

Abstract
Drug-induced burst suppression (DIBS) is bihemispheric and bisymmetric in adults and older children. However, asymmetric DIBS may occur if a pathological process is affecting one hemisphere only or both hemispheres disproportionately. The usual suspect is a destructive lesion; an irritative or epileptogenic lesion is usually not invoked to explain DIBS asymmetry. We report the case of a 66-year-old woman with new-onset seizures who was found to have a hemorrhagic cavernoma and periodic lateralized epileptiform discharges (PLEDs) in the right temporal region. After levetiracetam and before anesthetic antiepileptic drugs (AEDs) were administered, the electroencephalogram (EEG) showed continuous PLEDs over the right hemisphere with maximum voltage in the posterior temporal region. Focal electrographic seizures also occurred occasionally in the same location. Propofol resulted in bihemispheric, but not in bisymmetric, DIBS. Remnants or fragments of PLEDs that survived anesthesia increased the amplitude and complexity of the bursts in the right hemisphere leading to asymmetric DIBS. Phenytoin, lacosamide, ketamine, midazolam, and topiramate were administered at various times in the course of EEG monitoring, resulting in suppression of seizures but not of PLEDs. Ketamine and midazolam reduced the rate, amplitude, and complexity of PLEDs but only after producing substantial attenuation of all burst components. When all anesthetics were discontinued, the EEG reverted to the original preanesthesia pattern with continuous non-fragmented PLEDs. The fact that PLEDs can survive anesthesia and affect DIBS symmetry is a testament to the robustness of the neurodynamic processes underlying PLEDs.
Key words: PLEDs, Burst suppression, Seizure, Anesthesia, Propofol
Competing interest statement
Conflict of interest: the authors declare no potential conflict of interest.
Introduction
Pathological activation of a cortical region at a rate of about 1/s can be detected in the electroencephalogram (EEG) as periodic lateralized epileptiform discharges (PLEDs), a term introduced by Chatrian et al. in 1964.1 PLEDs consist of periodic sharp waves, slow waves, and/or multiwave complexes that are lateralized, i.e. electrodes near the cortical generator on one side of the head record the highest voltage while electrodes distant from the generator on the contralateral side may or may not pick up some of the volume conducted signal.1,2 When two cortical foci, one in each hemisphere, generate PLEDs independently, the pattern is referred to as bilateral independent PLEDs (BiPLEDs).3 Two PLEDs-generating foci may also be in the same hemisphere.4 Other less familiar variants include three or more PLEDs-generating foci,5,6 PLEDs with maximum voltage in the midline,7 and alternating PLEDs.8 Most PLEDs occur in patients with acute focal destructive brain lesions (e.g. stroke, herpes encephalitis, traumatic brain injury), chronic brain lesions (e.g. tumors, inflammatory lesions), and epilepsy (with or without discrete lesions).9-13 PLEDs and BiPLEDs have also been reported in a variety of toxic and metabolic encephalopathies, encephalitides, and neurodegenerative processes.2,3,9-13
PLEDs are considered epileptiform because patients with PLEDs have a high probability of experiencing seizures; in some series the probability is about 50-100%.12-14 Reiher et al. reported a higher incidence of seizures when PLEDs are accompanied by low-voltage fast rhythms (PLEDs-plus) than when they are not (PLEDs-proper).15 However, Chong et al. found that PLEDs-proper rarely occur in isolation and that the EEG often fluctuates between PLEDs-proper and PLEDs-plus.16 It has been debated for decades whether PLEDs are ictal or interictal. Those who argue that PLEDs are ictal point to the clinical correlates of PLEDs, such as motor, sensory, and cognitive changes,17 or to the presence of focal glucose hypermetabolism on positron emission tomography (PET)18 and focal hyperperfusion on single-photon emission computed tomography (SPECT),19 not to mention the resolution of these findings when PLEDs disappear. Nonetheless, some studies have shown that focal hypermetabolism and/or hyperperfusion may also occur during focal interictal spikes.20,21 Electrographic seizures may emerge during PLEDs, coexist with PLEDs, or evolve on top of PLEDs10,14 suggesting that seizures and PLEDs are distinct phenomena. PLEDs are resistant to treatment with antiepileptic drugs (AEDs).22 In this regard, PLEDs resemble focal interictal spikes more than seizures. It may very well be the case that PLEDs represent neurophysiological processes that are distributed along an interictal-ictal continuum.13 The underlying brain injury, preexisting seizure propensity, and coexisting acute metabolic derangements determine whether a patient develops interictal PLEDs, ictal PLEDs, electrographic seizures, or a combination of these patterns.16 Some authors have proposed a treatment algorithm for PLEDs and other EEG patterns based on the theory of an interictal-ictal continuum.23,24
There is also evidence that PLEDs are not always epileptiform. Gross et al reported chronic PLEDs during sleep in a patient with ipsilateral caudate nucleus atrophy25 and Wheless et al reported PLEDs in a patient with acute thalamic stroke.26 Because the presence of an underlying epileptic disturbance and the risk of neuronal injury are still unknown in some periodic and rhythmic EEG patterns (PLEDs included), the American Clinical Neurophysiology Society (ACNS) adopted a nomenclature for rhythmic and periodic EEG patterns, first published in 200527 and updated in 2012.28 The goal was to have a standard terminology for research and expert communication. Terms with clinicopathologic undertones, such as triphasic waves and epileptiform, were discouraged in favor of terms that are more descriptive of the EEG pattern and that are non-committal with respect to pathophysiology. Thus, the new term lateralized periodic discharges or LPDs is preferred to the old term PLEDs. ACNS is not necessarily suggesting that we completely abandon all of the old terms for clinical use.27,28 In this paper, we will continue using the old and familiar term PLEDs.
