

Abstract
Clostridium difficile consists of six clades but studies on Clade 3 are limited. Here, we report genome sequences of three Clade 3 C. difficile strains carrying genes encoding toxin A and B and the binary toxin. Isolates 103 and 133 (both of ST5) and isolate 106 (ST285) were recovered from three ICU patients. Whole genome sequencing using HiSeq 2500 revealed 4.1-Mb genomes with 28–29% GC content. There were ≥1,104 SNP between the isolates, suggesting they were not of a single clone. The toxin A and B gene-carrying pathogenicity locus (PaLoc) of the three isolates were identical and had the insertion of the transposon Tn6218. The genetic components of PaLoc among Clade 3 strains were the same with only a few nucleotide mutations and deletions/insertions, suggesting that the Tn6218 insertion might have occurred before the divergence within Clade 3. The binary toxin-genes carrying CDT locus (CdtLoc) of the three isolates were identical and were highly similar to those of other Clade 3 strains, but were more divergent from those of other clades. In conclusion, Clade 3 has an unusual clade-specific PaLoc characteristic of a Tn6218 insertion which appears to be the main feature to distinguish Clade 3 from other C. difficile.
Clostridium difficile, a Gram-positive spore-forming anaerobic bacterium, is the leading cause of antibiotic-associated diarrhea1 and also causes colitis which is not necessarily associated with antibiotic use2,3. Clinical manifestations of C. difficile infection (CDI) range between asymptomatic and mild or severe diarrhea which can even lead to death2,3. The main virulent factors of C. difficile are toxin A (enterotoxin, 308 kD) and toxin B (cytotoxin; 270 kD) encoded by genes tcdA and tcdB, respectively. These two genes together with three regulator genes tcdC (a negative regulator), tcdE (encoding a bacteriophage-like putative holin to facilitate the release of toxin A and B) and tcdR (encoding an alternative σ factor) are located on a pathogenicity locus called PaLoc (Fig. 1)4. PaLoc is typically 19.6 kb in length and is almost always integrated at the same location between genes cdu1 (encoding a transcriptional terminator) and cdd1 (encoding a putative ABC transporter) in the C. difficile genome. By contrast, in non-toxingenic strains such as CD37, the PaLoc is absent and there is a 115-bp non-coding sequence in its place (Fig. 1). At present, C. difficile consists of six distinct phylogenetic clades designated Clade 1, 2, 3, 4, 5 and C-I5,6 and the genetic composition of PaLoc appears to be clade-specific5. Among the six clades, Clade 3 has received relatively little interest. Currently, isolates of Clade 3 have been found to belong to ST5 (PCR ribotype 023 or 069), ST22 (PCR ribotype 023), ST25 (PCR ribotype 023), ST96 (PCR ribotype 058), ST162, ST201 and ST2215,7. It has been suggested that PCR ribotype 023 is able to cause CDI with the same severity of illness as PCR ribotype 0278, which is a well-characterized hypervirulent lineage causing outbreaks and severe illness such as toxic megacolon9. Nonetheless, although the infection caused by strains of PCR ribotype 023 is associated with high levels of biomarkers such as white blood cell count and C-reactive protein, the 14-day mortality (7%) in CDI cases caused by PCR ribotype 023 was lower than those (12–25%) caused by PCR ribotypes belonging to clade 1, 2, 4 and 510. PCR ribotype 023 is one of the most common ribotypes causing CDI in the world11. However, it is not one of the main ribotypes in mainland China12 as it only accounts for 0.86% (1/116)13 or 2.7% (2/74)14 of toxigeneic C. difficile.
Figure 1. Schematic representations of PaLoc and flanking genes.

The 5′ flanking gene cdu1, the 3′ flanking gene cdd1, positive regulator gene tcdR, negative regulator gene tcdC, and toxin genes tcdA and tcdB are shown. For Tn6218, the genetic components from left to right are int, xis, rep, xre, merR, oxidoreductase-encoding gene, flavodoxin-encoding gene, orf and σ70-encoding gene.
The PaLoc of Clade 3 is unusual as a transposon. Tn6218 is inserted between genes tcdE and tcdA, which results in a larger PaLoc5 (28.6 kb; Fig. 1). Tn6218 is related to the well-studied conjugative transposon Tn916 but is non-conjugative5. Tn6218 typically contains four common genes, i.e. int (encoding a tyrosine recombinase for catalyzing integration and excision), xis (encoding an excisionase), rep (encoding a putative topoisomerase as the replication initiation factor) and xre (a putative transcription regulatory gene involved in the xenobiotic response), and a variety of accessory genes including antimicrobial resistance determinants5.
Binary toxin, the third toxin that has been found to enhance the virulence15, is produced by 20% to 32% of C. difficile strains according to previous reports16,17. Genes encoding the binary toxin, cdtA and cdtB, together with the regulation gene cdtR are located on the CDT locus (CdtLoc, Figure S1 in the Supplementary Data)18. CdtLoc was distributed among C. difficile strains of Clades 2, 3 and 5. Although binary toxin gene-carrying C. difficile has been reported worldwide, such isolates remain uncommon in China. Previous studies demonstrated that binary toxin gene-carrying isolates accounted for no more than 7% of all toxigenic C. difficile14,19,20,21,22,23,24.
During a surveillance project on toxigenic C. difficile in our hospital, three Clade 3 isolates were recovered. All of them had genes encoding toxins A and B and the binary toxin. They were then subjected to whole genome sequencing. Here, we report their genome sequence and antimicrobial susceptibilities below.
Materials and Methods
Isolates
Three C. difficile isolates, 103, 133 and 106, were recovered from three different patients at West China Hospital, Sichuan University, Chengdu, southwest China during a preliminary active screening study of colonization by toxigenic C. difficile among ICU patients in 2014. Isolates 103 and 133 were recovered from the rectal swabs of two different patients in the General ICU ward in March and June, respectively, while isolate 106 was recovered from the stool sample of a patient in the Respiratory ICU ward in June. This study was conducted in accordance with the amended Declaration of Helsinki and was approved, under a waiver of consent, by the Ethics Committee of West China Hospital. After an initial alcohol shock treatment to kill vegetative cells, stool specimens were grown on cycloserine-cefoxifin-fructose agar (CCFA; Oxoid, Basingstoke, UK) with 10% sheep blood and incubated 48 h at 37 °C under anaerobic conditions. A single colony of each isolate was further cultured in Brain Heart Infusion (BHI; Oxoid) broth for 24 h. The three isolates were found to produce toxin A and B as determined using the C. DIFF QUIK CHEK COMPLETE enzyme immunoassay kit (Techlab, Blacksburg, VA, USA).
Genomic DNA was prepared from the three isolates using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). The three isolates were positive to tcdA, tcdB and binary toxin genes cdtA and cdtB by PCR as described previously25. The isolates were assigned to sequence types (ST) using the multilocus sequence typing (MLST) scheme targeting seven housekeeping genes, adk, atpA, dxr, glyA, recA, sodA and tpi (http://pubmlst.org/cdifficile/)26 by PCR and Sanger sequencing, which was also confirmed using their genome sequences to query the MLST database (see below).
Whole genome sequence and phylogenetic analysis
All three isolates were subjected to the 150 bp paired-end whole genome sequencing with a ca. 200× coverage using the HiSeq 2500 Sequencer (Illumina, San Diego, CA, USA) following the manufacturer’s protocol. Reads were filtered using the Sickle quality trimming tool (https://github.com/najoshi/sickle/) with 20 as the cutoff value of both quality and length. Filtered reads were then assembled into contigs using the SPAdes program27 with auto-cutoff and careful functions being turned on. Annotation of the genomic sequence was carried out using the Prokka program28 against its incorporated bacterial gene database. The coding sequence (CDS) of interest (e.g. unique genes) was then manually checked and modified using Protein BLAST (https://blast.ncbi.nlm.nih.gov/) against non-redundant protein sequences in GenBank. Settings of programs mentioned above, unless specifically noted, all remained as default.
