

Abstract
During mitosis, higher order chromatin structures are disrupted and chromosomes are condensed to achieve accurate chromosome segregation. CCCTC‐binding factor (CTCF) is a highly conserved and ubiquitously expressed C2H2‐type zinc finger protein which is considered to be involved in epigenetic memory through regulation of higher order chromatin architecture. However, the regulatory mechanism of CTCF in mitosis is still unclear. Here we found that the DNA‐binding activity of CTCF is regulated in a phosphorylation‐dependent manner during mitosis. The linker domains of the CTCF zinc finger domain were found to be phosphorylated during mitosis. The phosphorylation of linker domains impaired the DNA‐binding activity in vitro. Mutation analyses showed that amino acid residues (Thr289, Thr317, Thr346, Thr374, Ser402, Ser461, and Thr518) located in the linker domains were phosphorylated during mitosis. Based on these results, we propose that the mitotic phosphorylation of the linker domains of CTCF is important for the dissociation of CTCF from mitotic chromatin.
Keywords: higher order chromatin structure, mitosis, zinc finger protein
Abbreviations
- ChIP
chromatin immunoprecipitation
- CTCF
CCCTC‐binding factor
- ZF
zinc finger
Cis‐acting regulatory DNA elements such as insulators and enhancers are involved in the temporal and cell type‐specific control of gene expression through the formation of higher order chromatin structure. Higher order chromatin structure is tightly regulated throughout the cell cycle to achieve proper and dynamic chromosome processes. CCCTC‐binding factor (CTCF) is a transcription factor containing 11 highly conserved zinc finger motifs (ZF1‐ZF11; Fig. 1) responsible for its DNA‐binding activity 1, 2. CTCF also mediates higher order chromatin structure formation by modulation of chromatin loops that define the boundary between active and inactive chromatin 3, 4.
Figure 1.
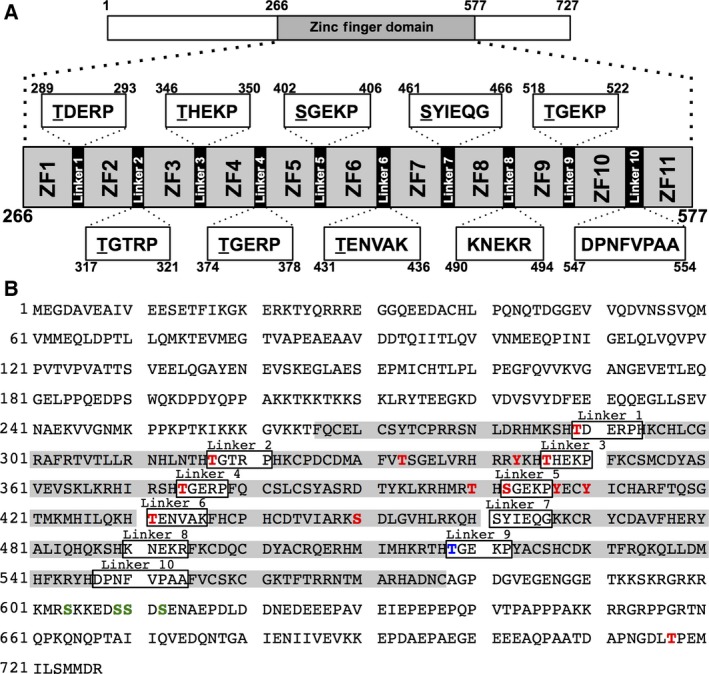
Schematic diagram of human CTCF. (A) Zinc finger domains. Amino acid sequences of each linker domain are indicated, and candidate amino acid residues for phosphorylation are underlined. (B) The amino acid sequence of human CTCF. Zinc finger motifs are highlighted in gray. All known phosphorylation amino acid positions are marked. Red characters indicate phosphorylation sites identified by proteomics analyses in 9 and 10. Blue and green characters indicate phosphorylation sites analyzed in 11 and 17, respectively.
More than 3% of the total number of human genes belongs to the zinc finger protein family. Most zinc finger proteins, including CTCF, have tandemly repeated multiple zinc finger motifs separated by a highly conserved short linker peptide sequence, TGEKP. This linker domain is important for the structural stabilization of the zinc finger motif by forming an alpha‐cap structure 5, and the DNA‐binding activity of zinc finger motifs is regulated by phosphorylation of threonine residues in the linker domain during mitosis 6, 7, 8. CTCF has 10 linker domains, but only linker domain 9 located between ZF9 and ZF10 contains the exact TGEKP sequence. It has been reported that CTCF is highly phosphorylated at multiple amino acid residues including T518 at the linker domain 9 9, 10, 11. However, the effect of mitotic phosphorylation on CTCF DNA‐binding activity with regard to each phosphorylation site is not well understood.
Materials and methods
Cell culture, synchronization, and transfection
HeLa S3 cells and MCF‐7 cells were maintained at 37 °C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. Cells were synchronized at mitotic phase by two cycles of “excess thymidine blockage followed by nocodazole arrest”. Briefly, at 12 h post treatment of 2.5 mm thymidine (Sigma‐Aldrich Co. LLC., St. Louis, MO, USA), cells were released into a fresh culture medium without an excess amount of thymidine for 10 h, and then synchronized again at G1/S boundary in growth medium with 2.5 mm thymidine for 14 h. At 6 h post release from thymidine treatment, cells were treated with 165 nm nocodazole (Sigma‐Aldrich Co. LLC.) for 6 h. Mitotic cells were collected by gentle shaking of cell culture dishes. Transient DNA transfection assays were performed using Gene Pulser Xcell System (Bio‐Rad Laboratories Inc., Hercules, CA, USA) according to the manufacturer's protocol.