Burst suppression was initially observed as a response of the EEG to high doses of anesthetic agents.29 Like PLEDs, drug-induced burst suppression (DIBS) is periodic with bursts of high-voltage activity alternating with periods of severe background attenuation or suppression.29,30 The cortical discharges in DIBS are clustered into bursts with variable amounts of slow waves, fast waves, and sharp waves (usually higher in amplitude than PLEDs) and the periods of suppression are characterized by isoelectric or severely attenuated EEG (usually more attenuated than the background in PLEDs).30 DIBS is used to monitor the effectiveness of anesthetic AEDs during treatment of refractory status epilepticus.31,32 The presence of DIBS in the EEG is often equated with adequate suppression of epileptic activity, including ictal and interictal PLEDs. However, whether DIBS is appropriate for all cases of status epilepticus and the duration and interburst interval of DIBS that is optimal for suppressing epileptic activity without causing hypotension and other anesthesia-related morbidities remains unknown.31-34
We report a case [note that the authors were involved with EEG monitoring but not with the clinical management of the patient] wherein PLEDs survived treatment with multiple AEDs, including three anesthetics (propofol, ketamine, and midazolam), and persisted during DIBS in the form of PLEDs remnants or fragments that resulted in DIBS asymmetry. The PLEDs reverted back to the original continuous (not fragmented) preanesthesia EEG pattern after withdrawal of anesthesia.
Case Report
A 66-year-old woman was found wandering aimlessly on the street with bruises on the extremities. Emergency responders suspected that she had an unwitnessed convulsion and was in a postictal state. She had a tonic-clonic seizure while in the emergency department (ED) and was treated with lorazepam 4-mg IV and levetiracetam 1500-mg IV load/750-mg q12. Her past medical history was significant for diabetes mellitus and hypertension but negative for seizure or epilepsy. The patient remained stuporous and was admitted to the intensive care unit (ICU). On admission, her vital signs, oxygen saturation, blood cell counts, glucose, electrolytes, liver and kidney function tests, urinalysis, and toxicology were all within normal limits.
Head CT was performed in the ED, followed 12 h later by a magnetic resonance imaging (MRI) scan of the brain (Figure 1). Contrast was not used in both studies. CT showed subtle punctate hyperintensity in the right temporal region (Figure 1CT). MRI revealed a well-circumscribed lesion in the inferior aspect of the right temporal lobe with the signal characteristics of a cavernous malformation or cavernoma, i.e. the lesion consisted of a core of extracellular methemoglobin indicating late subacute hemorrhage (age 1-4 weeks) and a rim of hemosiderin indicating old hemorrhage (age >1 month). Diffusion-weighted imaging (DWI) revealed a hyperintense lesion (Figure 1DWI). The apparent diffusion coefficient (ADC) of the core was high and a portion of the rim had low ADC (Figure 1-ADC). The core was hyperintense on T1-weighted (Figure 1T1a,c) and T2-weighted (Figure 1-T2a,c) images. The rim was isointense or slightly hypointense on T1-weighted (Figure 1T1a,c) and hypointense on T2-weighted (Figure 1T2a,c) images. These signal characteristics correspond to a type I cavernoma in the MRI-based classification of Zabramski et al.35 Pathologically, a type I cavernoma consists of a center with subacute hemorrhage surrounded by a rim of hemosiderin-stained macrophages and gliosis.35 MRI gradient echo (GRE) sequences showed a very dark rim around the bright core, consistent with blooming artifact (Figure 1GRE1). In addition to the main lesion, GRE revealed small asymptomatic hemorrhages near the midline and in the left hemisphere (Figure 1GRE2). These old hemorrhages could have been caused by cavernomas or other vascular lesions that bled in the past.
Figure 1.