Antimicrobial resistance genes were predicted using the ResFinder web-based tool from the Center for Genomic Epidemiology (http://genomicepidemiology.org/). Prophages were predicted using the PHASTER web server (http://phaster.ca/)29. The predicted intact and incomplete phage sequences were searched for similar regions, which were defined as ≥80% coverage and ≥90% highest nucleotide identity, on other C. difficile genomes using BLAST (http://blast.ncbi.nlm.nih.gov/).
To identify plasmids, reads filtered by Sickle were assembled into contigs using the PlasmidSPAdes30. Contigs assembled by SPAdes and PlasmidSPAdes were screened for plasmids using Microbial Genome BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch&BLAST_SPEC=MicrobialGenomes) against the complete plasmid database. In addition, sequences of all C. difficile plasmids (n = 6; Table S1 in the Supplementary Data) that have been deposited in GenBank were retrieved. Contigs assembled by SPAdes and PlasmidSPAdes of the three isolates in the present study were aligned with the C. difficile plasmid sequences using BLAST to further examine the presence of plasmids.
There were four Clade 3 strains with whole genome sequences available in GenBank. These were ZJCDC-S82 (ST5; clinical isolate, recovered in 2013, China, GenBank accession no. JYNK01000000), CD69 (ST221; clinical isolate, recovered in 2010, USA, GenBank accession no. AVHE00000000), VL-0391 (ST201; clinical isolate, recovered date not available, Canada, GenBank accession no. FALK01000001) and VL-0104 (ST201; clinical isolate, recovered date not available, Canada, GenBank accession no. FAAJ0100000). In addition, six strains of Clade 3 from the UK (Oxford) and Australia have been reported5 with short reads of genome sequences available in the Sequence Read Achieve (SRA) database. The short reads of these strains were retrieved and were assembled into contigs using the SPAdes program27, which were then used for comparison. All of the Clade 3 strains listed above carry genes encoding toxins A and B and the binary toxin.
Strains with whole genome sequence available in GenBank (http://www.ncbi.nlm.nih.gov/genome/genomes/535?, accessed by July 31, 2016) that belong to a Clade other than Clade 3 could be assigned to 44 ST. Whole genome sequences of all Clade 3 strains (n = 13; 10 from GenBank and SRA databases and 3 in the present study) and a representative strain of each of the 44 ST of other clades (Table 1) were included into the phylogenetic analysis. The representative genomes selected, were based on those that have been included in analyses to infer the phylogeny of C. difficile in two previous studies31,32. The genomes of 57 strains were aligned using the Harvest suite33 with the incorporated Phi test, which identifies recombination sites, being turned on and other settings remaining as default. Approximately 2 Mb sequences representing the core genome of C. difficile were identified. After removing single nucleotide polymorphisms (SNP) either on a recombination site or not tagged with ‘PASS’ using Harvest33, a total of 180,004 SNP sites in the core genome were kept for further analyses. The phylogenetic tree was inferred based on all filtered SNP by MEGA 7.034 using the neighbour-joining method with Jukes-Cantor and 1,000 bootstrap replicates as the nucleotide substitution model and method to test phylogeny, respectively. A circular plot was constructed using the Circleator tool35 to demonstrate the locations of SNPs among the three isolates in the present study. Pair-wise average nucleotide identity (ANI) based on BLAST was calculated for the genome sequence of the three isolates and all Clade 3 strains and the 44 representative strains of other clades (Table 1) using the JSpecies web program (http://imedea.uib-csic.es/jspecies/) with default settings36.
Table 1. ANI values (%) of isolates 103, 106 and 133 compared with other Clade 3 strains and representative strains of other clades.
| Clade | Strain | ST | Genome accession no. | Country | Year | 103 ANI value | 106 ANI value | 133 ANI value |
|---|---|---|---|---|---|---|---|---|
| 3 | 103 | 5 | China | |||||
| 106 | 285 | China | 99.52 | |||||
| 133 | 5 | China | 99.51 | 99.25 | ||||
| ZJCDC-S82 | 5 | JYNK01000000 | China | 2013 | 99.44 | 99.18 | 99.38 | |
| OX2183 | 5 | C00005010 | UK | 2009 | 99.43 | 99.16 | 99.36 | |
| OX1485 | 5 | C00004983 | UK | 2008 | 99.37 | 99.20 | 99.39 | |
| OX561 | 22 | C00001506 | UK | 2007 | 99.42 | 99.20 | 99.41 | |
| OX1232 | 25 | C00002723 | UK | 2007 | 99.46 | 99.18 | 99.49 | |
| L058 | 96 | C00002452 | UK | 2008 | 99.48 | 99.23 | 99.37 | |
| Q24 | 162 | C00006439 | Australia | 2008 | 99.43 | 99.23 | 99.28 | |
| VL-0104 | 201 | FAAJ01000000 | Canada | NA | 99.49 | 99.18 | 99.37 | |
| VL-0391 | 201 | FALK01000001 | Canada | NA | 99.48 | 99.28 | 99.36 | |
| CD69 | 221 | AVHE01000000 | USA | 2010 | 99.41 | 99.22 | 99.52 | |
| 1 | E14 | 2 | CAMS01000000 | France | NA | 97.71 | 97.77 | 97.68 |
| CD37 | 3 | AHJJ01000000 | USA | 1980 | 97.66 | 97.81 | 97.73 | |
| P49 | 4 | AVMN01000000 | USA | NA | 97.91 | 97.91 | 97.77 | |
| E25 | 6 | CAMJ01000000 | France | NA | 97.70 | 97.77 | 97.82 | |
| E16 | 7 | CAMH01000000 | France | NA | 97.90 | 97.88 | 97.74 | |
| P15 | 8 | AVLV01000000 | USA | 2005 | 97.92 | 97.90 | 97.90 | |
| P8 | 10 | AVLR01000000 | USA | 2001 | 97.68 | 97.83 | 97.73 | |
| T6 | 13 | CAMR01000000 | France | NA | 97.61 | 97.55 | 97.66 | |
| P64 | 14 | AWZL01000000 | USA | 2009 | 97.66 | 97.71 | 97.61 | |
| 5.3 | 15 | AZSH01000000 | Australia | 2008 | 97.97 | 97.93 | 97.95 | |
| DA00129 | 16 | AVIY01000000 | USA | 2010 | 97.52 | 97.46 | 97.77 | |
| CD8-15 | 17 | LYDP01000000 | Italy | 2015 | 97.83 | 97.52 | 97.60 | |
| P30 | 26 | AVME01000000 | USA | 2009 | 97.63 | 97.50 | 97.57 | |
| CD200 | 28 | AVIF01000000 | USA | 2010 | 97.54 | 97.56 | 97.54 | |
| P38 | 29 | AVMH01000000 | USA | 2009 | 97.67 | 97.55 | 97.55 | |
| P7 | 34 | AVLQ01000000 | USA | NA | 97.83 | 97.53 | 97.62 | |
| P29 | 35 | AVMD01000000 | USA | 2008 | 97.64 | 97.44 | 97.62 | |
| E12 | 42 | CAMZ01000000 | France | NA | 97.94 | 97.93 | 97.84 | |
| DA00126 | 43 | AVIW01000000 | USA | 2010 | 97.72 | 97.78 | 97.77 | |
| CD45 | 44 | AVGY01000000 | USA | 2010 | 97.60 | 97.65 | 97.67 | |
| CD113 | 45 | AVHN01000000 | USA | 2010 | 97.76 | 97.64 | 97.64 | |
| ATCC43255 | 46 | CM000604 | Canada | 1935 | 97.77 | 97.78 | 97.73 | |
| CD22 | 48 | AVGP01000000 | USA | 2010 | 97.73 | 97.82 | 97.60 | |
| T17 | 49 | CAMT01000000 | France | NA | 97.98 | 97.74 | 97.76 | |
| P11 | 53 | AVLT01000000 | USA | NA | 97.89 | 97.82 | 97.80 | |
| CD630 | 54 | CP010905 | Switzerland | 1982 | 97.94 | 97.65 | 97.66 | |
| T19 | 55 | CANA01000000 | France | NA | 98.09 | 97.82 | 97.87 | |
| P46 | 58 | AVML01000018 | USA | 2009 | 97.76 | 97.77 | 97.71 | |
| E7 | 63 | CAMV01000000 | France | NA | 97.75 | 97.73 | 97.63 | |
| Y343 | 83 | AVLG01000000 | USA | 2010 | 97.81 | 97.89 | 97.77 | |
| CD105KSE11 | 107 | FJVK01000000 | UK | NA | 98.05 | 97.77 | 97.22 | |
| DA00216 | 110 | AVJX01000000 | USA | 2010 | 97.82 | 97.65 | 97.71 | |
| DA00211 | 139 | AVJU01000000 | USA | 2010 | 97.87 | 97.47 | 97.52 | |
| 2 | CD196 | 1 | NC_013315 | France | 1985 | 97.89 | 97.66 | 97.76 |
| E19 | 62 | CAMO01000000 | France | 2012 | 97.94 | 97.59 | 97.79 | |
| T23 | 67 | CAMN01000000 | France | NA | 97.79 | 97.71 | 97.84 | |
| E15 | 95 | CAMM01000000 | France | NA | 97.51 | 97.54 | 97.58 | |
| P59 | 123 | AVMQ01000000 | USA | 2009 | 97.57 | 97.58 | 97.59 | |
| 4 | M68 | 37 | NC_017175 | UK | 2006 | 97.59 | 97.58 | 97.48 |
| CF5 | 86 | FN665652 | Belgium | 1995 | 97.78 | 98.08 | 97.85 | |
| CD002 | 170 | CAMG01000000 | France | 2010 | 97.74 | 98.06 | 97.71 | |
| 6503 | 320 | ADEI01000000 | USA | 2009 | 97.76 | 97.84 | 97.38 | |
| 5 | M120 | 11 | NC_017174 | UK | 2007 | 96.31 | 96.33 | 96.32 |
| C-I | CD105KSO8 | 297 | FJUI01000000 | UK | NA | 91.24 | 91.43 | 90.45 |
NA, not available.
