

Abstract
Hepatocellular carcinoma (HCC) is one of the most prevalent and malignant cancers with high inter- and intra-tumor heterogeneity. A central common signaling mechanism in cancer is proline-directed phosphorylation, which is further regulated by the unique proline isomerase Pin1. Pin1 is prevalently overexpressed in human cancers including ~70% of HCC, and promotes tumorigenesis by activating multiple cancer-driving pathways. However, it was challenging to evaluate the significance of targeting Pin1 in cancer treatment until the recent identification of all-trans retinoic acid (ATRA) as a Pin1 inhibitor. Here we systematically investigate functions of Pin1 and its inhibitor ATRA in the development and treatment of HCC. Pin1 knockdown potently inhibited HCC cell proliferation and tumor growth in mice. ATRA-induced Pin1 degradation inhibited the growth of HCC cells, although at a higher IC50 as compared with breast cancer cells, likely due to more active ATRA metabolism in liver cells. Indeed, inhibition of ATRA metabolism enhanced the sensitivity of HCC cells to ATRA. Moreover, slow-releasing ATRA potently and dose-dependently inhibited HCC growth in mice. Finally, chemical or genetic Pin1 ablation blocked multiple cancer-driving pathways simultaneously in HCC cells. Thus, targeting Pin1 offers a promising therapeutic approach to simultaneously stop multiple cancer-driving pathways in HCC.
Hepatocellular carcinoma (HCC) is highly prevalent especially in Asia and sub-Saharan Africa, and has an increasing trend in previously low incidence areas such as the US and Europe1. Despite significant advance in understanding molecular mechanisms of HCC tumorigenesis, there is still lack of efficacious therapeutics. Cytotoxic chemotherapeutic agents show little improvement of overall survival2, while the only FDA-approved HCC targeted drug Sorafenib increases overall survival modestly by 2.8 months in advanced cancer patients3. As a result, the majority of HCC patients die within 9–12 months after diagnosis and HCC has moved up the rank from the third most common cause of cancer death to the second according to the recently released World Cancer Report 2014, even though it is the sixth most common cancer4.
Targeted drugs have changed cancer treatment, but are often ineffective against aggressive solid tumors due to the activation and feedback of multiple interactive and/or redundant cancer-driving pathways5,6,7,8,9. A central common signaling mechanism in cell proliferation and transformation is proline-directed phosphorylation that is regulated by many kinases and phosphatases, but further controlled by a unique proline isomerase Pin110,11,12. Pin1 is prevalently overexpressed or overactivated in many cancer types13, Pin1 promotes cancer and cancer stem cells tumorigenesis by turning on more than 40 oncogenes/growth-promoting factors and off more than 20 tumor suppresses/growth-inhibitory factors12. Interestingly, although Pin1 is important for cancer cells growth, it is dispensable for normal cell growth. Pin1−/− mice develop normally and do not exhibit obvious defects at young ages14. However, these Pin1 null mice are highly resistant to Ras, Neu/HER2 or mutant p53-induced breast cancer15,16, Myc-induced Burkitt’s lymphoma17 or Notch3-induced T-cell acute lymphoblastic leukemia18, demonstrating an essential role for Pin1 in tumorigenesis of many cancer types. Thus, targeting Pin1 represents a novel anticancer strategy to block multiple cancer pathways simultaneously without general toxic effects on normal tissues. However, because the available Pin1 inhibitors lacked the required specificity and/or potency, or cannot enter cells19,20,21, it was challenging to evaluate the significance of targeting Pin1 for cancer treatment until our discovery of all-trans retinoic acid (ATRA) as a Pin1 inhibitor22.
Use of ATRA for acute promyelocytic leukemia (APL) is considered the first example of modern targeted cancer therapy23,24,25,26,27, but how it causes driven fusion oncogene PML–RAR-α degradation and inhibition of self-renewal of leukemia stem cells had remained elusive28,29. Unexpectedly, ATRA was discovered as a Pin1 inhibitor from a high throughput screening. ATRA was proved to be a potent submicromolar Pin1 inhibitor that specifically binds to, inhibits and ultimately degrades the active Pin1 selectively without cross-reacting with other isomerase members22. ATRA also shows detectable but not striking anticancer activity against many solid tumors30,31,32, which is likely due to its light sensitivity and a short half-life of 45 minutes in humans33. However, using slow-releasing ATRA pellets to maintain constant blood drug levels, we have shown that ATRA exerts potent anticancer activity against both APL and aggressive triple negative breast cancer by inhibiting and ablating Pin1 and thereby turning off and on numerous oncogenes and tumor suppressors, respectively, at the same time22. These results indicate that Pin1 inhibitors have the unique and promising property to effectively block multiple cancer-driving pathways at once12,22.
