

Abstract
We report a case of hemoglobin (Hb) Willamette (β51 Pro → Arg) in the Hematology Department of a tertiary hospital in Fortaleza, Northeast of Brazil. A literature review of the cases described in health sciences databases using as a descriptor Hb Willamette was performed, revealing 12 reported cases, of which only one presented with anemia. Herein, we describe a case of a female 29 years old, with hemoglobinopathy Willamette presenting clinically with anemia, having the lowest hemoglobin rate of the published cases. The relatives of the patient were evaluated andthe patient’s mother corresponded to the first description of the association between Hb Willamette and HbC. Among the hemoglobinopathies, hemoglobin Willamette is an extremely rare disease; therefore it is important to analyze its clinical and laboratory manifestations for accurate diagnosis and assessment of potential interactions with other genetic variants.
Key words: hemoglobins, hemoglobin Willamette, Anemia, hemolytic, Anemia, hemolytic, congenital
Competing interest statement
Conflict of interest: the authors declare no potential conflict of interest.
Introduction
Inherited hemoglobin (Hb) disorders are the most common monogenic disorders in humankind,1 with an estimated 300,000 children born annually with severe disease.2 Hereditary hemoglobinopathies originally appeared in tropical and subtropical regions, but are common nowadays worldwide as a result of migration.3 The correct identification and classification emerge with importance in the medical, genetics and biochemistry fields, for presenting a wide clinical spectrum with consequences ranging from barely noticeable to lethal.4
Within this context, although most cases are structural variants of β globin chains, changes of α chains, γ and δ are also relatively common. Some of these changes affect the stability and/or solubility of the molecule or alter its functional properties, leading to hemolytic anemias, increased number of red blood cells and cyanosis, respectively. There are elongated or extremely unstable variants that result in thalassemic phenotypes.5
Hb Willamette is a rare mutation in the β chain globin which, although does not produce significant chemical and hematological abnormalities is considered of interest by causing changes detected in vitro, which include higher thermal fragility, increased affinity to oxygen and reducing the Bohr effect.6
Given the rare occurrence of this mutation, this paper describes a case of Hb Willamette in a 29-year-old female accompanied due to anemia as an outpatient in a tertiary public hospital in Fortaleza, Northeast of Brazil, associated with a literature review of the previously reported cases.
Case Report
Woman, 29 years old, born in Fortaleza, Brazil, referring history of anemia since childhood. The patient reported episodes of breathlessness associated with asthma, as well as malaise, asthenia and difficulty concentrating which has worsened since 2010, when she sought medical assistance and was diagnosed with Hashimoto’s disease. She was referred to the hematology department of a tertiary hospital for persistent anemia, despite levothyroxine replacement therapy. Her previous history revealed hypertrophic allergic rhinitis, bronchial asthma and hypothyroidism in use of levothyroxine. Laboratory tests: hemoglobin: 8.4 g/dL, hematocrit: 23.4%, mean corpuscular volume: 75.0 fL, white blood cell count: 9745 mm3, platelet count: 384×109/L, reticulocyte count: 4.8%. peripheral smear featuring anisocromia, microcytosis and polychromasia, with normal serum iron, transferrin saturation index and ferritin.
Study of hemoglobin by high-performance liquid chromatography (HPLC): HbA: 54.4% HbF: 0% HbA2: 0.0% *HbS: 44.9%. The chromatographic profile was suggestive of Hb Willamette in heterozygosis (Figure 1) and a mutation survey for HbS by molecular analysis study (PCRRFLP) was negative.
Figure 1.
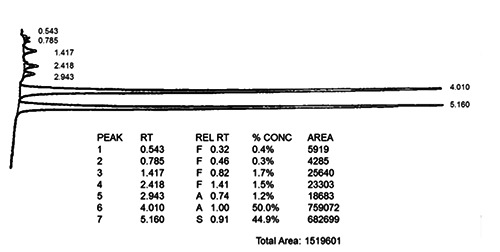
High-performance liquid chromatography demonstrating an abnormal peak that comprises approximately 44.9% of the total hemoglobin, consistent with an variant hemoglobin Willamette.