The pan-genomes of 13 strains of Clade 3 were constructed based on the amino acids of previously predicted genes (5,656 genes in total) and were then aligned against 44 annotated representative genomes of other clades (Table 1) by Roary37 with default settings. Genes present in all of 13 strains of Clade 3 but absent (arbitrarily defined as <80% nucleotide identity and <70% coverage) from strains of other clades were further examined whether they were unique to Clade 3 among C. difficile using Microbial Genome BLAST against all available C. difficile genomes. Genes present in only one of the three isolates were considered unique. This was also confirmed using Microbial Genome BLAST against all C. difficile genomes. The function of genes that was unique to one of the three isolates or Clade 3 was further predicted using Protein BLAST. The unique regions were searched for insertion sequences using ISFinder (https://www-is.biotoul.fr/) and integrative and conjugative elements (ICE) using ICEberg (http://db-mml.sjtu.edu.cn/ICEberg/).
The sequence of PaLoc and CdtLoc of the three isolates were aligned to genomes of all Clade 3 strains and representative strains of other clades. A phylogenetic tree of CdtLoc was constructed for all Clade 3 strains and several representative strains of Clade 2 or Clade 5 using the Harvest tool33.
Antimicrobial susceptibility testing
Minimum inhibitor concentrations (MICs) of clindamycin, fidaxomicin, metronidazole, moxifloxacin, rifampicin, tetracycline and vancomycin were determined using the agar dilution method following the recommendations of the Clinical and Laboratory Standard Institute (CLSI) guidelines38. Breakpoints defined by CLSI (clindamycin, fidaxomicin, metronidazole, moxifloxacin, rifampicin and tetracycline) or by the European Committee on Antimicrobial Susceptibility Testing (EUCAST; vancomycin) were applied.
Results
C. difficile isolates, MLST and general genome features
Isolates 103 and 133 belonged to ST5 (adk1, atpA6, dxr4, glyA7, recA2, sodA8, and tpi7), while the remaining isolate, 106, belonged to ST285 (adk1, atpA6, dxr4, glyA7, recA25, sodA8, and tpi7). ST5 and ST285 differ in a single allele (recA) with only one SNP. A total of 5,703,662 to 5,812,358 reads were generated by whole genome sequencing of the three isolates. These were assembled into 243, 303 and 276 contigs (61, 69 and 75 contigs ≥1,000 bp in length; N50, 131,308 to 137,573 bp) with a 28.38 to 28.74% GC content, respectively (Table 2). There were 3,764 to 4,000 CDS that were identified from the genome of the three isolates. Of note, isolate 133 had 227 or 236 more CDS than the other two isolates, which is due to a 150 kb phage present on 133 but absent from the other two (see below).
Table 2. General features of the three genomes.
| Isolate | ST | Clean reads | Genome size (bp) | GC content | No. of contigs | No. of contigs ≥1,000 bp | No. of coding sequences | No. of tRNA genes |
|---|---|---|---|---|---|---|---|---|
| 103 | 5 | 5,703,662 | 4,060,612 | 28.56% | 243 | 69 | 3,777 | 43 |
| 106 | 285 | 5,698,996 | 4,080,409 | 28.74% | 303 | 61 | 3,764 | 42 |
| 133 | 5 | 5,696,658 | 4,181,327 | 28.38% | 276 | 75 | 4,000 | 43 |
SNP and phylogenetic analysis
There were 1,267 SNPs between isolate 103 and 133 (both of ST5), while isolate 106 (ST285) had 1,364 and 1,104 SNPs compared with isolate 133 and 103, respectively (Figure S2 in the Supplementary Data). The phylogenetic tree constructed correlates well to the distribution of clades (Fig. 2, the number of SNPs is listed in Table S3 in the Supplementary Data). All Clade 3 strains were clustered together and were closer to Clade 4, 5 and C-I rather than Clade 1 and 2 (Fig. 2). In Clade 3, isolates 106 and 133 were clustered with strain CD69, while isolate 103 appeared to be more distinct from isolates 103 and 106 (Fig. 3).
Figure 2. Phylogenetic tree based on SNPs of genome sequences of the three isolates, other 10 strains of Clade 3 and 44 representative strains of other clades.

ST is shown after the strain names and PCR ribotype is indicated in parentheses with NA representing not available. The three isolates are highlighted with boxes. Bar, 0.02 changes per nucleotide position.
Figure 3. Phylogenetic tree based on SNP identified in the core genomes of Clade 3 strains.
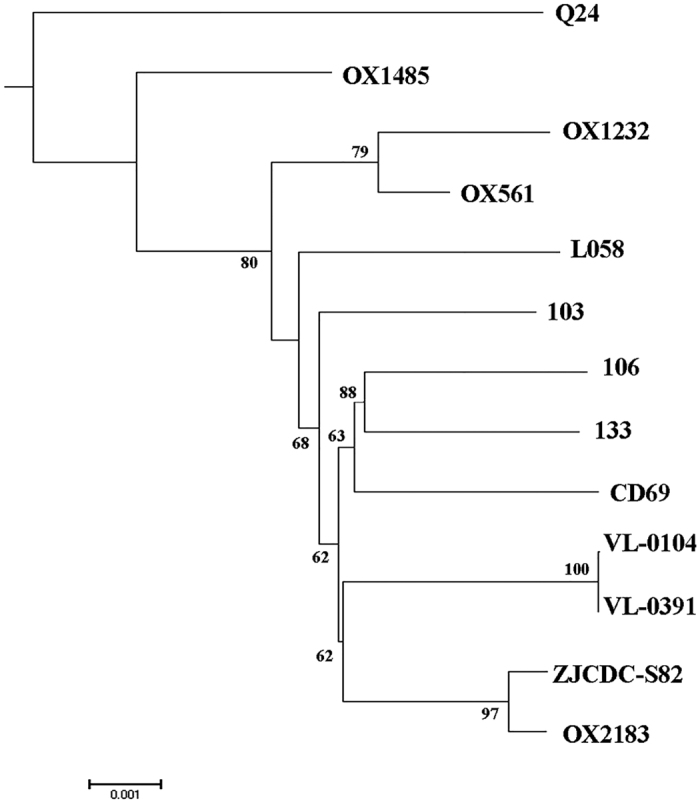
Bar, 0.01 changes per nucleotide position. Bootstrap values >50% (based on 1,000 resamplings) are shown.