The above property of Pin1 inhibitors are desirable for treating aggressive solid tumors such as HCC, because HCC caused by various etiologies has a high intertumor and intratumor heterogeneity, with multiple interactive and/or redundant cancer-driving pathways being activated simultaneously in the same patient34,35,36,37. Furthermore, Pin1 is overexpressed in about 70% of human HCC patients and it promotes heptocarcinogenesis in cell line PLC/PRF/538,39,40,41. Pin1 might play different roles in HCC cells depending on status of TP53 gene mutation42. Pin1 single nucleotide promoter and exon polymorphisms are associated with the susceptibility of HBV-related HCC43. Interestingly, Pin1 also physically interacts with and increases hepatitis B virus X-protein (HBx) protein stability to enhance hepatocarcinogenesis44. These results suggest that Pin1 plays an important role in hepatocarcinogenesis, functioning as novel anticancer target. However, the significance of targeting Pin1 for HCC treatment is not clear.
Here, we show that either chemical or genetic Pin1 inhibition potently suppresses HCC growth by blocking multiple cancer-driving pathways. Silencing Pin1 expression in multiple human HCC cell lines by a validated shRNA not only potently inhibited HCC cell proliferation and migration, but also suppressed HCC tumor growth in mice. ATRA induced Pin1 protein degradation in HCC cells, although at a higher IC50 as compared with breast cancer cells, possibly due to a high ATRA metabolic enzyme in liver cells. Indeed, inhibition of such enzyme significantly enhanced the sensitivity of HCC cells to ATRA. Moreover, chemical or genetic ablation of Pin1 blocked multiple cancer-driving pathways simultaneously. Finally, slow-releasing ATRA potently inhibited HCC growth in vivo without obvious side effects. These results demonstrate that Pin1 is a promising therapeutic target in HCC and provide a further rationale for developing longer half-life ATRA or more potent and specific Pin1-targeted ATRA variants for treating HCC and other cancers.
Results
Pin1 knockdown suppresses cell proliferation in various human HCC cell lines
Pin1 has been shown to be overexpressed in about 70% human HCC38, but its therapeutic potential in treating HCC is still not clear. To determine the importance of Pin1 in HCC, we first examined the effects of Pin1 loss of function by genetically suppressing Pin1 expression in various human HCC cell lines using lentiviruses expressing a validated Pin1 shRNA45. PLC/PRF/5 (HBV-positive) (Fig. 1A,B,E), Huh-7 (HBV-negative) (Fig. 1C,D,F) and several other HCC cells (including Hep3B and Sk-Hep-1, data not shown) became unhealthy after infection with lentiviruses expressing Pin1 shRNA, as compared with scrambled shRNA. PLC/PRF/5 cells growth was significantly retarded, as illustrated by growth curves over time (Fig. 1B). Huh-7 cells showed similar dependency on Pin1 but to a less extent (Fig. 1D). After Pin1 knockdown, overall cell morphology of both PLC/PRF/5 and Huh-7 cells were dramatically changed, with loss of protruding pseudopodia (Fig. 1E,F). Consistent with these morphological changes are that migration of Pin1 knockdown HCC cells was significantly suppressed, as shown in transwell assay (Fig. 1G,H). Since lack of Pin1 has little effect on normal tissues and cells, as demonstrated in Pin1−/− mice and MEFs14, these results indicate that human HCC cells likely develop addiction to Pin1, demonstrating a pivotal role for Pin1 in HCC cells in vitro.
Figure 1. Pin1 knockdown suppresses the proliferation of human HCC cells in vitro.

PLC/PRF/5 (A) or Huh-7 (C) cell lines were infected with lentiviruses expressing scrambled or Pin1 shRNA. Cell lysates were subject to western blot analysis with antibodies against Pin1 or internal control Actin. Growth curves of PLC/PRF/5 (B) and Huh-7 (D) cell lines with Pin1 knockdown were plotted over time, based on the cell numbers counted daily. 3 × 104 cells for both cell lines were placed in each well of 12-well plate with triplicate for each time point. Error bars represent standard deviations (N = 3). p values were derived from the cell numbers at the end point. (E,F) Morphological change of PLC/PRF/5 (E) and Huh-7 (F) cells after Pin1 knockdown. (G,H) Pin1 knockdown affects cells migration in PLC/PRF/5 (G) and Huh-7 (H), as evaluated by transwell assay. 5 × 104 cells for both cell lines were used for this assay. Error bars represent standard deviations (n = 2).