Patient’s parents were also investigated for mutated Hb with HPLC. Father Result: HbA: 97.5%, HbF: 0% Hb A2: 2.5%. Mother Result: HbF: 0%, HbA2: 0%, HbS : 55.1%, HbC: 41.0%. The chromatographic profile of the mother revealed Hb Willamette as heterozygosis, the study by technique of molecular analysis (PCRRFLP) showed no mutations for HbS, but Mutation for HbC.
This paper has been approved by an ethical committee and thus meets the standards of the Declaration of Helsinki.
Discussion
In 1976, as a result of a sickle cell disease screening program, a variant of hemoglobin with the same electrophoretic mobility of HbS was identified in three generations of an African-American family. Functional studies of hemoglobin showed the presence of arginine in the proline in position 51 residue of the abnormal β chain, and thus hemoglobin identified as Hb Willamette (β51 D2 Pro → Arg). Seven articles were found in the search for Hb Willamette in the PubMed database. From the total of seven publications, three reported the presence of new cases of mutation. Three other citations to Hb Willamette were found in SciVerse ScienceDirect database.
The three articles reporting the presence of twelve new cases of hemoglobinopathy also exhibited their structural description,6-8 the oxygen balance in Hb Willamette,9 and the successful use of the methods of electrospray mass spectrometry to identify variant Hb.10-12 Another paper assesses the structure of Hb Willamette through Activated Dissociation Collision and mass spectrometry.13 There are also reports of Hb variant in a review paper on unstable hemoglobins,14 and as a note for the advent of its discovery in another journal.15
As to the heritability and hematologic workup, the study of University of Oregon Health Sciences Center disclosed the presence of a heterozygous affected family member in each of the three generations analyzed. The patients did not have anemia, splenomegaly, history of jaundice, cholelithiasis, reticulocytes or other clinical evidence of significant hematologic abnormalities, except the presence of target red blood cells.6
In 1981, three cases in China were published describing the structural analysis of the abnormal hemoglobin molecule. There were also identified four relatives of these patients with the mutation. In a female patient, 10 years, anemia was diagnosed, which consisted of a new finding in relation to the series of American cases.7
In 1983, another case was diagnosed with Hb in Willamette woman, 15 years old, black, Cuban, assessed at the Hematology Institute of Havana, where it was referenced by the diagnosis of acute lymphoblastic leukemia. The variant found was also present in her father.
As in the first report, hematologic abnormalities were not found with the exception of target cells in the peripheral blood smear. The instability test was slightly positive, as reported in the American family, however Heinz bodies were found after three hours’ incubation at 37°C in the Cuban study, finding absent in the American study. The Cuban authors highlighted the possibility that the discrepancy was due to differences in efficiency of various lots of coloring dyes, which can be considerable. Furthermore, the percentage of hemoglobin variant determined by DEAE-Sephadex chromatography method was 35% in two patients.8
In the case reported, it is possible to observe anemia in association with Hb Willamette (Hb=8.4 g/dL). There was no clinical evidence of jaundice, splenomegaly and cholelithiasis, but the patient had the lowest Hb rates and the higher reticulocyte count between the published cases. Among the hemoglobinopathies, Hb Willamette is extremely rare, and may present with hemolytic anemia, due to its high reticulocyte index and RBC target, although often the patient is asymptomatic. Due to microcytosis an assessment for sideroblastic anemia on the basis of bone-marrow examination with Prussian blue stain was made with negative result. The percentage of Hb variant surpasses those described in previous articles, Willamette Hb=44.9% in the proband and Hb Willamette=55.1% in the mother. In addition, the mother of the patient corresponds to the first description of an association between Hb Willamette and HbC.
The description of sex, RBC, MCV, RETIC and relative amount of Hb Willamette of patients already detailed in the literature and presented in this paper can be observed in Table 1.
Table 1.