Construction of plasmids
To make 3 × FLAG‐tagged CTCF WT expression vector, CTCF cDNA fragment from pBS‐FLAG‐CTCF 12 was cloned into pCAGGS‐3 × FLAG 13. To construct 4A, T518A, 7A, 8A, and 8D expression vectors, cDNA fragments containing each point mutation were amplified by PCR using the primers shown in Table 1. 3 × FLAG‐tagged cDNA fragments were cloned into pCAGGS vector.
Table 1.
Primers for plasmids construction
| Primer Name | Sequence |
|---|---|
| T289A/D rev | cGTGGCTTTTCATGTGACG |
| T289A for | CTGATGAGAGACCACACAAG |
| T289D for | acGATGAGAGACCACACAAG |
| T317A/D rev | cGTGTGTGTTAAGGTGATTC |
| T317A for | CAGGTACTCGTCCTCACAAG |
| T317D for | acGGTACTCGTCCTCACAAG |
| T346A/D rev | cGTGTTTGTAACGACGATGC |
| T346A for | CCCACGAGAAGCCATTCAAG |
| T346D for | acCACGAGAAGCCATTCAAG |
| T374A/D rev | cATGAGAGCGAATGTGACG |
| T374A for | CTGGAGAGCGTCCGTTTCAG |
| T374D for | acGGAGAGCGTCCGTTTCAG |
| S402A/D rev | cATGGGTTCTCATGTGCC |
| S402A for | CAGGGGAAAAGCCTTATGAA |
| S402D for | acGGGGAAAAGCCTTATGAA |
| T431A/D rev | cGTGCTTCTGTAAAATGTGC |
| T431A for | CAGAAAATGTGGCCAAATTTC |
| T431D for | acGAAAATGTGGCCAAATTTC |
| S461A/D rev | cATGCTGCTTTCGCAAGTGG |
| 1382A for | CCTATATTGAGCAAGGCAAG |
| 1382D for | acTATATTGAGCAAGGCAAG |
| T518A/D rev | cGTGGGTGCGCTTGTGCATG |
| T518A for | CCGGGGAGAAGCCTTACGCC |
| T518D for | acGGGGAGAAGCCTTACGCC |
| S4A rev | ATCTTCTTTCTTAGcGCGCATCTTTC |
| S4A for | gCCgCTGACgcTGAAAATGCTGAACCAGATC |
Antibodies
A rabbit polyclonal anti‐CTCF antibody was prepared as previously described 12. Anti‐FLAG (M2, Sigma‐Aldrich Co. LLC.), rabbit polyclonal anti‐histone H3 (ab1791, Abcam plc., Cambridge, UK) and anti‐phospho‐histone H3 antibodies (06‐570, Merck Millipore, Billerica, MA, USA) were purchased.
Subcellular fractionation
Cells were washed with cold PBS(−), and kept on ice for 5 min in a cold hypotonic buffer (10 mm HEPES‐NaOH, pH 8.0, 10 mm KCl, 1.5 mm MgCl2, 0.1% Triton X‐100, 5 mm Na3VO4, 10 mm NaF, 25 mm glycerol 2‐phosphate) on ice for 5 min. Supernatant and pellet fractions were obtained by centrifugation at 2300 g for 5 min.
ChIP assay
Cells were washed with cold PBS(−), and kept on ice for 5 min in a cold CSK buffer (10 mm PIPES‐NaOH, pH 6.8, 100 mm NaCl, 3 mm MgCl2, 1 mm EGTA, 300 mm sucrose, 0.1% Triton X‐100, 1 mm Na3VO4, 1 mm NaF, 5 mm glycerol 2‐phosphate). After centrifugation at 2300 g for 5 min, pellet fractions were incubated at 25 °C for 10 min in CSK buffer containing 0.5% formaldehyde, and then incubated with 25 °C for 5 min in 0.125 m glycine/PBS. Immunoprecipitation assays were carried out according to the manufacturer's protocol (Chromatin Immunoprecipitation Assay Kit; Merck Millipore). The quantitative PCR (qPCR) was performed with primers 5′‐GGTCCACGGGCCGCCCTGCCAG‐3′ and 5′‐CGCAGCTCCGGAAGCCGAGAGC‐3′ corresponding to a part of rRNA gene between nucleotide positions −961 and −851, where +1 is set to be the transcription start site 14 using FastStart SYBR Green Master (Roche Molecular Systems, Inc., Hacienda, CA, USA) and Thermal Cycler Dice Real Time System (TaKaRa Bio Inc., Shiga, Japan) according to the manufacturers’ protocols.