Pin1 knockdown suppresses HCC growth in vivo
To examine whether Pin1 affects HCC tumorigenesis in vivo, we performed xenograft tumor growth assay in nude mice. After expansion in vitro (<6 passages after lentiviral infection), 2 × 106 Huh-7 cells expressing Pin1 shRNA or scrambled shRNA were subcutaneously injected into the left and right flanks of the same mice, respectively. Tumor growth curves were established over time by measuring the tumor size every week. When tumor diameter reached 1.5 cm, all of tumors were dissected from the nude mice, photographed and weighed. The tumor growth was significantly suppressed after Pin1 knockdown, as illustrated by the tumor growth curve (Fig. 2A) and tumor weight (Fig. 2B,C) n = 9, P = 0.035). Thus, Pin1 represents a promising therapeutic target in HCC.
Figure 2. Pin1 knockdown suppresses tumor growth of human HCC cells in vivo.
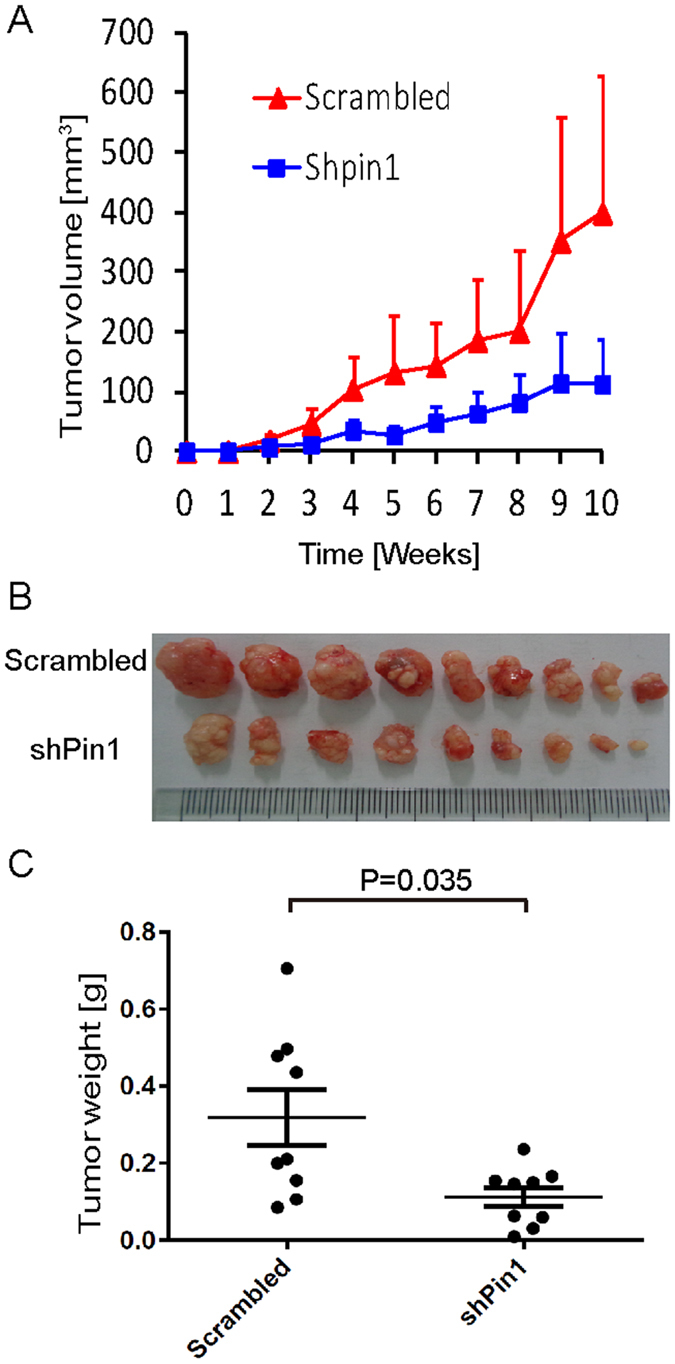
2 × 105 Huh-7 cells infected with lentiviruses expressing scrambled or Pin1 shRNA were inoculated subcutaneously into nude mice. (A) Huh-7 tumor volumes were measured weekly and the tumor growth curves were plotted over time. Error bars represent standard deviations. (B) Photographic illustration of 9 pairs of tumors harvested from nude mice at the end point (10 weeks). Each scale of the ruler represents 1 mm. (C) Weights of tumors harvested from nude mice at the end point. Error bar represents SEM (n = 9).