When compared with other Clade 3 strains and 44 representative strains of each clade, isolate 103 had the highest ANI value with isolates 106 (99.52%) and 133 (99.51%; Table 1). The ANI values between any two strains of Clade 3 were all above 99%, while those between a Clade 3 strain and strains of Clade 1, 2, 4 and 5 were in the range of 96.3% to 98.1%. The ANI value between Clade 3 and Clade C-I was only 90% to 91.4%.
Antimicrobial susceptibility and antimicrobial resistance genes
All three isolates were susceptible to clindamycin (MIC, 0.125 to 2 μg/mL), fidaxomicin (0.015 to 0.06 μg/mL), metronidazole (<0.03 μg/mL), moxifloxacin (<0.03 to 2 μg/mL), rifampicin (<0.03 μg/mL), tetracycline (<0.03 to 0.06 μg/mL) and vancomycin (<0.03 to 0.25 μg/mL). Correspondingly, no known antimicrobial resistance genes were identified from their genome sequences.
Sequence analysis of PaLoc
All three isolates had the identical complete sequence of PaLoc and the following comparison was used the consensus PaLoc sequence of the three isolates. PaLoc of the three isolates had the same genetic organization as other Clade 3 PaLoc but with a few SNPs and insertions or deletions (Table 3). Like the PaLoc of all other Clade 3 strains, those of the three isolates had the insertion of Tn6218 between genes tcdE and tcdA (Fig. 1). Tn6218 in Clade 3 PaLoc had four common genes (int, xis, rep and xre) and five accessory genes, i.e., merR (a transcription regulator gene), a genes encoding oxidoreductase, a gene encoding flavodoxin, an open reading frame (orf) encoding a hypothetical protein containing cupin domain, and a gene encoding RNA polymerase σ70 (Fig. 1 and Table 3).
Table 3. PaLoc of isolates 103, 106 and 133 compared with other Clade 3 strains and a representative strain of each Clade 1, 2 and 5.
| Clade | Strain | tcdR | tcdB | tcdE | Tn6218 | tcdA | tcdC | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| int | xis | rep | xre | merR | oxidoreductase | flavodoxin | orf | σ70 | |||||||
| 1 | CD630 | 12 (6NS) | 92 (48NS) | 5 + 31nt del + 1nt in (7NS + 10 del) | — | — | — | — | — | — | — | — | — | 116 (44NS) | 5 + 318nt del (1NS + 106 del) |
| 2 | CD196 | 12 (6NS) | 446 (175NS) | 3 + 31nt del + 1nt in (6NS + 10 del) | — | — | — | — | — | — | — | — | — | 89 + 63nt del (34NS + 21 del) | 4 + 36nt del (12 del) |
| 5 | M120 | 18 (8NS) | 173 (87NS) | 4 + 42nt in (2NS + 14 in) | — | — | — | — | — | — | — | — | — | 89SNP + 63nt del (44NS + 21 del) | 3 + 15nt del (3NS + 5 del)a |
| 3 | ZJCDC-S82 | ID | 6 (3NS) | ID | ID | ID | 5 (3NS) | ID | ID | ID | ID | ID | 11 + 210nt in (5NS + 70 del) | 18 (11NS) | 36nt del (12 del) |
| CD69 | ID | 12 (7NS) | ID | 3 (4NS) | ID | 5 (3NS) | ID | ID | ID | ID | ID | ID | 153 (73NS) | ID | |
| VL-0391 | ID | 2 (2NS) | ID | 7 (4NS) | ID | 3 (3NS) | 1 (1NS) | ID | 1 (1NS) | 1 (1NS) | ID | 1 (0NS) | 11 + 36nt del (5NS + 12 del) | 36nt del (12 del) | |
| VL-0104 | ID | 2 (2NS) | ID | 8 (4NS) | ID | 3 (3NS) | 1 (1NS) | ID | 1 (1NS) | 1 (1NS) | ID | 1 (0NS) | 11 + 36nt del (5NS + 12 del) | 36nt del (12 del) | |
| L058 | 1 (1NS) | 5 (2NS) | ID | 5 (3NS) | ID | 3 (2NS) | ID | ID | 2 (2NS) | ID | ID | ID | 8 (4NS) | ID | |
| OX561 | 1 (1NS) | 6 (3NS) | ID | 9 (3NS) | 1 (1NS) | 2 (2NS) | ID | ID | 1 (1NS) | ID | ID | ID | 7 + 63nt del (5NS + 21 del) | ID | |
| OX1232 | 1 (1NS) | 6 (3NS) | ID | 8 (3NS) | ID | 2 (2NS) | ID | ID | 1 (1NS) | ID | ID | ID | 14 (10NS) | ID | |
| OX1485 | 1 (1NS) | 5 (3NS) | ID | 8 (3NS) | ID | 2 (2NS) | ID | ID | 1 (1NS) | ID | ID | ID | 37 (21NS) | ID | |
| OX2183 | ID | 3 (3NS) | ID | ID | 47nt in (9 in) | 5 (3NS) | ID | ID | ID | ID | ID | ID | 51 (23NS) | 36nt del (12 del) | |
| Q24 | ID | 8 (4NS) | ID | 7 (2NS) | 1 (1NS) | 2 + 108nt in (1NS + 36 in) | 2 (1NS) | 1 (1NS) | 1 (1NS) | 3 (0NS) | 2 (0NS) | ID | 62 (35NS) | 36nt del (12 del) | |
The results shown in the Table are that the PaLoc sequence of isolate 103, 106 and 133 queries those of strains listed in the Strain column. The number of SNP is shown in the column with the resulted amino acid substitutions and deletions being indicated in parentheses. ID, identical; in, insertion; del, deletion; -, absent; nt, nucleotide; NS, non-synonymous SNP. The nucleotide deletions and insertions are consecutive. atcdC of strain NAP08 has a nonsense mutation, resulting in 37 aa deletions of the C-terminal end compared to those of isolates 103, 106 and 133.
The toxin B gene tcdB of the three isolates were highly similar to those of all other Clade 3 strains with only 2 to 12 SNPs, but they were more distinct from those of other clades as there were 92 to 446 SNPs compared to the representative strains of Clade 1, 2 and 5 (Table 3 and Figure S3 in the Supplementary Data). Toxin A gene tcdA of the three isolates had 1 to 5 SNPs from several Clade 3 strains (such as OX561) but had a number of nucleotide insertions or deletions when compared with those of other Clade 3 strains such as strains CD69 and ZJCDC-S82 (Table 3 and Figure S3).
Compared with the typical PaLoc of strain CD630 (Clade 1), tcdC of the three isolates contained a 54-bp consecutive deletion that resulted in a truncated TcdC protein. The 54-bp deletion was also present in some Clade 3 PaLoc such as that of strain CD69 (Table 3) and Clade 1 strains AIP2005162 (GenBank accession no. EU271784), strain 55767 (GenBank accession no. FJ409552), KK296 (GenBank accession no. EF447033) and CD3980 (GenBank accession no. JN944624). In contrast, tcdC of other Clade 3 PaLoc such as that of strain ZJCDC-S82 had a 36-bp consecutive deletion compared with that of strain CD630.