Cases of hemoglobin Willamette described in literature.
| Case | Sex | Hemoglobin (g/dL) |
Red blood cell (×1012/L) |
Mean corpuscolar volume (fL) |
Reticulocytes (%) |
Hb Willamette (%) |
|---|---|---|---|---|---|---|
| 16 | Male | 15.2 | 5.3 | NA | 1.3 | 33.9 |
| 26 | Female | 14.2 | 4.5 | NA | 2.2 | 32.1 |
| 36 | Male | 14.9 | 5.3 | NA | 1.9 | 35.2 |
| 47 | Male | 13.5 | 4.5 | 109 | NA | 30.3 |
| 57 | Male | 12.5 | 4.2 | 107 | NA | 35.8 |
| 67 | Male | 10.5 | 4.3 | 90 | NA | 33.1 |
| 78 | Male | 12.2 | 4.3 | NA | 1.0 | 35.2 |
| 88 | Male | 15.8 | 5.7 | NA | 0.8 | 35.0 |
| 9* | Female | 8.4 | 3.1 | 75 | 4.8 | 44.9 |
| 10* | Female | NA | NA | NA | NA | 55.1 |
*Cases of this article. NA = Data not available.
Conclusions
Indeed, given the diversity of hemoglobin variants found in the population, an accurate laboratory diagnosis, able to elucidate possible interactions between genetic variants is necessary. The correct diagnosis is essential for treatment strategies and genetic counseling.
References
- 1.Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ 2001;79:704-12. [PMC free article] [PubMed] [Google Scholar]
- 2.Weatherall D. The inherited disorders of haemoglobin: an increasingly neglected global health burden. Indian J Med Res 2011;134:493-7. [PMC free article] [PubMed] [Google Scholar]
- 3.Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ 2008;86:480-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leoneli GG, Imperial RE, Marchi-Salvador DP, et al. Hemoglobinas anormais e dificuldade diagnóstica. Rev Bras Hematol Hemoter 2000;22:396-403. [Google Scholar]
- 5.Kimura EM, Oliveira DM, Jorge SEDC, et al. Identificação e caracterização de variantes novas e raras da hemoglobina humana. Rev Bras Hematol Hemoter 2008;30:316-9. [Google Scholar]
- 6.Jones RT, Koler RD, Duerst ML, Dhindsa DS. Hemoglobin Willamette [α2β2 51 PRO→ARG (D2)] a new abnormal human hemoglobin. Hemoglobin 1976;1:45-57. [DOI] [PubMed] [Google Scholar]
- 7.Chen S. [Three cases of Hb Willamette (beta 51(D2)Pro leads to Arg) and their structural analysis (author’s transl)]. Zhonghua Yi Xue Za Zhi 1981;61:733-5. [Article in Chinese]. [PubMed] [Google Scholar]
- 8.Martínez G, Canizares ME, Colombo B. A second family with Hemoglobin Willamette. Hemoglobin 1984;8:193-5. [DOI] [PubMed] [Google Scholar]
- 9.Quarum M, Shih T, Jones RT. Oxygen equilibrium studies of Hb Willamette α2β251(D2)Pro→Arg. Hemoglobin 1983;7:57-69. [DOI] [PubMed] [Google Scholar]
- 10.Shackleton CHL, Falick AM, Green BN, Witkowska HE. Electrospray mass spectrometry in the clinical diagnosis of variant hemoglobins. J Chromatogr A 1991;562:175-90. [DOI] [PubMed] [Google Scholar]
- 11.Falick AM, Shackleton CHL, Green BN, Witkowskat HE. Tandem mass spectrometry in the clinical analysis of variant hemoglobins. Rapid Commun Mass Spectrom 1990;4:396-400. [DOI] [PubMed] [Google Scholar]
- 12.Oliver RWA, Green BN. The application of electrospray mass spectrometry to the characterization of abnormal or variant hemoglobins. Trends Anal Chem 1991;10:85-91. [Google Scholar]
- 13.Light-Wahl KJ, Loo JA, Edmonds CG, et al. Collisionally activated dissociation and tandem mass spectrometry of intact hemoglobin β-chain variant proteins with electrospray ionization. Biol Mass Spectrom 1993;22:112-20. [DOI] [PubMed] [Google Scholar]
- 14.Williamson D. The unstable haemoglobins. Blood Rev 1993;7:146-63. [DOI] [PubMed] [Google Scholar]
- 15.No Authors listed Notes and News. The Lancet 1976;308:1315-6. [Google Scholar]
- 16.Couto GK, Lorenzini PF, Pilger DA, et al. Association of hemoglobin E-Saskatoon with Hemoglobin S: report of the first case found in Brazil. Acta Haematol 2014;131:84-7. [DOI] [PubMed] [Google Scholar]