ATRA promotes Pin1 degradation in HCC cell lines
Since shRNA could have off-target effects and that it is still very challenging to deliver shRNA to tumors for cancer therapy, we used a small molecular Pin1 inhibitor, ATRA, which has been identified through a mechanism-based screening from a compound library and found to bind to the Pin1 active site and thereby induce degradation of active Pin122. In PLC/PRF/5 HCC cells, ATRA began to induce Pin1 degradation at 5 μM (Fig. 3A), a little higher in comparison with ~1 μM in the breast cancer and APL cells22. However, in Huh-7 HCC cells, Pin1 was only degraded efficiently at 50 μM, a much higher dose in comparison with the breast cancer and APL cells (Fig. 3B). Since ATRA is known to be metabolized in the liver by enzymes including cytochrome P45046, lower sensitivity of Huh-7 HCC cells to ATRA might be due to high activity of ATRA metabolism. To examine this possibility, we used liarozole that inhibits the cytochrome P450-dependent 4-hydroxylation of retinoic acid, thereby reducing ATRA drug metabolism46. After Huh-7 HCC cells were treated with ATRA or liarozole or their combination for various times, Pin1 degradation was assayed. Although liarozole alone did not significantly induced Pin1 degradation and might even slightly elevate Pin1 protein levels (Fig. 3C), its combination with ATRA potently increased the ability of ATRA to induce Pin1 degradation (Fig. 3D,E). When the concentration of ATRA was fixed at 10 μM, Pin1 was degraded in a dose dependent manner depending on increasing concentrations of liarozole (Fig. 3D), with IC50 ~24.5 μM. In the other hand, when the concentration of liarozole was fixed at 25 μM, Pin1 was degraded in a dose dependent manner depending on increasing concentrations of ATRA (Fig. 3E), with IC50 ~7.3 μM. These results indicate that liarozole functions on Pin1 through stabilizing ATRA and further support the specificity of Pin1 degradation induced by ATRA. Taking together, these results demonstrate that ATRA targets Pin1 in HCC, as in APL and breast cancer22.
Figure 3. The Pin1 inhibitor ATRA induces Pin1 degradation in HCC cell lines.

PLC/PRF/5 or Huh-7 cells were treated with chemicals for 72 hours, followed by subjecting cell lysates to immunoblotting with Pin1 antibody. (A) PLC/PRF/5 cells were treated with different doses of ATRA. (B,C) Huh-7 cells were treated with different doses of ATRA (B) or liarozole (C) alone. (D) Huh-7 cells were treated with fixed 10 μM ATRA combined with different doses of liarozole. (E) Huh-7 cells were treated with fixed 25 μM liarozole combined with different doses of ATRA. The graphics were derived from quantification of Pin1 relative intensity normalized with Actin intensity. The error bars represent standard deviations from two to four independent blots. *p < 0.05 vs control.
ATRA inhibits HCC cells growth in vitro
The above results indicate that Pin1 genetic knockdown inhibits HCC cells growth and that ATRA inhibits and degrades Pin1 in HCC cells. We then asked whether ATRA would mimic Pin1 genetic inhibition to suppress HCC cell growth. ATRA alone effectively suppressed the cell growth in PLC/PRF/5 cells (P = 0.002), and also showed inhibitory activity in Huh-7 cells (P = 0.005) (Fig. 4A). We then used liarozole to inhibit ATRA metabolism in Huh-7 cells, as described above. Although liarozole alone had some inhibitory effects (P = 0.014), its combination with ATRA further increased the ability of ATRA to inhibit cell proliferation of Huh-7 cells (P = 0.003) (Fig. 4B). Similarly, we examined ATRA effects on HCC cells migration. ATRA alone suppressed cells transwell both in PLC/PRF/5 and Huh-7 cell lines (Fig. 4C,D). A combination of ATRA with liarozole further increased the inhibitory effects on Huh-7 cells migration, which is consistent with the results that liarozole helps ATRA to induce Pin1 degradation in Huh-7 cells (Fig. 4D). These results show that the Pin1 inhibitor ATRA inhibits HCC cell growth and further support that Pin1 concentration is critical for HCC cell growth and migration.
Figure 4. ATRA suppresses HCC cell growth and migration in vitro.