Sequence analysis of CdtLoc
Unlike the PaLoc of Clade 3, there were no insertions of mobile genetic elements in CdtLoc. The CdtLoc of the three isolates was identical to that of strain ZJCDC-S82 and was also highly similar to those of other Clade 3 strains. The exception to this was strain Q24 with only several SNP (Table S4 in the Supplementary Data). There were a number of SNPs between the CdtLoc of the three isolates, those of Q24 (Clade 3) and the representative strains of Clade 2 and 5 (Table S4). Phylogenetic tree of CdtLoc (Figure S4 in the Supplementary Data) revealed that the CdtLoc of all Clade 3 strains, with the exception of strain Q24, formed a cluster, while those of Q24, a Clade 5 strain and the Clade 2 strains belonged to three separate clusters.
Prophages and plasmids
Isolate 103 contained two intact, 9 incomplete and one questionable phage, while isolate 106 did not carry any intact, but had 8 incomplete phages (Table 4). Isolate 133 contained two intact and 6 incomplete phages. Among the intact and incomplete phages, a 27.2 kb phage region was common to all of the three isolates and very similar (96.4% to 97.7% identity; 100% coverage) regions were also present on genomes of several C. difficile strains of other clades such as CD196 (Clade 1). The three isolates did not carry any known plasmid replicons. Among genome sequences of the three isolates, none except a 2.4 kb fragment of an 8.4 kb contig, which were seen in all of the three isolates by both SPAdes and PlasmidSPAdes, matched any sequences of known C. difficile plasmids. The 2.4 kb fragment matched (98% identity) with two genes of unknown function on C. difficile plasmid pCD6 (GenBank accession no. AY350745). The remaining part of the 8.4 kb contig did not match any known sequences on either chromosomes or plasmids of any bacteria. Therefore, whether the contig belongs to a plasmid is still questionable and needs further study.
Table 4. Predicted phages in the three isolates.
| Phage region | Region length | Completeness | Total no. of orf | Mostly matched Phagea | GC % |
|---|---|---|---|---|---|
| Isolate103 | |||||
| 1 | 27.2 kb | Incomplete | 30 | phiCDHM19_NC_028996(11) | 27.9 |
| 2 | 18.3 kb | Incomplete | 25 | phiC2_NC_009231(6) | 28.3 |
| 3 | 24.7 kb | Questionable | 27 | phiCD505_NC_028764(11) | 28.3 |
| 4 | 17.4 kb | Incomplete | 22 | phiCD27_NC_011398(6) | 27.2 |
| 5 | 15.1 kb | Incomplete | 10 | phiCD505_NC_028764(2) | 28.8 |
| 6 | 7.3 kb | Incomplete | 12 | phiCDMH11_NC_029001(4) | 28.3 |
| 7 | 8 kb | Incomplete | 9 | vB_Bans_Tsamsa_NC_023007(2) | 29.3 |
| 8 | 19.2 kb | Incomplete | 9 | c_st__NC_007581(6) | 27.4 |
| 9 | 23.6 kb | Incomplete | 32 | phiMMP02_NC_019421(10) | 27.7 |
| 10 | 13.8 kb | Incomplete | 21 | CDMH1_NC_024144(8) | 27.5 |
| 11 | 36.1 kb | Intact | 51 | phiMMP03_NC_028959(23) | 32.1 |
| 12 | 39.4 kb | Intact | 61 | CDMH1_NC_024144(16) | 33.8 |
| Isolate106 | |||||
| 1 | 27.2 kb | Incomplete | 30 | phiCDHM19_NC_028996(11) | 28.04 |
| 2 | 26.5 kb | Incomplete | 34 | phiMMP03_NC_028959(11) | 29.86 |
| 3 | 8.3 kb | Incomplete | 9 | vB_Bans_Tsamsa_NC_023007(2) | 29.19 |
| 4 | 21.9 kb | Incomplete | 9 | c_st__NC_007581(3) | 26.62 |
| 5 | 18.5 kb | Incomplete | 22 | CDMH1_NC_024144(12) | 30.13 |
| 6 | 15.1 kb | Incomplete | 11 | phiCD505_NC_028764(2) | 28.86 |
| 7 | 12.5 kb | Incomplete | 21 | phiMMP02_NC_019421(15) | 29.12 |
| 8 | 25.1 kb | Incomplete | 13 | phiARI0746_NC_031907(3) | 39.23 |
| Isolate133 | |||||
| 1 | 27.2 kb | Incomplete | 29 | phiCDHM19_NC_028996(11) | 28 |
| 2 | 32.8 kb | Incomplete | 27 | phiCD27_NC_011398(6) | 28 |
| 3 | 10 kb | Incomplete | 15 | phiMMP02_NC_019421(7) | 28.8 |
| 4 | 22 kb | Incomplete | 10 | c_st__NC_007581(6) | 26.6 |
| 5 | 55.9 kb | Intact | 59 | phiCD6356_NC_015262(40) | 28.4 |
| 6 | 21.2 kb | Incomplete | 10 | phiCD505_NC_028764(2) | 28.8 |
| 7 | 18.1 kb | Incomplete | 24 | phiMMP02_NC_019421(14) | 28.5 |
| 8 | 166.8 kb | Intact | 231 | phiCDHM19_NC_028996(46) | 30 |
aThe phage (phage name_GenBank accession no.) with the highest number of orf most similar to those in the region being indicated in parentheses.
Unique genes
Compared with other clades, all isolates of Clade 3, including the three in the present study and those with genomic sequence available in the GenBank and SRA, had 15 unique genes (Table 5). These unique genes included 4 of the 5 accessory genes (except the gene encoding RNA polymerase σ70) of Tn6218 in PaLoc (see above), genes encoding metallo-β-lactamase (MBL) fold metallo-hydrolyase, transcriptional regulator and methylthioadenosine/S-adenosylhomocysteine (MTA/SAH) nucleosidase, and 8 genes of unknown function.
Table 5. Unique genes of Clade 3 and isolates 103, 106 and 133.
| Locus | Producta | Sizeb | Location | Query Coverage |
|---|---|---|---|---|
| Clade 3c | ||||
| 103_01388 | Transcriptional regulator | 184 | Contig 5c | 100% |
| 103_01956 | Methylthioadenosine/S-adenosylhomocysteine nucleosidase | 181 | Contig 10c | 97% |
| 103_02913d | Hypothetical protein | 133 | Contig 19c | |
| 103_02915d | Putative flavodoxin | 222 | Contig 19c | 100% |
| 103_02916d | Putative oxidoreductase | 286 | Contig 19c | 100% |
| 103_02917d | MerR Transcriptional regulator | 135 | Contig 19c | 100% |
| 103_03125 | Hypothetical protein | 255 | Contig 22c | |
| 103_03128 | Hypothetical protein | 200 | Contig 22c | |
| 103_03130 | Hypothetical protein | 198 | Contig 22c | |
| 103_03132 | Hypothetical protein | 233 | Contig 22c | |
| 103_03133 | Hypothetical protein | 214 | Contig 22c | |
| 103_03507 | Hypothetical protein | 372 | Contig 32c | |
| 103_03634 | Hypothetical protein | 110 | Contig 34c | |
| 103_03635 | MBL fold metallo-hydrolase | 253 | Contig 34c | 100% |
| 103_03735 | Hypothetical protein | 282 | Contig 63 | |
| Isolate 103 | ||||
| 103_01535 | Cobalamin-binding protein | 220 | Contig 7 | 95% |
| 103_01536 | Uroporphyrinogen decarboxylase | 288 | Contig 7 | 75% |
| 103_01537 | Iron-sulfur protein | 574 | Contig 7 | |
| 103_01538 | Methylcobamide–CoM methyltransferase | 344 | Contig 7 | 97% |
| 103_01539 | Uroporphyrinogen-III decarboxylase | 362 | Contig 7 | 98% |
| 103_01540 | Membrane transport protein | 303 | Contig 7 | |
| 103_01541 | Metal ABC transporter substrate-binding protein | 273 | Contig 7 | 99% |
| 103_01542 | Methionine ABC transporter permease | 222 | Contig 7 | 100% |
| 103_01543 | ABC transporter ATP-binding protein | 335 | Contig 7 | 97% |
| 103_01544 | IS30 family transposase | 318 | Contig 7 | 99% |
| Isolate 106 | ||||
| 106_00822 | Multidrug-efflux transporter 1 regulator | 275 | Contig 4 | |
| 106_00823 | Cytidylate kinase | 204 | Contig 4 | 100% |
| 106_00824 | MATE family efflux transporter | 442 | Contig 4 | 100% |
| 106_00827 | RNA polymerase sigma factor FliA | 138 | Contig 4 | 100% |
| 106_01206 | Antirestriction protein ArdA | 167 | Contig 6 | 98% |
| 106_01208 | Hypothetical protein | 188 | Contig 6 | |
| 106_01209 | Nucleotidyltransferase, polymerase | 294 | Contig 6 | 98% |
| 106_01213 | Hypothetical protein | 234 | Contig 6 | |
| 106_02320 | Endonuclease | 367 | Contig 14 | 99% |
| 106_03618 | Ribonuclease/integrase | 407 | Contig 35 | |
| 106_03620 | Transposase | 252 | Contig 35 | 94% |
| 106_03734 | Membrane protein | 716 | Contig 48 | 98% |
| Isolate 133 | ||||
| 133_02100 | GIY-YIG catalytic domain-containing protein | 401 | Contig 11 | |