DNA‐binding assay
Asynchronously growing MCF‐7 cells were incubated on ice for 5 min in the hypotonic buffer. Supernatant and pellet fractions were separated by centrifugation at 2300 g for 5 min, and the pellet fractions were incubated on ice for 5 min in a buffer (10 mm Tris/HCl, pH 7.9, 275 mm NaCl, 1 mm MgCl2, 0.1% Triton X‐100, 1 mm Na3VO4, 1 mm NaF, 5 mm glycerol 2‐phosphate). Nocodazole‐arrested MCF‐7 cells were incubated on ice for 5 min in the hypotonic buffer. CTCF was immunoprecipitated from the lysates using the anti‐CTCF antibody. After washing with a buffer containing 10 mm Tris/HCl, pH 7.9, 500 mm NaCl, 1 mm MgCl2, 0.1% Triton X‐100, 1 mm Na3VO4, 1 mm NaF, 5 mm glycerol 2‐phosphate, immunoprecipitated proteins were incubated with or without lambda protein phosphatase (P7053, New England Biolabs Inc., Ipswich, MA, USA) at 30 °C for 2 h, and then incubated in a DNA‐binding buffer (20 mm Tris/HCl, pH 7.4, 150 mm NaCl, 2 mm MgCl2, 0.01% NP‐40, 6.25% glycerol, 1 mm Na3VO4, 1 mm NaF, 5 mm glycerol 2‐phosphate) with 0.15 ng of the rRNA gene upstream region fragment and 50 ng of poly (dIdC). The rRNA gene upstream region fragment used here is amplified from genomic DNA and purified from agarose gel. The exact sequence of the fragment corresponding to the region between nucleotide positions −961 and −851, where +1 is set to be the transcription start site 14 is as follows: 5′‐ GGTCCACGGGCCGCCCTGCCAGCCGGATCTGTCTCGCTGACGTCCGCGGCGGTTGTCGGGCTCCATCTGGCGGCCGCTTTGAGATCGTGCTCTCGGCTTCCGGAGCTGCG‐3′, where the CTCF‐binding site is underlined. The amounts of coimmunoprecipitated DNA were analyzed by qPCR and normalized by the protein amount of CTCF measured by imagej software (developed at the National Institutes of Health, Bethesda, MD, USA) from western blotting results.
Results
CTCF is dissociated from mitotic chromatin
It is reported that most of the C2H2 zinc finger family proteins are excluded from mitotic chromosomes 11. To determine the amount of CTCF bound to mitotic chromatin, subcellular fractionation was performed using a hypotonic buffer. HeLa S3 cells were synchronized at mitosis as described above. The amount of CTCF in the supernatant fraction obtained from mitotic cells was increased compared to asynchronous cells (Fig. 2A). It has been reported that CTCF binds upstream of the rRNA gene promoter 14, so that next we examined the amount of CTCF bound to the rRNA gene locus during mitosis by chromatin immunoprecipitation (ChIP) assays. The amount of CTCF on the rRNA gene locus in mitotic cells was 80% less than that in asynchronous cells (Fig. 2B). After release from the mitotic block, the amount of CTCF on the rRNA gene was restored (Fig. 2B). These results suggest that CTCF is dissociated from chromatin during mitosis and reassociated upon G1 entry.
Figure 2.
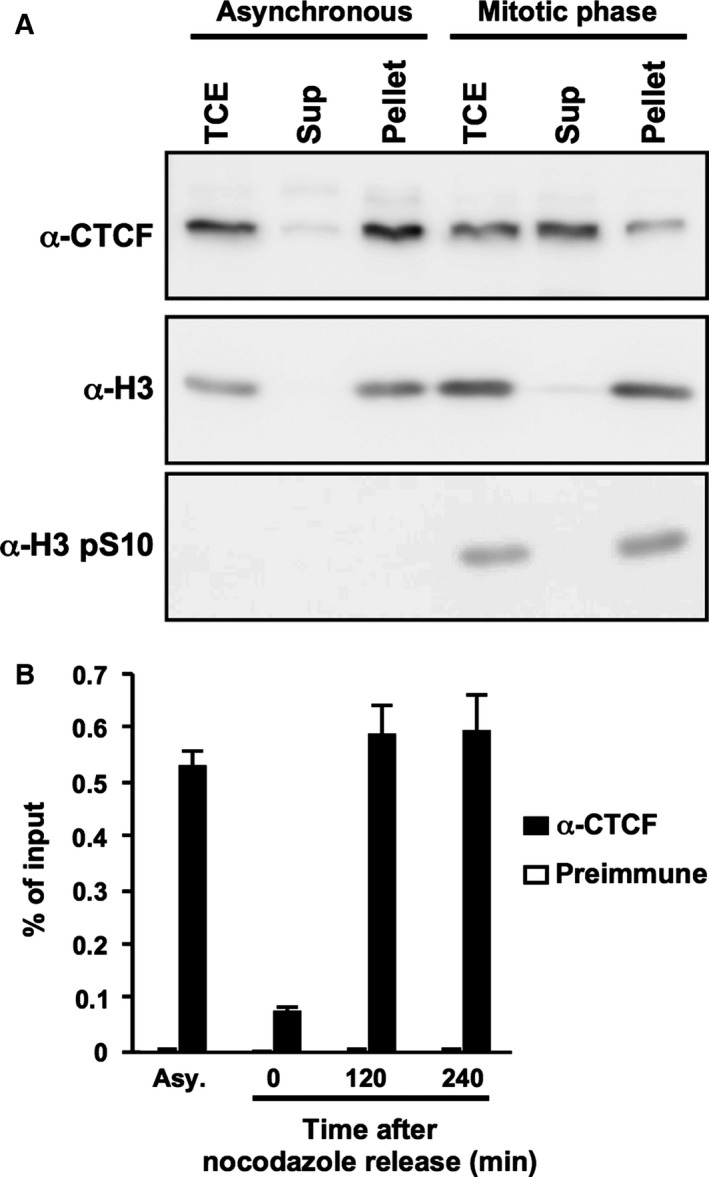
CTCF is dissociated from mitotic chromatin. (A) Fractionation of asynchronous and nocodazole‐arrested HeLa S3 cells. Total cell extracts (TCE), and supernatant and pellet fractions were subjected to western blotting using anti‐CTCF, anti‐histone H3, and anti‐phospho‐histone H3 antibodies. Phosphorylated histone H3 was used as a marker of the mitotic chromatin fraction. (B) Chromatin‐binding activity of mitotic CTCF. HeLa S3 cells were collected at 2 or 4 h post release from nocodazole treatment. The cell lysates were subjected to ChIP assays with anti‐CTCF antibody (filled bars) or rabbit preimmune serum (open bars). qPCR was performed with primers specific for the upstream region of the rRNA gene. The amount of DNA coprecipitated with each antibody was shown as % of input.