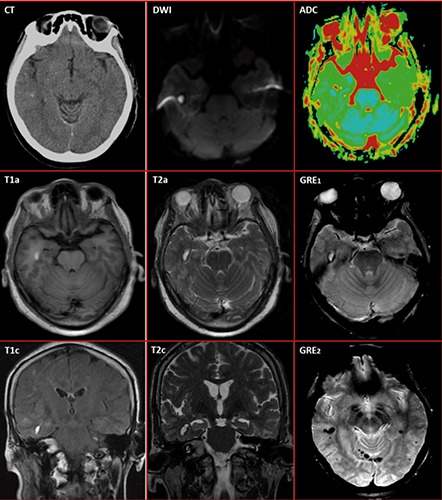
Brain structural imaging showing a circumscribed lesion in the inferior aspect of the right temporal lobe. Non-contrast CT (CT) shows a punctate hyperintensity in the right temporal area suggestive of calcification. The magnetic resonance imaging (MRI) signal characteristics of the lesion are consistent with a cavernous malformation or cavernoma with late subacute hemorrhage (core) and old chronic hemorrhage (rim) corresponding to a type I cavernoma in the MRI-based classification of Zabramski et al.35 The lesion is hyperintense (bright) on diffusion-weighted imaging (DWI) with high apparent diffusion coefficient (ADC) in the core and low ADC in a portion of the rim. The core is hyperintense on T1-weighted (T1a, T1c) and T2-weighted (T2a, T2c) images, indicating extracellular methemoglobin deposits from late subacute hemorrhage (age of 1 to 4 weeks). The rim is isointense on T1a, slightly hypointense on T1c, and moderately hypointense on T2-weighted sequences. On gradient echo sequences (GRE1), the bright lesion core is surrounded by a very dark rim (blooming artifact), indicating peripheral deposits of hemosiderin (a paramagnetic compound) from an old hemorrhage (age>1 month). Peripheral calcium deposits may also account for some of the blooming since hyperintensity was present on CT (see above). GRE sequences (GRE2) also revealed additional lesions with hypointense (dark) spots in other brain locations, which may represent multiple asymptomatic hemorrhages in the past, perhaps from other cavernomas.
Continuous EEG monitoring was performed over a five-day stretch (Figure 2). After levetiracetam was loaded, but before anesthetic AEDs were administered, the EEG showed PLEDs in the right hemisphere with maximum voltage in the right posterior temporal electrode (P8) and rarely in the right mid-temporal (T8) or anterior temporal (F8) electrodes (Figure 2A). The PLEDs were not associated with motor or behavioral change. Propofol was started at 50 mcg/kg/minute IV and DIBS with 5-10 bursts/minute was achieved with an infusion rate of 60 mcg/kg/minute. PLEDs survived as remnants or fragments mixed with other burst components or as discharges outside the bursts in the interburst intervals (Figure 2B). Surviving PLEDs remnants gave DIBS an asymmetric appearance. The EEG also showed recurrent focal electrographic seizures arising from the same PLEDs focus (not shown in the figure). The tenacity of PLEDs to propofol and the occasional occurrence of electrographic seizures motivated her physicians to escalate antiepileptic therapy. Phenytoin 1300-mg IV load/100-mg IV q8 and lacosamide 200-mg IV load/100-mg IV q12 were added. Ketamine 25 mcg/kg/minute IV was also started and incrementally titrated to 65 mcg/kg/minute. Electrographic seizures ceased, the burst rate, amplitude, and complexity of DIBS decreased, and most fast rhythms disappeared, but PLEDs remnants and DIBS asymmetry persisted (Figure 2C). The addition of midazolam up to 1.2 mcg/kg/minute IV and topiramate 200-mg per NGT q12 reduced the burst rate, amplitude, and complexity of DIBS further. PLEDs remnants were attenuated but did not disappear (Figure 2D).
Figure 2.

Electroencephalogram (EEG) tracings represent different time points (A-E) during treatment with antiepileptic and anesthetic drugs. Display settings: 16-channel longitudinal bipolar montage; filters at 1-Hz high-pass, 70-Hz low-pass, 60-Hz notch; sensitivity at 7µV/cm; voltage-time scale: 100 µV/1 sec for the full tracings and 50 µV/1 sec for the magnified epochs. The EEG is shown on the left as a 16-channel trace with the channels separated from top to bottom into the following groups: left temporal, left parasagittal, right parasagittal, and right temporal. For clarity, the final 6 seconds of each tracing are magnified and displayed as two 8-channel traces: parasagittal (left over right) derivations in the middle column and temporal (left over right) derivations in the right column. A) After levetiracetam was loaded, but before infusion of anesthetic antiepileptic drugs, periodic lateralized epileptiform discharges (PLEDs) were present on the right and maximum in the right posterior temporal region. B) Propofol resulted in asymmetric drug-induced burst suppression with surviving remnants or fragments of PLEDs on the right that are mixed with other burst components increasing the overall amplitude and complexity of bursts on the right. C) The addition of phenytoin, lacosamide, and ketamine reduced the rate and complexity of bursts and caused fast rhythms to disappear but failed to completely suppress the PLEDs remnants. D) The addition of midazolam and topiramate resulted in further reduction of burst rate and complexity and attenuation of burst components. The PLEDs remnants were also attenuated but not completely suppressed. E) After withdrawal of anesthesia, the PLEDs started to reacquire their preanesthesia configuration. The post-anesthesia trace in this figure shows continuous PLEDs with interdischarge interval of 2-3 sec.