| 133_02107 | Hypothetical protein | 120 | Contig 11 | |
| 133_02109 | Hypothetical protein | 318 | Contig 11 | |
| 133_02112 | Hypothetical protein | 102 | Contig 11 | |
| 133_02117 | HaeIII-like C-5 cytosine methyltransferase | 340 | Contig 11 | 97% |
| 133_02118 | BspRI-like C-5 cytosine methyltransferase | 350 | Contig 11 | 96% |
| 133_03129 | Flp pilus assembly protein CpaB | 261 | Contig 24 | 91% |
| 133_03131 | ATPase involved in chromosome partitioning/Flp pilus assembly protein, ATPase CpaE | 261 | Contig 24 | 99% |
| 133_03131 | Type II secretion system protein E/Flp pilus assembly protein, ATPase CpaF | 256 | Contig 24 | |
| 133_03133 | Hypothetical protein | 287 | Contig 24 | |
| 133_03134 | Hypothetical protein | 138 | Contig 24 | |
| 133_03137 | Hypothetical protein | 176 | Contig 24 | |
| 133_03174 | Hypothetical protein | 177 | Contig 24 | |
| 133_03175 | Transcriptional regulator | 116 | Contig 24 | 95% |
| 133_03177 | radC-like JAB domain protein | 127 | Contig 24 | |
| 133_03181 | Chromosome/plasmid-partitioning protein ParA | 276 | Contig 24 | 97% |
| 133_03182 | Chromosome/plasmid-partitioning protein ParB | 450 | Contig 24 | |
| 133_03183 | Transcriptional regulator | 194 | Contig 24 | |
| 133_03185 | Adenosine monophosphate/protein transferase VbhT | 112 | Contig 24 | 100% |
aAnnotation was curated using Protein BLAST (https://blast.ncbi.nlm.nih.gov) against non-redundant protein sequences in GenBank. bSize, the number of amino acids. cLoci and locations refer to those of isolate 103. dThese genes are accessory genes of Tn6218.
Isolate 103 had 10 unique genes, all of which were clustered together in a 10.2 kb region of a single contig (Table 5). Isolate 106 had 12 unique genes, some of which formed two clusters (Table 5). Isolate 133 had 20 unique genes, which were clustered on either the 131.4-kb Contig 11 or the 75.0-kb Contig 24 (Table 5). None of these genes unique to each of the three isolates were predicted as part of a phage. Among the unique genes of Clade 3 and the three isolates, none belonged to an ICE and only one gene that is unique to isolate 103 (locus 103_01544; Table 5) was predicted to belong to an insertion sequence.
Discussion
The binary toxin has been increasingly recognized as an important virulence factor. Although binary toxin genes have been found in 23% toxigenic C. difficile in Europe39, the prevalence of binary toxin genes in toxigenic C. difficile was less than 7% in China14,19,20,21,22,23,24. Such a low prevalence of binary toxin gene-carrying toxigenic C. difficile in China resembles the prevalence (<10%) before the emergence of hypervirulent PCR ribotype 02717. It has been found that binary toxin gene-carrying C. difficile belonged to several STs including ST1 (Clade 2), ST5 (Clade 3), ST11 (Clade 5), ST22 (Clade 3), ST201 (Clade 3) and ST220 (Clade 1) in China7,19,20,40. In the present study, we also reported a binary toxin gene-carrying strain of ST285 (Clade 3) in China. Outside of China, binary toxin gene-carrying C. difficile of ST5 has been reported in Oxford, UK5.
The estimated evolutionary rate of C. difficile was 0.6 to 2.3 mutations per genome per year41. It has been proposed that ≤2 SNPs are defined for transmission and >10 SNPs are defined for genetically distant strains31. There were 1,267 SNPs between isolate 103 and 133 (both of ST5), while isolate 106 (ST285) had 1,104 and 1,364 SNPs compared with isolate 133 and 103, respectively. This suggests that the three strains were not part of a common clone. Isolates 103 and 133 were recovered from the same ward, but the SNP analysis of genome sequences did not indicate the inter-patient transfer of a common strain.
ANI has been increasingly employed to determine bacterial species. The ≥95% ANI cutoff corresponds to the ≥70% DNA-DNA hybridization value42, which is the gold standard for defining a bacterial species. Therefore, the ≥95% ANI cutoff has been proposed to define species. Clade 3 strains had ≥95% ANI values with clades 1, 2, 4 and 5, indicating that these clades were truly of a single species. In contrast, the newly identified Clade C-I shared only 90% to 91.6% ANI values with Clade 3 strains and those of other clades (Table S2 in the Supplementary Data), suggesting that Clade C-I may represent a new bacterial species other than C. difficile. In addition, Clade 3 strains had >99% ANI values when compared with strains of the same clade and had <99% values with strains of other clades. This raises the question whether the >99% ANI cutoff can be employed to define clades of C. difficile or not. However, some strains of Clade 1 or Clade 4 had <99% ANI values with strains of the same clade (Table S2 in the Supplementary Data) and no certain ANI values were found to correctly assign all C. difficile strains to a clade.
The major feature of PaLoc in Clade 3 is the insertion of Tn6218 between tcdE and tcdA, which is distinct from PaLoc in other clades. Strains belonging to Clade 3 strains were found in Australia, Bulgaria, Canada, China, UK and USA5,7,43, suggesting that the insertion of Tn6218 in PaLoc is clade-specific rather than geographical location-specific. The insertion of Tn6218 in PaLoc has also been suggested to be associated with the relatively low virulence of Clade 35, as some previous studies have shown that 14-day mortality in CDI cases caused by strains of Clade 3 (PCR ribotype 023; 7%) was lower than those caused by Clade 1 (12%)10. However, there were additional differences between Clade 3 and Class 1 PaLoc such as SNP and/or insertions/deletions in tcdA, tcdB and their regulatory genes. The exact impact of Tn6218 insertion on the virulence remains undetermined and warrants further investigations.
Previous studies suggested that Tn6218 elements are widespread among the C. difficile isolates5. Compared to Tn6218 elements located outside of PaLoc, those in the Clade 3 PaLoc did not carry known antimicrobial resistance genes5. Instead, Tn6218 in Clade 3 PaLoc carried four accessory genes that were absent from Tn6218 elements located outside of PaLoc, and appeared to be the main feature to distinguish Clade 3 from other C. difficile. In Clade 3 PaLoc, Tn6218 is always flanked by an 863-bp sequence on one side and a 263 bp sequence on the other. The two sequences do not contain any orfs and their origins remain unclear, although 169 bp of the 863 bp sequence shows some similarity (81% identity) to several Clostridium phages such as phiMMP03 (GenBank accession no. LN681542). Tn6218 requires AT rich insertion sites5 and the two sequences are AT rich (78% AT content for the two sequences as a whole), suggesting that the two sequences might have inserted into Clade 3 PaLoc prior to the insertion of Tn6218 and therefore provide the insertion site of Tn6218 like a Trojan horse. The two sequences are absent from almost all PaLoc of other clades and therefore there are no insertion sites of Tn6218 in those PaLoc, which could explain that the insertion of Tn6218 in PaLoc is restricted to Clade 3 so far. There is only one exception at present. In the PaLoc of strain 8864 of Clade 2 (GenBank accession no. AF035716), the two sequences are jointed together with a single copy of the 36-bp terminal repeat of Tn6218, suggesting that Tn6218 might have been excised.