(A,B) PLC/PRF/5 (A) or Huh-7 (B) cells were treated with 10 μM ATRA and/or 25 μM P450 inhibitor liarozole and the growth curves were plotted over time. 5 × 104 of PLC/PRF/5 cells or 3 × 104 of Huh-7 were placed in each well of 12-well plate with triplicate for each time point. Error bars represent standard deviations (n = 3). p values were derived from the cell numbers at the end point. (C,D) PLC/PRF/5 (C) or Huh-7 (D) cells were treated with ATRA and/or P450 inhibitor liarozole, as evaluated by transwell assay. 5 × 104 cells for both cell lines were used for this assay. Error bars represent standard deviations (n = 2).
Genetic or chemical inhibition of Pin1 blocks multiple cancer-driving pathways simultaneously in HCC
Multiple cancer-driving pathways can be activated in the highly heterogeneous HCC34,35,36,37. As a major regulator of oncogenesis, Pin1 activates more than 40 tumor-promoting regulators and inhibits more than 20 tumor-suppressive regulators12. Since Pin1 might regulate different cancer substrates in different cancer types12, we asked whether inhibition of Pin1 by genetic or chemical ablation would affect protein levels of a selected set of Pin1 substrates or downstream factors, whose protein stability and abundance have been shown to be regulated by Pin1. We found that many growth promoting factors such as CyclinD147, CDK6, pAKT48, c-Jun49, B-Raf, Notch intracellular domain NICD50, and β-catenin51 were significant decreased after Pin1 knockdown, both in Huh-7 and PLC/PRF/5 cells (Fig. 5A). Interestingly, LC3B, an autophage marker, and detoxification enzyme GSTP1 were also significantly changed (Fig. 5A). More importantly, a similar inhibition of multiple Pin1 downstream cancer-driving pathways was also found after chemical inhibition of Pin1 by its inhibitor ATRA (Fig. 5B). Thus, inhibition of Pin1 by chemical inhibitors or genetic knockdown has a unique property to inhibit multiple cancer-driving pathways at the same time in the complex cancer type HCC.
Figure 5. Both genetic and chemical Pin1 inhibition blocks multiple cancer-driving pathways simultaneously in human HCC cells.

PLC/PRF/5 or Huh-7 cell lines were infected with lentivirus expressing scrambled or Pin1 shRNA (A), or treated with 10 μM ATRA (PLC/PRF/5) or 10 μM ATRA combined with 25 μM liarozole (Huh-7) (B). Cell lysates were subjected to Western blot analysis with specific antibodies.
Slow-releasing Pin1 inhibitor ATRA exerts potent anticancer activity against HCC in vivo
Given the obvious effects of ATRA on Pin1 protein levels, multiple cancer-driving pathways and cell growth in HCC in vitro, a critical question is whether ATRA suppresses HCC tumor growth in vivo. We thus examined ATRA in mouse models xenografted with HBV-positive PLC/PRF/5 and HBV-negative Huh-7 cells. ATRA is extremely light-sensitive and can be metabolized quickly in the liver, with a half life of 45 minutes in humans33,52,53. To improve drug activity against HCC, we implanted slow-releasing ATRA pellets, which maintain a constant drug level in the blood, as described previously22. 2 × 105 PLC/PRF/5 together with matrigel or 2 × 106 Huh-7 cells were injected subcutaneously onto both flanks of nude mice to generate tumors. When tumor volumes reached 60 mm3 in PLC/PRF/5 xenograft model or after 3 weeks in Huh-7 model, mice were randomly grouped to receive 5 or 10 mg 21-day slow-releasing ATRA pellets or placebo pills implanted under the skin in the back of the neck of each mouse, followed by measuring tumor growth every week. The tumor growth was potently suppressed by ATRA treatment in a dose dependent manner in PLC/PRF/5 model. As revealed by tumor growth curves or final tumor weights, while 5 mg slightly inhibited HCC tumor growth (P = 0.384), 10 mg ATRA completely blocked the tumor growth (P = 0.004) (Fig. 6A,C,E). Similar results were obtained in Huh-7 tumor model, with tumor inhibitory effects in 5 mg ATRA treatment group (P = 0.181) and more potent in 10 mg treatment group (P = 0.020) (Fig. 6B,D,F). The body weight of adult mice didn’t change during drug treatment, indicating a low cytotoxicity of ATRA at therapeutic dosage (data not shown). As expected, Pin1 protein was also downregulated in xenograft tumors from nude mice treated with slow-releasing ATRA (Fig. 6G). These results demonstrate for the first time that ATRA has potent anti-tumor activity against HCC through targeting Pin1.
