first quartile, second quartile, third quartile, 95% CI, will be used to summarise the change in BP, 24 hour proteinuria, ACR, vWF, SA and ES at V12. They will also be analysed with a linear mixed-effects model as a function of treatment group, and month, with adjustment for age, sex, race, baseline eGFR, diabetes or non-diabetes, baseline ACR and clinical site. We will test slopes for overall 24-month changes; 24 hour proteinuria will be transformed with the use of the natural logarithm.
The incidence of renal events will be evaluated using the time-to-event analysis. The cumulative incidence rates will be calculated using the Kaplan-Meier method, defining the random allocation date as the starting point. The comparison between treatment arms is conducted using the log-rank test. The HRs and 95% CIs will be calculated from the univariate Cox regression of treatment and Cox regression with adjustment of important prognostic factors as above. AEs will be analysed using χ2 tests.
This study will define subgroups according to diabetes or non-diabetes, and baseline ACR >300 or ≤300. Subgroup divisions will be applied to the comparisons of the two treatment arms for primary and secondary endpoints.
Ethics and dissemination
BEAHIT is approved by the Changzheng Hospital Ethics Committee. We will publish the outcomes in peer-reviewed journals and they will be presented at national or international meetings. The ethics committee approved the revisions to the original protocol before the start of the trial.
Intra-study data sharing
The DMC will monitor the intra-study data sharing procedure. All principal investigators will be given access to the cleaned datasets. Data dispersed to trial team members will be blinded as to any patient identifying information. Project data sheets will be stored on a separate website protected with a password. The principal investigators will have direct access to datasets of their own site. The access to data at other sites needs a request.
Ancillary and post-trial care
All cases are covered by the Chinese Ping An indemnity during the trial. If patients suffer harm as a result of their participation in the trial, there will be free treatment and sufficient compensation.
Sponsorship
The trial is sponsored by Second Military Medical University and Changzheng Hospital Foundation.
Contact name: Mr Sun. Address: Xiangyin Road 800, Yangpu District, Shanghai, China; telephone: +86-21-81885114; email: syh001@smmu.edu.
Monitoring
The study investigators will allow authorised third parties (DMC, ethics committee and trial-related monitors) access to the study site, ethics, medical records and audits. The DMC will conduct more than one monitoring visit per 6 months over the duration of this trial. The objectives of the monitors will be to check the protocol in detail, solve any problems, identify any weaknesses and provide support. The DMC will audit the completeness and overall quality of the data, interview coordinators and investigators, examine the source documents, and check that the centre has complied with the protocol. Monitors will confirm that all AEs are consistent with the definition and have been correctly recorded.
Consent and confidentiality
This trial will be performed in accordance with the Declaration of Helsinki. Each patient will need to agree with a fully informed consent form and sign it. All study-related information will be stored securely in locked file cabinets at Changzheng Hospital. All laboratory specimens, data collection, reports, administrative forms and the process itself will be identified by a coded ID to maintain patient confidentiality. All records that include the name or personal identifier will be stored separately from records identified by ID. Datasets on the website will be protected by password. The principal investigators will need to sign agreements to maintain the confidentiality of all patients. Study information relating to any patient will not be released externally without the written permission of the patient.
Footnotes
Contributors: MCL conceived of this study. XC, ZCC, DB, YB, YSQ, LJY, ZGR, and MCL designed the trial. WY and ZGR helped with its implementation. MCL, YSQ, ZGR, and WY constituted the steering committee. XC and ZCC drafted the protocol aided by YB and DB. LJY and ZCC advised on the statistical plan. All the authors contributed to the refinement of this protocol, and decided to submit this final version.
Funding: The main phase is funded by the Second Military Medical University and Changzheng Hospital Foundation. Funding for this trial covers central organisational costs, meetings, and insurance of the included patients. The funders and sponsors had no role in the design of this trial, and will have no role in the analyses, execution, data interpretation and decisions forthcoming from the results.
Competing interests: None declared.
Ethics approval: This protocol was approved by the Changzheng Hospital Ethics Committee (CZ-20160504-16) and registered at clinicaltrials.gov (identifier: NCT02646397).
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Wang H, Zhang L, Lv J. Prevention of the progression of chronic kidney disease: practice in China. Kidney Int Suppl 2005;94:S63–7. [DOI] [PubMed] [Google Scholar]
- 2.Wheeler DC, Becker GJ. Summary of KDIGO guideline. What do we really know about management of blood pressure in patients with chronic kidney disease? Kidney Int 2013;83:377–83. [DOI] [PubMed] [Google Scholar]
- 3.Kloke HJ, Branten AJ, Huysmans FT et al. . Antihypertensive treatment of patients with proteinuric renal diseases: risks or benefits of calcium channel blockers? Kidney Int 1998;53:1559–73. 10.1046/j.1523-1755.1998.00912.x [DOI] [PubMed] [Google Scholar]
- 4.Bakris GL, Toto RD, McCullough PA et al. . Effects of different ACE inhibitor combinations on albuminuria: results of the GUARD study. Kidney Int 2008;73:1303–9. 10.1038/ki.2008.102 [DOI] [PubMed] [Google Scholar]
- 5.Hayashi M, Uchida S, Kawamura T et al. . Prospective randomized study of the tolerability and efficacy of combination therapy for hypertensive chronic kidney disease: results of the PROTECT-CKD study. Clin Exp Nephrol 2015;19:925–32. 10.1007/s10157-015-1091-5 [DOI] [PubMed] [Google Scholar]
- 6.Ishimitsu T, Ohno E, Nakano N et al. . Combination of angiotensin II receptor antagonist with calcium channel blocker or diuretic as antihypertensive therapy for patients with chronic kidney disease. Clin Exp Hypertens 2011;33:366–72. 10.3109/10641963.2010.503299 [DOI] [PubMed] [Google Scholar]
- 7.Bakris GL, Sarafidis PA, Weir MR et al. . Renal outcomes with different fixed-dose combination therapies in patients with hypertension at high risk for cardiovascular events (ACCOMPLISH): a prespecified secondary analysis of a randomised controlled trial. Lancet 2010;375:1173–81. 10.1016/S0140-6736(09)62100-0 [DOI] [PubMed] [Google Scholar]
- 8.Abe M, Okada K, Maruyama T et al. . Comparison of the antiproteinuric effects of the calcium channel blockers benidipine and amlodipine administered in combination with angiotensin receptor blockers to hypertensive patients with stage 3–5 chronic kidney disease. Hypertens Res 2009;32:270–5. 10.1038/hr.2009.11 [DOI] [PubMed] [Google Scholar]
- 9.Chan AW, Tetzlaff JM, Gotzsche PC et al. . SPIRIT 2013 explanation and elaboration: guidance for protocols of clinical trials. BMJ 2013;346:e7586 10.1136/bmj.e7586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taler SJ, Agarwal R, Bakris GL et al. . KDOQI US commentary on the 2012 KDIGO clinical practice guideline for management of blood pressure in CKD. Am J Kidney Dis 2013;62:201–13. 10.1053/j.ajkd.2013.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levey AS, Stevens LA, Schmid CH et al. . A new equation to estimate glomerular filtration rate. Ann Intern Med 2009;150:604–12. 10.7326/0003-4819-150-9-200905050-00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coresh J, Turin TC, Matsushita K et al. . Decline in estimated glomerular filtration rate and subsequent risk of end-stage renal disease and mortality. JAMA 2014;311:2518–31. 10.1001/jama.2014.6634 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjopen-2016-013672supp.pdf (15.5KB, pdf)