Phosphorylation of CTCF in mitosis
It has been reported that a variety of DNA‐binding factors including transcription factors are phosphorylated and released from highly condensed chromosomes during mitosis 15. To examine whether CTCF is also phosphorylated in the mitotic phase, we carried out phos‐tag SDS/PAGE 16 which relies on the fact that phosphorylated proteins migrate slower than unphosphorylated proteins in the phos‐tag gel. CTCF in mitotic cell lysates migrated slower than that in asynchronous cell lysates on phos‐tag SDS/PAGE (Fig. 3A), suggesting that mitotic CTCF is phosphorylated and is therefore slower to migrate. To confirm the phosphorylation of CTCF during mitosis, the immunoprecipitated CTCF was treated with lambda protein phosphatase and subjected to phos‐tag electrophoresis. The phosphatase treatment resulted in the increased migration rate of mitotic CTCF (Fig. 3B, lanes 3, 4). These results suggest that CTCF is phosphorylated during mitosis. Both mitotic and asynchronous CTCF showed a slight increase in migration rate after lambda protein phosphatase treatment (Fig. 3B, lanes 1, 2). This result suggests phosphorylation of CTCF in interphase. It has been reported that the amino acid residues (Ser604, Ser609, Ser610, Ser612) of the C‐terminal domain of CTCF are phosphorylated by CK2 17. Phosphorylation of these amino acid residues, especially Ser612, is involved in functional switching of CTCF from transcriptional repressor to activator in c‐myc 18. Thus, CTCF is phosphorylated in not only mitosis but also interphase.
Figure 3.
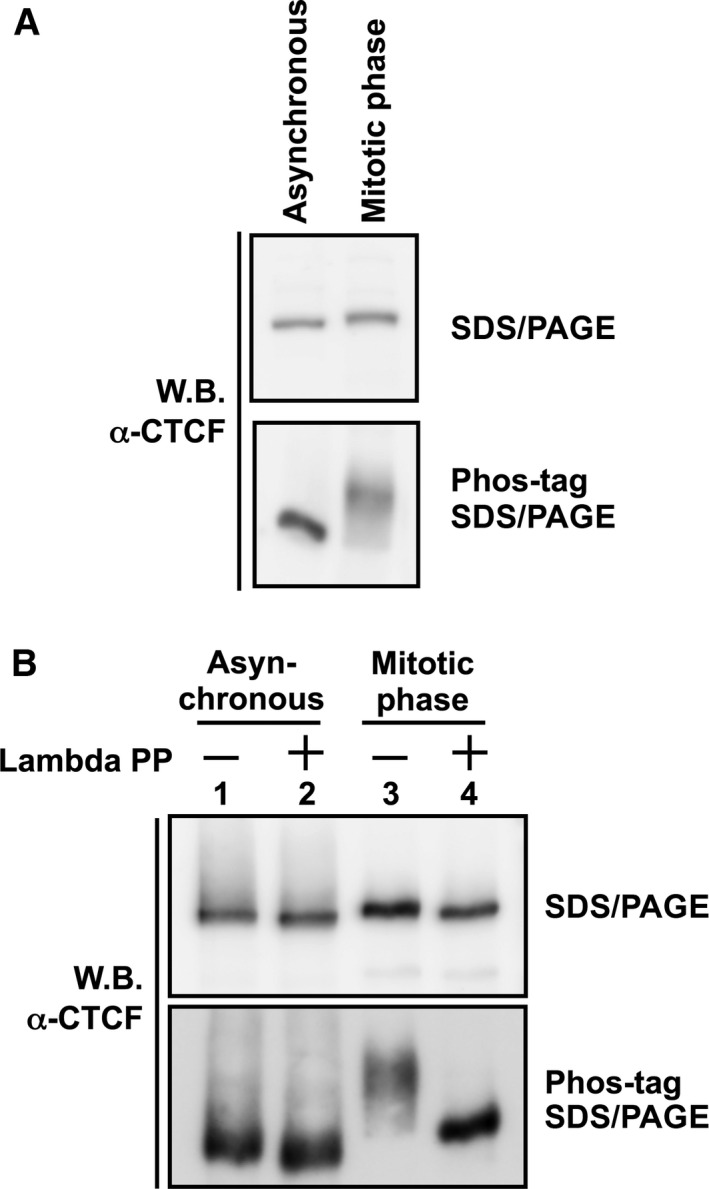
Phosphorylation of mitotic CTCF. (A) Analysis by phos‐tag SDS/PAGE. Whole cell lysates were separated on a 7.5% SDS/PAGE (upper panel) and a 5% SDS/PAGE containing 20 μm phos‐tag (lower panel), followed by western blotting with anti‐CTCF antibody. (B) Lambda phosphatase treatment of mitotic CTCF. Cell lysates prepared from asynchronous cells (lane 1) or mitotic‐arrested cells (lane 2) were subjected to immunoprecipitation assays using anti‐CTCF antibody. The immunoprecipitated CTCF was incubated in the absence or presence of lambda protein phosphatase and subjected to phos‐tag SDS/PAGE as described above.