By day 5, all anesthetic AEDs have been discontinued and the patient passed the extubation test. Approximately 12 h after all anesthetic agents were discontinued, the same PLEDs started appearing in the same location (Figure 2E). At first, the PLEDs were very fragmented and far apart with an interdischarge interval >3 s and minimal periodicity. With time, the PLEDs became more continuous and exhibited a more stable interdischarge interval of 2-3 sec. EEG monitoring was discontinued at this point. The patient was successfully extubated but her clinical course was regrettably complicated with metabolic disturbances and cardiac arrhythmias: she expired after 9 days in the ICU.
Discussion
This case shows that PLEDs can survive anesthesia and result in asymmetric DIBS. In general, DIBS asymmetry is attributed to a loss of function, i.e. burst components are attenuated in one hemisphere due to a destructive lesion. An irritative or epileptogenic lesion resulting in a gain of function in one hemisphere is not usually invoked to explain DIBS asymmetry. The patient developed PLEDs secondary to a hemorrhagic cavernoma in the right temporal lobe (Figure 1). During DIBS, the PLEDs persisted as PLEDs fragments that were mixed with the burst components of DIBS in the right hemisphere (Figure 2). The waveform morphology, polarity, and spatial distribution (maximum voltage was often at P8 and occasionally at T8 or F8) indicate that these burst-embedded fragments are the remnants of PLEDs that were originally present in the EEG prior to anesthesia. These PLEDs remnants increased the overall amplitude and complexity of bursts in the right hemisphere giving DIBS an asymmetric appearance.
PLEDs are the expression of neural network dysfunction producing cortical hypersynchrony at a rate of about 1/s. It is not clear what neurodynamic perturbations give rise to PLEDs, much less whether these perturbations are interictal, ictal, or nonepileptic in nature. It is for this reason that a non-committal term, such as LPDs, is preferred over PLEDs.27,28 In 1950, Cobb et al. attributed periodic discharges to a disconnection of the cerebral cortex from subcortical structures.36 After studying autopsy specimens, Chatrian et al. found no anatomic isolation of cortical areas involved in generating PLEDs and the putative lesions were frequently far from the cortical generator.1 Gloor et al. suggested a form of cortical-subcortical interaction in which subcortical discharges are projected to the cortex and cortical recovery time determines discharge rate (~1/s).37 Raroque et al. reviewed the CT/MRI of 39 patients with PLEDs/BiPLEDs and concluded that structural lesions have a primary role in the pathogenesis of PLEDs but a role for metabolic abnormalities cannot be excluded.38 Brain lesions in patients with PLEDs vary in their age and location; however, the majority are acute structural lesions in the cortical gray and the adjacent white matter.39 It is also a fact that some patients with PLEDs have no CT/MRI evidence of a causative brain lesion.9-11 PLEDs may be triggered by systemic conditions that lower the seizure threshold.12,15 Some authors hypothesized that PLEDs are the expression of a change in excitatory neurotransmission due to an acute brain lesion producing partial functional denervation in a localized area of the cortex.8 Studies of EEG phase reversals suggest that PLEDs originate in hyperexcitable cortex at the margins of the lesion.40
Serial EEG recordings have shown that PLEDs often disappear spontaneously in 2 to 3 weeks.14,15 There are however reports of PLEDs persisting for months or years41 or resolving within a few days of onset.42 BiPLEDs tend to be more transitory because they are triggered by pathological processes that progress relentlessly, with other abnormal EEG patterns replacing BiPLEDs, or by physiological disturbances that are highly reversible, e.g. BIPLEDs appeared with nafcillin treatment and disappeared three days after the drug was discontinued.43 Even though PLEDs and BiPLEDs are self-limited, they are often resistant to treatment with AEDs if the causative pathological processes are still active. In terms of AED resistance, PLEDs tend to surpass seizures and resemble focal IEDs. In-vitro experiments showed that AEDs abolish prolonged seizure-like discharges at concentrations that do not influence shorter interictal-like events.44-46 In rodent hippocampal slices, AEDs resulted in truncation of long-lasting seizure-like discharges elicited by long-duration high-frequency stimulation, but did not affect short-lived IED-like activity induced by stimulation every 5 to10 seconds.47 Some well-controlled studies showed that AED levels that control seizures do not suppress focal IEDs.48,49 Recently, Guida et al. published a review of past studies that sought to determine the effects of AEDs (alone or in combination) on focal IEDs and concluded that the data is scarce or conflicting for classical AEDs and absent or limited for newer AEDs.50 In any case, they considered their findings consistent with the prevailing view that focal IEDs are not influenced by chronic AED therapy in patients with focal epilepsy. There are few reports of successful control of PLEDs using carbamazepine, midazolam, pentobarbital, sodium valproate, and felbamate,22 but these studies must be replicated. While the resolution (or progression) of the underlying pathology (not the direct effects of AEDs) is most likely the reason PLEDs disappear, most experts still recommend using nonanesthetic AEDs to decrease the probability of seizures in patients with PLEDs.