As for other components of PaLoc, tcdB appears to be more conserved in sequence than tcdA in Clade 3. It is well known that there are many variations in the amino acid sequence of TcdA and TcdB toxins44,45. However, the relevance of the amino acid variations of TcdA and TcdB to clinical illness remains largely undetermined as there is little evidence that the variations have significant impact on the outcome of diseases44,45. Nonetheless, a previous study has identified 8 amino acid variations in TcdB that are associated with reduced toxicity of host strains46. However, none of the 8 amino acid variations were present in the three isolates in the present study. Both TcdA and TcdB have four domains (glucosyltransferase, autoprotease, delivery and receptor-binding)47. The SNPs in tcdA and tcdB in the three isolates compared to the reference strain CD630 were distributed in all of the four domains (Figure S3 in the Supplementary Data) but their impact on the function of TcdA and TcdB remains unclear48, which may warrant further characterizations.
TcdC protein contains three domains. These are Hyd (Hydrophobic membrane anchor), Dim (Coiled-coil dimerization domain) and OB-fold (conserved C-terminal domain containing a predicted oligonucleotide-binding)49. It has been reported that mutations of tcdC contribute to the hypervirulence of ST1/PCR ribotype 02750. Compared with that of strain CD630, tcdC of ST1/PCR ribotype 027 has three major differences including 1) a deletion at location 117, resulting in frameshift and a truncated tcdC with the absence of Hyd; 2) an 18-bp deletion in the Dim-encoding region, resulting in the missing of 6 amino acids of the Dim domain; and 3) a T660A point mutation, resulting in an amino acid substitution (N219K) of the OB-fold domain (All of the above locations refer to those of tcdC of strain CD630). TcdC of all three isolates in the present study had two common features, which are the absence of Hyd and the N219K amino acid substitution. The two features are also seen in ST1/PCR ribotype 027. However, there was an additional deletion of 12-amino acids in the Dim domain as there is 54-bp deletion of tcdC in all three isolates in the present study compared with the 18-bp deletion in ST1/PCR ribotype 027. Of note, although the SNP and insertions/deletions in tcdA, tcdB and tcdC may provide useful information to untangle the evolution of PaLoc, their impact on the virulence of host strains remains undetermined. The sequence of CdtLoc appears to be more conserved than that of PaLoc. However, the sequence of CdtLoc like that of PaLoc also appears to be clade-specific. This suggests that the acquisition of CdtLoc may be prior to that C. difficile diverged into clades.
Among products of Clade 3-unique genes, MTA/SAH has multiple functions in bacterial metabolism and involves in quorum sensing as it produces the universal quorum sensing signal, autoinducer-251. The gene encoding MBL-fold metallo-hydrolase, which is unique to Clade 3 among C. difficile, was also present (100 coverage, 99% identity) on the chromosome of Clostridium symbiosum ATCC 14940 (GenBank accession no. AWSU00000000). C. symbiosum is non-toxin-producing strict anaerobe as part of the human intestinal bacterial flora52. This suggests inter-species horizontal transfer of this gene. The exact roles of MTA/SAH and MBL-fold metallo-hydrolase in Clade 3 C. difficile, however, remain unclear.
The 10 unique genes of isolate 103 encoded cobalamin-binding protein, uroporphyrinogen decarboxylase (an enzyme involved in the production of heme), methylcobamide--CoM methyltransferase, iron-sulfur protein and ABC transporter proteins. It therefore appears that these genes were formed be a methionine transport system. For unique genes of isolate 106, one cluster contained 4 genes and appeared to encode a transporter, while the other cluster contained 4 genes and appeared to be associated with DNA transfer (Table 5). The unique genes on the Contig 11 of isolate 133 encoded a protein with the GIY-YIG domain and two modification methylases. The GIY-YIG domain has been implicated in a variety of cellular processes involving DNA cleavage, from self-propagation with or without introns, to the restriction of foreign DNA, to DNA repair and maintenance of genome stability53. The unique genes on the Contig 24 of isolate 133 encoded pilus assembly proteins and the par chromosome or plasmid partitioning system, raising the suspicion that this Contig may belong to an unrecognized new plasmid, although no plasmid replicon was identified. As there is no reference plasmid sequence available, long-read genome sequencing such as using PacBio may be needed to confirm whether Contig 24 truly belonged to a plasmid.
Of note, resistance to fluoroquinolones and rifampicin are usually mediated by chromosomal mutations, which are unable to be identified by ResFinder. Nonetheless, the three isolates were all susceptible to moxifloxacin and rifampicin.
Conclusions
Although the three C. difficile isolates in the present study were recovered in the same hospital, it was apparent that they did not belong to a common clone. Clade 3 strains have unusual clade-specific PaLoc characteristic of Tn6218 insertion, which appears to be the main feature to distinguish Clade 3 from other C. difficile. A ≥99% ANI value with a known genome of Clade 3 may be useful to assign a C. difficile isolate to Clade 3 without the need to construct a phylogenetic tree. Clade C-I of C. difficile actually appears to be a different species.
Nucleotide sequence accession numbers
The Whole Genome Shotgun Sequencing projects of C. difficile isolates 103, 106 and 133 have been deposited into DDBJ/EMBL/GenBank under accession MBMH00000000, MBGC00000000 and MBGB00000000, respectively.
Additional Information
How to cite this article: Chen, R. et al. Whole genome sequences of three Clade 3 Clostridium difficile strains carrying binary toxin genes in China. Sci. Rep. 7, 43555; doi: 10.1038/srep43555 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
We thank Dr. Björn Espedido from School of Medicine, the Western Sydney University, Australia and Ms. Heather Yannacos from Prospect, South Australia, Australia for their critical proofreading and corrections. This work was supported by a grant from the National Natural Science Foundation of China (project no. 81572030), the New Century Excellent Talents Program, Ministry of Education, China (project no. NCET-13-0399) and grants from Sichuan provincial Science and Technology Agency, China (project no. 2011FZ0111 and 2015SZ0234-4).
Footnotes
The authors declare no competing financial interests.
Author Contributions Concept and design: X.L., Z.Z.; Acquisition of data: R.C., Y.F., J.Y., X.Z.; Analysis/Interpretation of data: R.C., Y.F., X.W., Z.Z.; Draft of manuscript: R.C., Z.Z.