The linker domains of CTCF zinc finger domain are phosphorylated during mitosis
It has been reported that serine residues at amino acid positions 604, 609, 610, and 612 of CTCF are phosphorylated by protein kinase CK2 17. Thr518, which is located in the linker domain 9, is phosphorylated during mitosis 11. Thus, we constructed alanine‐substituted mutants of these respective amino acid residues (Fig. 4A; designated 4A and T518A). Similar to the results of 3 × FLAG‐CTCF WT (Fig. 4B, lanes 3 and 4), ectopically expressed 3 × FLAG‐CTCF 4A and T518A were still phosphorylated during mitosis (Fig. 4B, lanes 5–8), suggesting that these residues are not critical as a mitotic phosphorylation signal. To determine the location of the phosphorylated amino acid residues of CTCF during mitosis, we substituted serine and threonine residues at amino acid positions 289, 317, 346, 374, 402, 431, 461, and 518 of the linker domains (8A mutant, Figs 1 and 4A). 3 × FLAG‐CTCF 8A in mitotic cells migrated similarly to asynchronous cells (Fig. 4B lanes 9 and 10), suggesting that some of these amino acid residues are phosphorylated during mitosis. Furthermore, serine residues at amino acid position 604, 609, 610, and 612 are not phosphorylated in mitosis. Next, to identify the phosphorylated amino acid residues of linker domains in mitosis, we introduced respective alanine substitutions to seven of eight amino acid residues. Among them, only the 3 × FLAG‐CTCF 7A + T431 mutant shows the same migration rate as 3 × FLAG‐CTCF 8A mutant (Fig. 4C lanes 2 and 8). These results strongly suggest that each candidate amino acid residue of the linker domains except for T431 is phosphorylated during mitosis.
Figure 4.
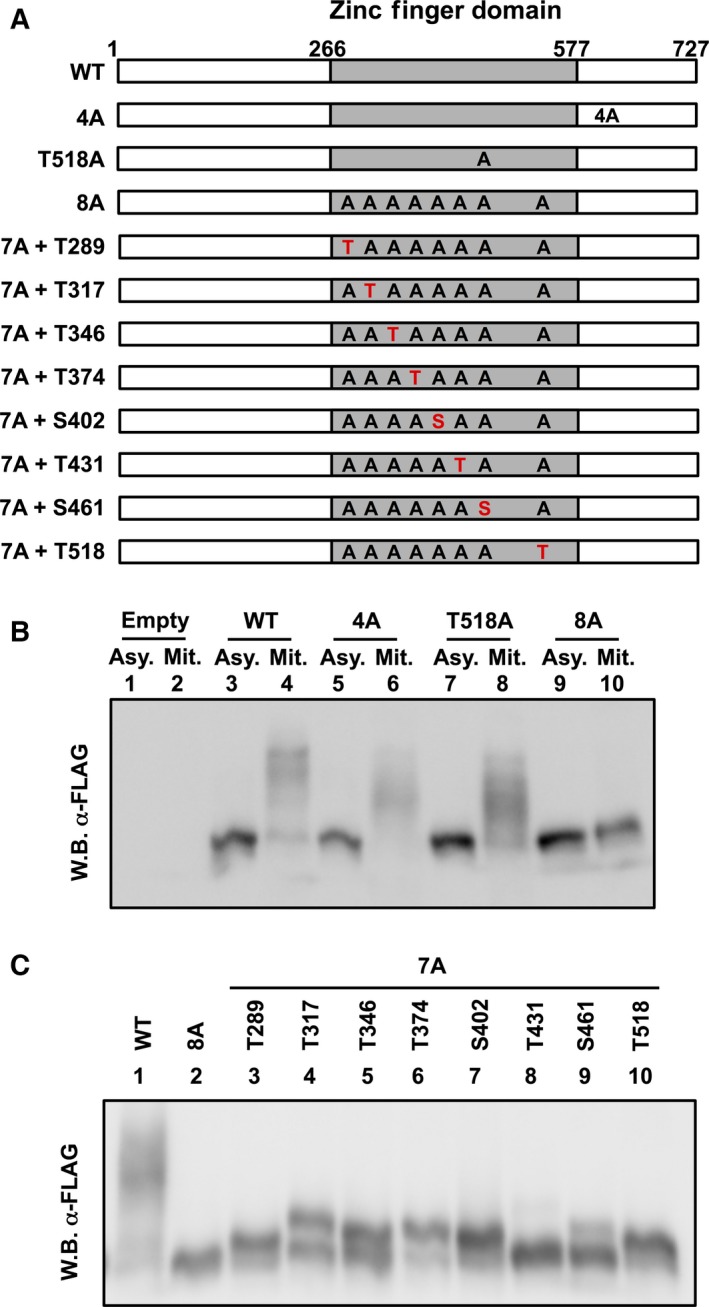
The linker domains of CTCF zinc finger domain are phosphorylated during mitosis. (A) Schematic representation of the CTCF mutants. The gray boxes indicate the zinc finger domains. 4A mutant has S604A, S609A, S610A, and S612A mutations. 8A mutant has T289A, T317A, T346A, T374A, S402A, T431A, S461A, and T518A mutations. 7A mutants have alanine substitutions in seven of eight amino acid residues, respectively. (B) Phosphorylation of exogenously expressed CTCF mutants by phos‐tag SDS/PAGE. At 72 h post transfection of CTCF mutants, asynchronously cultured or nocodazole‐arrested HeLa S3 cells were subjected to phos‐tag SDS/PAGE, followed by western blotting with anti‐FLAG M2 antibody. (C) At 72 h post transfection of CTCF mutants, nocodazole‐arrested HeLa S3 cells were subjected to phos‐tag SDS/PAGE, followed by western blotting with anti‐FLAG M2 antibody.
Mitotic phosphorylation of CTCF decreases the DNA‐binding activity