When anesthetics are administered to treat status epilepticus, it is often recommended to titrate the anesthetic to achieve a DIBS interburst interval of 2-20 s over a period of 24-48 h.51 However, a recent study where a cohort of 19 patients received anesthetics for refractory status epilepticus, showed that treatment success was not influenced by the interburst interval or the duration of DIBS.52 In the case presented, focal electrographic seizures ceased completely during DIBS but PLEDs persisted despite uptitration of anesthetic dosage (propofol up to 60 µg/kg/m, ketamine up to 65 µg/kg/m, and midazolam up to 1.2 µg/kg/m) to maintain a DIBS interburst interval of 6-12 s (burst rate of 5-10/m). The surviving PLEDs remnants were mixed with burst components, occasionally spilling over the interburst intervals (Figure 2B-D). The original pre-anesthesia pattern of continuous PLEDs (Figure 2A) was restored when anesthesia was discontinued (Figure 2E). Fishman and Legatt reported a case wherein PLEDs were resistant, not only to AEDs, but also to hypoxia.53 In the course of progressive cerebral dysfunction, the patient’s brain continued producing PLEDs until the last minutes of her life; the PLEDs disappeared only 40 seconds before the EEG turned isoelectric. The resistance of PLEDs to AEDs, anesthesia, and hypoxia reminds us of the robustness of the processes driving PLEDs. Anesthetizing the patient, increasing the depth of anesthesia, or combining anesthetics in order to suppress PLEDs may therefore be more harmful than beneficial to the patient.
As a rule, DIBS is bihemispheric, bisymmetric, and bisynchronous in adults. Asymmetric and/or asynchronous DIBS has been reported in patients with disorders involving the corpus callosum.54,55 However, true unihemispheric burst suppression (UBS) is rare. Recently, we reported two cases in which administration of propofol to terminate ongoing status epilepticus resulted in the appearance of UBS.56 In both patients, UBS occurred ipsilateral to a structural brain lesion and disappeared when the propofol infusion rate was increased indicating an epileptic mechanism.56 As an EEG finding, UBS challenges the prevailing view that burst suppression is global with widespread synchronous and homogenous cortical activation during burst episodes and inactivation during periods of suppression.57 The theory that DIBS is rooted in local neurodynamics was put forth as early as 1952.58 That DIBS is an expression of local cortical dynamics is supported by a recent intracranial EEG study of patients under propofol anesthesia which showed asynchronous expression of DIBS in different cortical areas and restriction of DIBS in some cortical areas with other areas exhibiting continuous activity.59
Abnormal neuronal network dynamics underlies cortical hypersynchrony in patients with epilepsy.60,61 The idea of an interictal-ictal continuum was contrived because some forms of cortical hypersynchrony could not be classified as interictal or ictal.12,15 Some studies showed the risk of seizures to be higher in PLEDs-plus than in PLEDs-proper.14,15 It is possible (there are no studies) that UBS carries a higher risk of seizures than PLEDs-plus and that the unihemispheric periodic patterns – PLEDs-proper, PLEDs-plus, and UBS – represent an interictal-ictal continuum. We also believe that DIBS is rooted in local cortical dynamics and that different periodic EEG patterns involve the same fundamental oscillatory mechanisms. Kalamangalam et al. reviewed the CT/MRI of 106 patients with PLEDs and found PLEDs to be longer in duration and more variable in morphology when the cause is a cortical lesion than when it is a subcortical lesion.62 The authors offered a unified view of periodic EEG patterns – they proposed that PLEDs from subcortical lesions and generalized periodic EEG patterns from severe metabolic diseases share common pathogenetic mechanisms. In another study, Kalamangalam et al. compared the frequency components of PLEDs and the EEG background and concluded that pathological processes can lead to the emergence of PLEDs from the background by means spectral condensation.62 Normal or pathological states that influence the spatiotemporal organization of local field cortical potentials and cause functional cortical domains to expand or contract can alter the frequencies and rhythms of the EEG. Pathological processes that enhance excitatory or reduce inhibitory neurotransmission in the cortex may lead to coupling of cortical domains, both in spatial terms and in terms of their intrinsic frequencies. When rhythms merge, the multi-frequency mix gives way to global synchrony at some smaller set of intermediate frequencies. This process, which Kalamangalam et al. refer to as spectral condensation, can serve as a framework for understanding all normal and abnormal, rhythmic and periodic, synchronous cortical activity.62,63
Conclusions
PLEDs can survive anesthesia and persist during DIBS as PLEDs fragments or remnants that give DIBS an asymmetric appearance. This may be subtle and can be easily overlooked if the EEG during DIBS is not carefully inspected and compared with the preanesthesia EEG. The PLEDs fragments are mixed with other burst components but continue to exhibit some of the original characteristics of PLEDs, such as spatial distribution and morphology. This finding suggests that PLEDs, DIBS, and other periodic EEG patterns share some basic neurodynamic mechanisms. Physicians, who are aware of the difficulty of suppressing PLEDs during the acute phase of a brain disorder, can avoid exposing patients to the risk of anesthesia. The pathophysiological and therapeutic significance of burst-embedded PLEDs fragments and their tenacity to anesthesia deserve further investigation.