References
- Bartlett J. G., Chang T. W., Gurwith M., Gorbach S. L. & Onderdonk A. B. Antibiotic-associated pseudomembranous colitis due to toxin-producing clostridia. N Engl J Med 298, 531–534 (1978). [DOI] [PubMed] [Google Scholar]
- Leffler D. A. & Lamont J. T. Clostridium difficile infection. N Engl J Med 372, 1539–1548 (2015). [DOI] [PubMed] [Google Scholar]
- Smits W. K., Lyras D., Lacy D. B., Wilcox M. H. & Kuijper E. J. Clostridium difficile infection. Nat Rev Dis Primers 2, 16020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun V., Hundsberger T., Leukel P., Sauerborn M. & von Eichel-Streiber C. Definition of the single integration site of the pathogenicity locus in Clostridium difficile. Gene 181, 29–38 (1996). [DOI] [PubMed] [Google Scholar]
- Dingle K. E. et al. Evolutionary History of the Clostridium difficile Pathogenicity Locus. Genome Biol Evol 6, 36–52 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabler R. A. et al. Macro and micro diversity of Clostridium difficile isolates from diverse sources and geographical locations. PLoS One 7, e31559 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y. et al. Genome Sequence and Analysis of Peptoclostridium difficile Strain ZJCDC-S82. Evol Bioinform Online 12, 41–49 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wren M. W., Kinson R., Sivapalan M., Shemko M. & Shetty N. R. Detection of Clostridium difficile infection: a suggested laboratory diagnostic algorithm. Br J Biomed Sci 66, 175–179 (2009). [DOI] [PubMed] [Google Scholar]
- Valiente E., Cairns M. D. & Wren B. W. The Clostridium difficile PCR ribotype 027 lineage: a pathogen on the move. Clin Microbiol Infect 20, 396–404 (2014). [DOI] [PubMed] [Google Scholar]
- Walker A. S. et al. Relationship between bacterial strain type, host biomarkers, and mortality in Clostridium difficile infection. Clin Infect Dis 56, 1589–1600 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox M. H. et al. Changing epidemiology of Clostridium difficile infection following the introduction of a national ribotyping-based surveillance scheme in England. Clin Infect Dis 55, 1056–1063 (2012). [DOI] [PubMed] [Google Scholar]
- Tang C. et al. The incidence and drug resistance of Clostridium difficile infection in Mainland China: a systematic review and meta-analysis. Sci Rep 6, 37865 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J. W. et al. Molecular Epidemiology and Antimicrobial Susceptibility of Clostridium difficile Isolates from a University Teaching Hospital in China. Front Microbiol 7, 1621 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F., Wu T., Xu S. & Huang H. Ribotyping and antimicrobial resistance patterns of Clostridium difficile during two periods with an interval of 5 years. Chin J Infect Chemother 14, 116–120 (2014). [Google Scholar]
- Cowardin C. A. et al. The binary toxin CDT enhances Clostridium difficile virulence by suppressing protective colonic eosinophilia. Nat Microbiol 1, 16108 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert C. et al. Prevalence and pathogenicity of binary toxin-positive Clostridium difficile strains that do not produce toxins A and B. New Microbes New Infect 3, 12–17 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerding D. N., Johnson S., Rupnik M. & Aktories K. Clostridium difficile binary toxin CDT: mechanism, epidemiology, and potential clinical importance. Gut Microbes 5, 15–27 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelle S., Gibert M., Bourlioux P., Corthier G. & Popoff M. R. Production of a complete binary toxin (actin-specific ADP-ribosyltransferase) by Clostridium difficile CD196. Infect Immun 65, 1402–1407 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. B. et al. Molecular epidemiology of Clostridium difficile in a tertiary hospital of China. J Med Microbiol 63, 562–569 (2014). [DOI] [PubMed] [Google Scholar]
- Cheng J. W. et al. The first two Clostridium difficile ribotype 027/ST1 isolates identified in Beijing, China-an emerging problem or a neglected threat? Sci Rep 6, 18834 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Cai L., Yu R., Huang W. & Zong Z. ICU-Onset Clostridium difficile infection in a university hospital in China: a prospective cohort study. PLoS One 9, e111735 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. et al. Detection and analysis of Clostridium difficile in hospitalized patients with diarrhea. Chin J Infect Control 11, 293–296+299 (2012). [Google Scholar]
- Dong D. et al. Genetic analysis of Tn916-like elements conferring tetracycline resistance in clinical isolates of Clostridium difficile. Int J Antimicrob Agents 43, 73–77 (2014). [DOI] [PubMed] [Google Scholar]
- Yang J. Study on the molecular epidemiology and antibiotic resistance of Clostridium difficile isolates. Hebei Medical University, (2012). [Google Scholar]
- Lemee L. et al. Multiplex PCR targeting tpi (triose phosphate isomerase), tcdA (Toxin A), and tcdB (Toxin B) genes for toxigenic culture of Clostridium difficile. J Clin Microbiol 42, 5710–5714 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths D. et al. Multilocus sequence typing of Clostridium difficile. J Clin Microbiol 48, 770–778 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19, 455–477 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014). [DOI] [PubMed] [Google Scholar]
- Arndt D. et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44, W16–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antipov D. et al. plasmidSPAdes: assembling plasmids from whole genome sequencing data. Bioinformatics (2016). [DOI] [PubMed] [Google Scholar]
- Knight D. R., Elliott B., Chang B. J., Perkins T. T. & Riley T. V. Diversity and Evolution in the Genome of Clostridium difficile. Clin Microbiol Rev 28, 721–741 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monot M. et al. Clostridium difficile: New Insights into the Evolution of the Pathogenicity Locus. Sci Rep 5, 15023 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen T. J., Ondov B. D., Koren S. & Phillippy A. M. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol 15, 524 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Stecher G. & Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol 33, 1870–1874 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree J. et al. Circleator: flexible circular visualization of genome-associated data with BioPerl and SVG. Bioinformatics 30, 3125–3127 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M. & Rossello-Mora R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA 106, 19126–19131 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page A. J. et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Third Informational Supplement. M100-S23. Clinical and Laboratory Standards Institute, (2013).
- Bauer M. P. et al. Clostridium difficile infection in Europe: a hospital-based survey. Lancet 377, 63–73 (2011). [DOI] [PubMed] [Google Scholar]
- Li C. et al. Emergence of a novel binary toxin-positive strain of Clostridium difficile associated with severe diarrhea that was not ribotype 027 and 078 in China. Infect Control Hosp Epidemiol 36, 1112–1114 (2015). [DOI] [PubMed] [Google Scholar]
- Didelot X. et al. Microevolutionary analysis of Clostridium difficile genomes to investigate transmission. Genome Biol 13, R118 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goris J. et al. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57, 81–91 (2007). [DOI] [PubMed] [Google Scholar]
- Dobreva E. G. et al. Advances in molecular surveillance of Clostridium difficile in Bulgaria. J Med Microbiol 62, 1428–1434 (2013). [DOI] [PubMed] [Google Scholar]
- Hunt J. J. & Ballard J. D. Variations in Virulence and Molecular Biology among Emerging Strains of Clostridium difficile. Microbiol Mol Biol Rev 77, 567–581 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupnik M. Heterogeneity of large clostridial toxins: importance of Clostridium difficile toxinotypes. FEMS Microbiol Rev 32, 541–555 (2008). [DOI] [PubMed] [Google Scholar]
- Zhang Z. et al. Translocation domain mutations affecting cellular toxicity identify the Clostridium difficile toxin B pore. Proc Natl Acad Sci USA 111, 3721–3726 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt R. N., Chambers M. G., Ng K. K., Ohi M. D. & Lacy D. B. Structural organization of the functional domains of Clostridium difficile toxins A and B. Proc Natl Acad Sci USA 107, 13467–13472 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P. et al. Sequence variation in tcdA and tcdB of Clostridium difficile: ST37 with truncated tcdA is a potential epidemic strain in China. J Clin Microbiol 52, 3264–3270 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen H. C., Bakker D., Steindel P., Kuijper E. J. & Corver J. Clostridium difficile TcdC protein binds four-stranded G-quadruplex structures. Nucleic Acids Res 41, 2382–2393 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter G. P. et al. The anti-sigma factor TcdC modulates hypervirulence in an epidemic BI/NAP1/027 clinical isolate of Clostridium difficile. PLoS Pathog 7, e1002317 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parveen N. & Cornell K. A. Methylthioadenosine/S-adenosylhomocysteine nucleosidase, a critical enzyme for bacterial metabolism. Mol Microbiol 79, 7–20 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneuchi C., Watanabe K., Terada A., Benno Y. & Mitsuoka T. Taxonomic study of Bacteroides clostridiiformis subsp. clostridiiformis (Burri and Ankersmit) Holdeman and Moore and of related organisms: proposal of Clostridium clostridiiformis (Burri and Ankersmit) comb. nov. and Clostridium symbiosum (Stevens) comb. nov Int J Syst Bacteriol 26, 195–204 (1976). [Google Scholar]
- Dunin-Horkawicz S., Feder M. & Bujnicki J. M. Phylogenomic analysis of the GIY-YIG nuclease superfamily. BMC Genomics 7, 98 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.