To determine whether the mitotic phosphorylation of linker domains affects the DNA‐binding activity, CTCF was purified using anti‐CTCF antibody from either asynchronous or mitotic MCF‐7 cell extracts and subjected to in vitro DNA‐binding assays. The purified CTCF, bound to protein A agarose beads, was treated with or without lambda protein phosphatase, and incubated with a fragment of the rRNA gene that contains the CTCF‐binding site. Quantification of fragments that interacted with CTCF was accomplished by qPCR and normalized against the protein amount of CTCF that was eluted from protein A agarose beads and quantified by western blotting. The DNA‐binding activity of mitotic CTCF was 80% lower than asynchronous CTCF (Fig. 5A). However, phosphatase treatment restored the DNA‐binding activity of mitotic CTCF (Fig. 5A). These results indicate that CTCF decreases its DNA‐binding activity in a phosphorylation‐dependent manner during mitosis. Next, we examined the DNA‐binding activity of a phosphomimetic mutant, termed CTCF‐8D, in which eight putative mitotic phosphorylation sites were changed to aspartic acid. 3 × FLAG‐tagged WT and 8D mutants of CTCF gave very similar expression levels in HeLa S3 cells (Fig. 5B). To analyze the DNA‐binding activity of the phosphomimetic mutant of CTCF, we carried out ChIP assays of exogenously expressed CTCF. The amount of the CTCF‐binding site in the rRNA gene locus bound to 3 × FLAG‐CTCF 8D was reduced by 30% compared to wild‐type (Fig. 5C). These results suggest that the mitotic phosphorylation of CTCF decreases its DNA‐binding activity.
Figure 5.
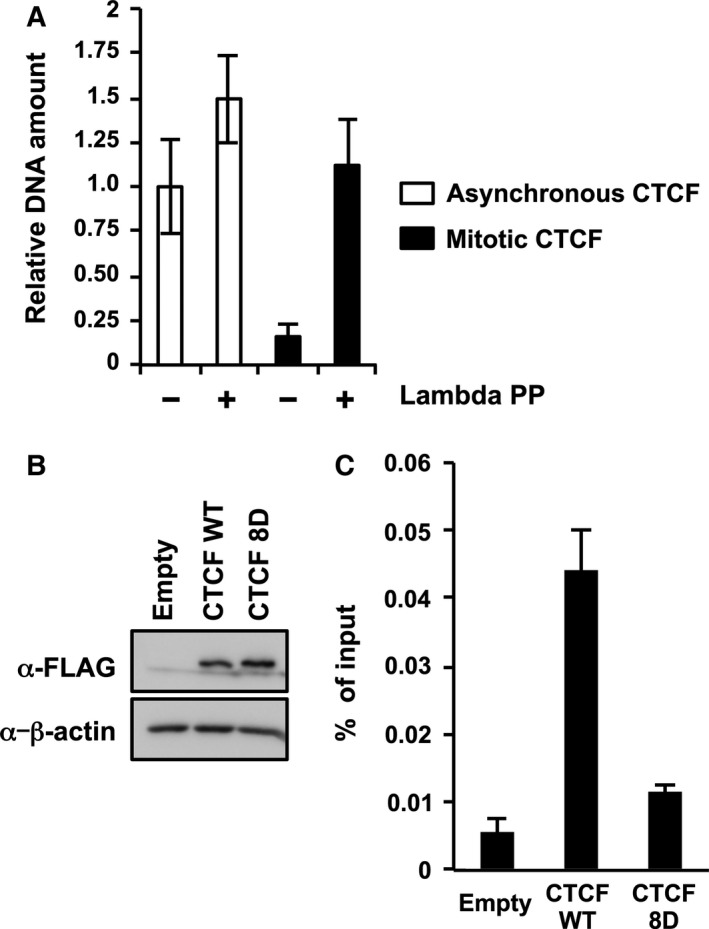
The DNA‐binding activity of CTCF is regulated by phosphorylation of linker domains during mitosis. (A) In vitro DNA‐binding assays. Immunoprecipitated CTCF prepared from asynchronous cell extracts or mitotic cell extracts were treated in the absence or presence of lambda protein phosphatase, and then incubated with the DNA fragments of the rRNA gene upstream region between nucleotide positions −60 and −851. The amount of coimmunoprecipitated DNA fragments was quantified by qPCR and normalized with the amount of CTCF protein. The mean value and standard deviations determined from three independent experiments are shown. (B) Expression level of exogenous CTCF in transfected cells. HeLa S3 cells were transiently transfected with plasmids expressing either CTCF WT or 8D mutant. At 72 h post transfection, cell lysates were subjected to SDS/PAGE, followed by western blotting. (C) Chromatin‐binding activity of CTCF phosphomimetic mutant. The cell lysates were subjected to ChIP assays with anti‐FLAG M2 antibody. qPCR was performed with primers specific for the upstream region of the rRNA gene. The amount of DNA coprecipitated with each antibody was shown as % of input. The mean value and standard deviations determined from three independent experiments are shown.