Acknowledgments
We are grateful to our EEG technologists Thomas Miller, Sheryl Wagamonte, and Lisa Keppard for recording high-quality electroencephalograms.
References
- 1.Chatrian GE, Shaw CM, Leffman H. The significance of periodic lateralized epileptiform discharges in EEG: an electrographic, clinical and pathological study. Electroencephalogr Clin Neurophysiol 1964;17:177-93. [DOI] [PubMed] [Google Scholar]
- 2.Brenner RP, Schaul N. Periodic EEG patterns: classification, clinical correlation, and pathophysiology. J Clin Neurophysiol 1990;7:249-67. [PubMed] [Google Scholar]
- 3.de la Paz D, Brenner RP. Bilateral independent periodic lateralized epileptiform discharges. Clinical significance. Arch Neurol 1981;38:713-5. [DOI] [PubMed] [Google Scholar]
- 4.Silbert PL, Radhakrishnan K, Sharbrough FW, Klass DW. Ipsilateral independent periodic lateralized epileptiform discharges. Electroencephalogr Clin Neurophysiol 1996;98:223-6. [DOI] [PubMed] [Google Scholar]
- 5.Hughes JR, Taber J, Uppal H. TRIPLEDs: a case report. Clin Electroencephalogr 1998;29:106-8. [DOI] [PubMed] [Google Scholar]
- 6.Lawn ND, Westmoreland BF, Sharbrough FW. Multifocal periodic lateralized epileptiform discharges (PLEDs): EEG features and clinical correlations. Clin Neurophysiol 2000;111:2125-9. [DOI] [PubMed] [Google Scholar]
- 7.Westmoreland BF, Frere RC, Klass DW. Periodic epileptiform discharges in the midline. J Clin Neurophysiol 1997;14:495-8. [DOI] [PubMed] [Google Scholar]
- 8.Bertolucci PH, Silva AB. Alternating periodic lateralized epileptiform discharges (cerebral bigeminy). Clin Electroencephalogr 1992;23:177-9. [DOI] [PubMed] [Google Scholar]
- 9.García-Morales I, García MT, Galán-Dávila L, et al. Periodic lateralized epileptiform discharges: etiology, clinical aspects, seizures, and evolution in 130 patients. J Clin Neurophysiol 2002;19:172-7. [DOI] [PubMed] [Google Scholar]
- 10.Fitzpatrick W, Lowry N. PLEDs: clinical correlates. Can J Neurol Sci 2007;34:443-50. [PubMed] [Google Scholar]
- 11.Andraus ME, Andraus CF, Alves-Leon SV. Periodic EEG patterns: importance of their recognition and clinical significance. Arq Neuropsiquiatr 2012;70:145-51. [DOI] [PubMed] [Google Scholar]
- 12.Baykan B, Kinay D, Gökyigit A, Gürses C. Periodic lateralized epileptiform discharges: association with seizures. Seizure 2000;9:402-26. [DOI] [PubMed] [Google Scholar]
- 13.Pohlmann-Eden B, Hoch DB, Cochius JI, Chiappa KH. Periodic lateralized epileptiform discharges: a critical review. J Clin Neurophysiol 1996;13:519-30. [DOI] [PubMed] [Google Scholar]
- 14.Garzon E, Fernandes RM, Sakamoto AC. Serial EEG during human status epilepticus: evidence for PLED as an ictal pattern. Neurology 2001. 9;57:1175-83. [DOI] [PubMed] [Google Scholar]
- 15.Reiher J, Rivest J, Grand’Maison F, Leduc CP. Periodic lateralized epileptiform discharges with transitional rhythmic discharges: association with seizures. Electroencephalogr Clin Neurophysiol 1991;78:12-7. [DOI] [PubMed] [Google Scholar]
- 16.Chong DJ, Hirsch LJ. Which EEG patterns warrant treatment in the critically ill? Reviewing the evidence for treatment of periodic epileptiform discharges and related patterns. J Clin Neurophysiol 2005;22:79-91. [DOI] [PubMed] [Google Scholar]
- 17.Sen-Gupta I, Schuele SU, Macken MP, et al. “Ictal” lateralized periodic discharges. Epilepsy Behav 2014;36:165-70. [DOI] [PubMed] [Google Scholar]
- 18.Handforth A, Cheng JT, Mandelkern MA, et al. Markedly increased mesiotemporal lobe metabolism in a case with PLEDs: further evidence that PLEDs are a manifestation of partial status epilepticus. Epilepsia 1994;35:876-81. [DOI] [PubMed] [Google Scholar]
- 19.Assal F, Papazyan JP, Slosman DO, et al. SPECT in periodic lateralized epileptiform discharges (PLEDs): a form of partial status epilepticus? Seizure 2001;10:260-5. [DOI] [PubMed] [Google Scholar]
- 20.Bittar RG, Andermann F, Olivier A, et al. Interictal spikes increase cerebral glucose metabolism and blood flow: a PET study. Epilepsia 1999;40:170-8. [DOI] [PubMed] [Google Scholar]
- 21.Tousseyn S, Dupont P, Goffin K, et al. Correspondence between large-scale ictal and interictal epileptic networks revealed by single photon emission computed tomography (SPECT) and electroencephalography (EEG)-functional magnetic resonance imaging (fMRI). Epilepsia 2015;56:382-92. [DOI] [PubMed] [Google Scholar]
- 22.Hughes JR. Periodic lateralized epileptiform discharges: do they represent an ictal pattern requiring treatment? Epilepsy Behav 2010;18:162-5. [DOI] [PubMed] [Google Scholar]
- 23.Sivaraju A, Gilmore EJ. Understanding and managing the ictal-interictal continuum in neurocritical care. Curr Treat Options Neurol 2016;18:8. [DOI] [PubMed] [Google Scholar]
- 24.Rodríguez V, Rodden MF, LaRoche SM. Ictal-interictal continuum: a proposed treatment algorithm. Clin Neurophysiol 2016;127:2056-64. [DOI] [PubMed] [Google Scholar]
- 25.Gross DW, Quesney LF, Sadikot AF. Chronic periodic lateralized epileptiform discharges during sleep in a patient with caudate nucleus atrophy: insights into the anatomical circuitry of PLEDs. Electroencephalogr Clin Neurophysiol 1998;107:434-8. [DOI] [PubMed] [Google Scholar]
- 26.Wheless JW, Holmes GL, King DW, et al. Possible relationship of periodic lateralized epileptiform discharges to thalamic stroke. Clin Electroencephalogr 1991;22:211-6. [DOI] [PubMed] [Google Scholar]
- 27.Hirsch LJ, Brenner RP, Drislane FW, et al. The ACNS subcommittee on research terminology for continuous EEG monitoring: proposed standardized terminology for rhythmic and periodic EEG patterns encountered in critically ill patients. J Clin Neurophysiol 2005;22:128-35. [DOI] [PubMed] [Google Scholar]
- 28.Hirsch LJ, LaRoche SM, Gaspard N, et al. American Clinical Neurophysiology Society’s standardized critical care EEG terminology: 2012 version. J Clin Neurophysiol 2013;30:1-27. [DOI] [PubMed] [Google Scholar]
- 29.Niedermeyer E. The burst-suppression electroencephalogram. Am J Electroneurodiagnostic Technol 2009;49:333-41. [PubMed] [Google Scholar]
- 30.Urrego JA, Greene SA, Rojas MJ. Brain burst suppression activity. Psychol Neurosci 2014;7:531-43. [Google Scholar]
- 31.Bergey GK. Refractory status epilepticus: is EEG burst suppression an appropriate treatment target during drug-induced coma? What is the Holy Grail? Epilepsy Curr 2006;6:119-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rossetti AO, Lowenstein DH. Management of refractory status epilepticus in adults: still more questions than answers. Lancet Neurol 2011;10:922-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shorvon S, Ferlisi M. The treatment of super-refractory status epilepticus: a critical review of available therapies and a clinical treatment protocol. Brain 2011;134:2802-18. [DOI] [PubMed] [Google Scholar]
- 34.Kang BS, Jung KH, Shin JW, et al. Induction of burst suppression or coma using intravenous anesthetics in refractory status epilepticus. J Clin Neurosci 2015;22:854-8. [DOI] [PubMed] [Google Scholar]
- 35.Zabramski JM, Wascher TM, Spetzler RF, et al. The natural history of familial cavernous malformations: results of an ongoing study. J Neurosurg 1994;80:422-32. [DOI] [PubMed] [Google Scholar]
- 36.Cobb W, Hill D. Electroencephalogram in subacute progressive encephalitis. Brain 1950;73:392-404. [DOI] [PubMed] [Google Scholar]
- 37.Gloor P, Kalabay O, Giard N. The electroencephalogram in diffuse encephalopathies: electroencephalographic correlates of gray and white matter lesions. Brain 1968;91:779-802. [Google Scholar]
- 38.Raroque HG, Jr, Purdy P. Lesion localization in periodic lateralized epileptiform discharges: gray or white matter. Epilepsia 1995;36:58-62. [DOI] [PubMed] [Google Scholar]
- 39.Gurer G, Yemisci M, Saygi S, Ciger A. Structural lesions in periodic lateralized epileptiform discharges (PLEDs). Clin EEG Neurosci 2004;35:88-93. [DOI] [PubMed] [Google Scholar]
- 40.Schwartz MS, Prior PF, Scott DF. The occurrence and evolution in the EEG of a lateralized periodic phenomenon. Brain 1973;96:613-22. [DOI] [PubMed] [Google Scholar]
- 41.Westmoreland BF, Klass DW, Sharbrough FW. Chronic periodic lateralized epileptiform discharges. Arch Neurol 1986;43:494-6. [DOI] [PubMed] [Google Scholar]
- 42.Marino D, Vatti G, Rufa A, et al. Transient periodic lateralised epileptiform discharges (PLEDs) following internal carotid artery stenting. Epileptic Disord 2012;14:85-9. [DOI] [PubMed] [Google Scholar]