Discussion
It has been reported that the phosphorylation of linker domains reduces the DNA‐binding activity of C2H2 zinc finger proteins 6, 7, 8. Structural analyses indicate that the threonine residues of linker domains are required for the stabilization of the interaction between DNA and zinc finger proteins by providing the capping structure for the alpha‐helix of the zinc finger motif 5. Our results suggest that the DNA‐binding activity of CTCF is similarly regulated by the phosphorylation of linker domains during mitosis. It has been reported that serine/threonine kinases TOPK/PBK 19 and Cdk1 20 phosphorylate the threonine residues of the conserved linker domains. Compared with other phosphorylated residues, the phosphorylation level of Ser461 in the linker domain 7 was weak, but a significant amount of band shift was observed during mitosis (Fig. 4). The peptide sequence of the linker domain 7 is different from the consensus motif (Fig. 1). Thus, it is possible that the DNA‐binding activity of CTCF is regulated by an unknown protein kinase in addition to TOPK/PBK and Cdk1.
It has been proposed that mitotic chromosomes exist in a highly condensed state to ensure stable and integral chromosome segregation 21. Because transcription‐related factors are excluded from mitotic chromatin, mitotic chromosomes are transcriptionally inactive 15. In contrast, some markers for epigenetic gene control are retained during mitosis 22. It is possible that transcription and the formation of higher order chromatin structures mediated by CTCF might result in a disadvantage to achieve accurate chromosome segregation; however, we found that a part of CTCF still interacts with mitotic chromatin as reported previously (Fig. 2A) 23, 24. It is known that CTCF recognizes a wide variety of target gene loci using various combinations of the zinc finger domains 25, 26. We found that mitotic CTCF is detected on phos‐tag SDS/PAGE as a smeared migration pattern (Figs 3 and 4), suggesting that CTCF is phosphorylated heterogeneously in mitosis. Therefore, it is possible that the partial phosphorylation of CTCF allows for a partial interaction with gene loci even during mitosis, possibly for the probable bookmarking of chromatin domains to achieve proper gene expression at the subsequent G1 phase.
Conclusions
Here we have shown that CTCF dissociates from mitotic chromosomes. Mutation analyses indicated that CTCF is phosphorylated in mitosis at Thr289, Thr317, Thr346, Thr374, Ser402, Ser461, and Thr518, all of which are located in linker domains. The mitotic phosphorylation of CTCF resulted in the reduction of the DNA‐binding activity to a CTCF‐binding site on rRNA upstream regions. These results suggest that the DNA‐binding activity of CTCF could be regulated through the phosphorylation of linker domains during mitosis.
Author contributions
TS conceived the research strategies, performed experiments, analyzed the data, and wrote the manucsript. KS designed the experiments and analyzed the data. KK and AK designed the experiments, analyzed the data, and wrote the manuscript. KN supervised the research and wrote the manuscript.
Acknowledgements
We thank Dr Bryan Mathis (Medical English Communications Center, University of Tsukuba) for grammatical review. This work was supported in part by grant‐in‐aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan Grant Number 16H05192 and 24115002 (to AK and KN).