- 43.Mader E, Olejniczak P, Fisch B. BIPLEDs with complete and rapid recovery in a patient with AIDS encephalopathy. Am J END Technol 2000;40:177-84. [Google Scholar]
- 44.Fueta Y, Avoli M. Effects of antiepileptic drugs on 4-aminopyridine-induced epileptiform activity in young and adult rat hippocampus. Epilepsy Res 1992;12:207-15. [DOI] [PubMed] [Google Scholar]
- 45.Brückner C, Stenkamp K, Meierkord H, Heinemann U. Epileptiform discharges induced by combined application of bicuculline and 4-aminopyridine are resistant to standard anticonvulsants in slices of rats. Neurosci Lett 1999;268:163-5. [DOI] [PubMed] [Google Scholar]
- 46.Brückner C, Heinemann U. Effects of standard anticonvulsant drugs on different patterns of epileptiform discharges induced by 4-aminopyridine in combined entorhinal cortex-hippocampal slices. Brain Res 2000;859:15-20. [DOI] [PubMed] [Google Scholar]
- 47.D’Antuono M, Köhling R, Ricalzone S, et al. Antiepileptic drugs abolish ictal but not interictal epileptiform discharges in vitro. Epilepsia 2010;51:423-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gotman J, Marciani MG. Electroencephalographic spiking activity, drug levels, and seizure occurrence in epileptic patients. Ann Neurol 1985;17:597-603. [DOI] [PubMed] [Google Scholar]
- 49.Spencer SS, Goncharova II, Duckrow RB, et al. Interictal spikes on intracranial recording: behavior, physiology, and implications. Epilepsia 2008;49:1881-92. [DOI] [PubMed] [Google Scholar]
- 50.Guida M, Iudice A, Bonanni E, Giorgi FS. Effects of antiepileptic drugs on interictal epileptiform discharges in focal epilepsies: an update on current evidence. Expert Rev Neurother 2015;15:947-59. [DOI] [PubMed] [Google Scholar]
- 51.Brophy GM, Bell R, Claassen J, et al. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care 2012;17:3-23. [DOI] [PubMed] [Google Scholar]
- 52.Johnson EL, Martinez NC, Ritzl EK. EEG characteristics of successful burst suppression for refractory status epilepticus. Neurocrit Care 2016. (in press). [DOI] [PubMed] [Google Scholar]
- 53.Fishman O, Legatt AD. PLEDs following control of seizures and at the end of life. Clin EEG Neurosci 2010;41:11-4. [DOI] [PubMed] [Google Scholar]
- 54.Lambrakis CC, Lancman ME, Romano C. Asynchronous and asymmetric burst-suppression in a patient with a corpus callosum lesion. Clin Neurophysiol 1999;110:103-5. [DOI] [PubMed] [Google Scholar]
- 55.Lazar LM, Milrod LM, Solomon GE, Labar DR. Asynchronous pentobarbital-induced burst suppression with corpus callosum hemorrhage. Clin Neurophysiol 1999;110:1036-40. [DOI] [PubMed] [Google Scholar]
- 56.Mader EC, Jr, Villemarette-Pittman NR, Rogers CT, et al. Unihemispheric burst suppression. Neurol Int 2014;6:5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clark DL, Rosner BS. Neurophysiologic effects of general anesthetics. I. The electroencephalogram and sensory evoked responses in man. Anesthesiology 1973;38:564-82. [PubMed] [Google Scholar]
- 58.Henry CE, Scoville WB. Suppression-burst activity from isolated cerebral cortex in man. Electroencephalogr Clin Neurophysiol 1952;4:1-22. [DOI] [PubMed] [Google Scholar]
- 59.Lewis LD, Ching S, Weiner VS, et al. Local cortical dynamics of burst suppression in the anaesthetized brain. Brain 2013;136:2727-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spencer SS. Neural networks in human epilepsy: evidence of and implications for treatment. Epilepsia 2002;43:219-27. [DOI] [PubMed] [Google Scholar]
- 61.Da Silva FH, Gorter JA, Wadman WJ. Epilepsy as a dynamic disease of neuronal networks. Handb Clin Neurol 2012;107:35-62. [DOI] [PubMed] [Google Scholar]
- 62.Kalamangalam GP, Diehl B, Burgess RC. Neuroimaging and neurophysiology of periodic lateralized epileptiform discharges: observations and hypotheses. Epilepsia 2007;48:1396-405. [DOI] [PubMed] [Google Scholar]
- 63.Kalamangalam GP, Slater JD. Periodic lateralized epileptiform discharges and afterdischarges: common dynamic mechanisms. J Clin Neurophysiol 2015;32:331-40. [DOI] [PMC free article] [PubMed] [Google Scholar]