References
- 1. Klenova EM, Nicolas RH, Paterson HF, Carne AF, Heath CM, Goodwin GH, Neiman PE and Lobanenkov VV (1993) CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c‐myc gene, is an 11‐Zn‐finger protein differentially expressed in multiple forms. Mol Cell Biol 13, 7612–7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vostrov AA and Quitschke WW (1997) The zinc finger protein CTCF binds to the APBβ domain of the amyloid β‐protein precursor promoter. Evidence for a role in transcriptional activation. J Biol Chem 272, 33353–33359. [DOI] [PubMed] [Google Scholar]
- 3. Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS and Ren B (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ong CT and Corces VG (2014) CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet 15, 234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Laity JH, Dyson HJ and Wright PE (2000) DNA‐induced α‐helix capping in conserved linker sequences is a determinant of binding affinity in Cys(2)‐His(2) zinc fingers. J Mol Biol 295, 719–727. [DOI] [PubMed] [Google Scholar]
- 6. Dovat S, Ronni T, Russell D, Ferrini R, Cobb BS and Smale ST (2002) A common mechanism for mitotic inactivation of C2H2 zinc finger DNA‐binding domains. Genes Dev 16, 2985–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jantz D and Berg JM (2004) Reduction in DNA‐binding affinity of Cys2His2 zinc finger proteins by linker phosphorylation. Proc Natl Acad Sci USA 101, 7589–7593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rizkallah R and Hurt MM (2009) Regulation of the transcription factor YY1 in mitosis through phosphorylation of its DNA‐binding domain. Mol Biol Cell 20, 4766–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ and Gygi SP (2008) A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci USA 105, 10762–10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA et al (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal 3, ra3. [DOI] [PubMed] [Google Scholar]
- 11. Rizkallah R, Alexander KE and Hurt MM (2011) Global mitotic phosphorylation of C2H2 zinc finger protein linker peptides. Cell Cycle 10, 3327–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Komatsu T, Sekiya T and Nagata K (2013) DNA replication‐dependent binding of CTCF plays a critical role in adenovirus genome functions. Sci Rep 3, 2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Asaka MN, Kawaguchi A, Sakai Y, Mori K and Nagata K (2016) Polycomb repressive complex 2 facilitates the nuclear export of the influenza viral genome through the interaction with M1. Sci Rep 6, 33608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van de Nobelen S, Rosa‐Garrido M, Leers J, Heath H, Soochit W, Joosen L, Jonkers I, Demmers J, van der Reijden M, Torrano V et al (2010) CTCF regulates the local epigenetic state of ribosomal DNA repeats. Epigenetics Chromatin 3, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gottesfeld JM and Forbes DJ (1997) Mitotic repression of the transcriptional machinery. Trends Biochem Sci 22, 197–202. [DOI] [PubMed] [Google Scholar]
- 16. Kinoshita E, Kinoshita‐Kikuta E, Takiyama K and Koike T (2006) Phosphate‐binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics 5, 749–757. [DOI] [PubMed] [Google Scholar]
- 17. Klenova EM, Chernukhin IV, El‐Kady A, Lee RE, Pugacheva EM, Loukinov DI, Goodwin GH, Delgado D, Filippova GN, Leon J et al (2001) Functional phosphorylation sites in the C‐terminal region of the multivalent multifunctional transcriptional factor CTCF. Mol Cell Biol 21, 2221–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. El‐Kady A and Klenova E (2005) Regulation of the transcription factor, CTCF, by phosphorylation with protein kinase CK2. FEBS Lett 579, 1424–1434. [DOI] [PubMed] [Google Scholar]
- 19. Rizkallah R, Batsomboon P, Dudley GB and Hurt MM (2015) Identification of the oncogenic kinase TOPK/PBK as a master mitotic regulator of C2H2 zinc finger proteins. Oncotarget 6, 1446–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Suzuki K, Sako K, Akiyama K, Isoda M, Senoo C, Nakajo N and Sagata N (2015) Identification of non‐Ser/Thr‐Pro consensus motifs for Cdk1 and their roles in mitotic regulation of C2H2 zinc finger proteins and Ect2. Sci Rep 5, 7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wei Y, Yu L, Bowen J, Gorovsky MA and Allis CD (1999) Phosphorylation of histone H3 is required for proper chromosome condensation and segregation. Cell 97, 99–109. [DOI] [PubMed] [Google Scholar]
- 22. Delcuve GP, He S and Davie JR (2008) Mitotic partitioning of transcription factors. J Cell Biochem 105, 1–8. [DOI] [PubMed] [Google Scholar]
- 23. Burke LJ, Zhang R, Bartkuhn M, Tiwari VK, Tavoosidana G, Kurukuti S, Weth C, Leers J, Galjart N, Ohlsson R et al (2005) CTCF binding and higher order chromatin structure of the H19 locus are maintained in mitotic chromatin. EMBO J 24, 3291–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nekrasov M, Amrichova J, Parker BJ, Soboleva TA, Jack C, Williams R, Huttley GA and Tremethick DJ (2012) Histone H2A.Z inheritance during the cell cycle and its impact on promoter organization and dynamics. Nat Struct Mol Biol 19, 1076–1083. [DOI] [PubMed] [Google Scholar]
- 25. Filippova GN, Qi CF, Ulmer JE, Moore JM, Ward MD, Hu YJ, Loukinov DI, Pugacheva EM, Klenova EM, Grundy PE et al (2002) Tumor‐associated zinc finger mutations in the CTCF transcription factor selectively alter tts DNA‐binding specificity. Cancer Res 62, 48–52. [PubMed] [Google Scholar]
- 26. Nakahashi H, Kwon KR, Resch W, Vian L, Dose M, Stavreva D, Hakim O, Pruett N, Nelson S, Yamane A et al (2013) A genome‐wide map of CTCF multivalency redefines the CTCF code. Cell Rep 3, 1678–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]