Figure 6. Slow-releasing Pin1 inhibitor ATRA potently inhibits HCC growth in mice.

2 × 105 PLC/PRF/5 cells together with matrigel or 2 × 106 Huh-7 cells were inoculated subcutaneously into nude mice. When tumors were obvious (tumor size 30–70 mm3), mice were randomly grouped to receive 5 or 10 mg 21-day slow-releasing ATRA or placebo pellets embedded under the neck skin. (A,B) PLC/PRF/5 (A) or Huh-7 (B) tumor volumes were measured weekly and the tumor growth curves were plotted over time. Error bars represent standard deviations. (C,D) Photographic illustration of PLC/PRF/5 (C) or Huh-7 (D) tumor nodules harvested from nude mice at the end point. Each scale of the ruler represents 1 mm. (E,F) Weights of Huh-7 (E) or PLC/PRF/5 (F) tumors harvested from nude mice at the end point. Error bars represent SEM. (G) Immunoblots of Pin1 expressed in xenograft tumors from nude mice inoculated with 4 × 106 HuH7 cells and treated with placebo or 5 mg 21-day ATRA slow-releasing pellet for 3 weeks.
Discussion
We systematically evaluated the importance of Pin1 in HCC growth using genetic Pin1 knockdown and chemical Pin1 inhibition. Pin1 knockdown significantly inhibits both cells proliferation and tumorigenesis in various HCC cell lines in vitro and in vivo. Slow-releasing Pin1 inhibitor ATRA exhibits very potent inhibitory effects on HCC growth both in vitro and in vivo. Mechanistically, both genetic and chemical inhibition of Pin1 downregulates multiple cancer-driving pathways. Thus, Pin1 represents a novel promising therapeutic target for treating highly heterogeneous cancer HCC.
As the organ of metabolism and detoxifying, liver is exposed to many chemicals including carcinogens. HCC is a highly heterogeneous tumor in which cancer cells are able to use redundant pathways to escape from specific target/pathway blocking by drug treatment34,35,36,37. Although HCC is one of the most prevalent and malignant cancers, there are not many options for patients to choose against this disease54. As the only approved molecular therapeutic drug, Sorafenib only extends overall survival of patients by 2.8 months3. There is an urgent need to find novel efficacious therapeutics to control this disease.
As a global regulator of phosphor-proteins with Ser/Thr-Pro motif, Pin1 regulates multiple cancer-driving pathways in various HCC cell lines, making Pin1 as an ideal target for treating this heterogeneous cancer. Pin1 inhibitor ATRA is used in clinical to treat APL, but shows detectable but not striking anticancer activity against solid tumors30,31,32, very likely due to its light sensitivity and short half life of 45 min in humans33,52,53. By improving its stability in packaged slow-releasing pellet, we have shown for the first time a very potent anticancer activity of ATRA against HCC. As an acid form of vitamin A, ATRA only exhibits minor side effects on animals at experimental dose. In contrast, we noticed more severe side effects of Sorafenib in a side by side comparison experiment (unpublished data). Thus ATRA has unique advantages over current drugs, with characteristics of targeting multiple cancer-driving pathways and low side effects.
ATRA mimics the pSer/Thr-Pro motif in Pin1 substrates and its carboxylic and aromatic moieties can bind to the substrate phosphate- and proline-binding pockets of the Pin1 active site, respectively, thereby inhibit Pin1 catalytic activity and somehow induce Pin1 degradation22. Potentially due to high metabolism of ATRA in Huh-7 cells, ATRA alone could not induce Pin1 degradation efficiently in vitro. By blocking ATRA metabolism pathway, Cytochrome P450 inhibitor liarozole significantly enhances ATRA activity of inducing Pin1 degradation in vitro. This indicates that liarozole and ATRA combination might have additive effects on HCC therapy in vivo.
ATRA is light sensitive and unstable in vivo, which significantly decreases its activity against solid tumors. Although regular ATRA has moderate but detectable efficacy against solid tumors in some clinical trials, new generations of supposedly much more potent retinoid derivatives targeting their receptors RARs or RXRs show little efficacy30,31,55,56,57, which is likely due to the failure of these retinoids to inhibit Pin122. Structural modification of ATRA molecule based on the Pin1-ATRA co-crystal structure22 with improved stability needs to be developed to increase ATRA anticancer activity.
We have shown a potent anticancer activity when using slow-releasing ATRA pellets to treat mice bearing HCC tumors. In contrast to free ATRA, these pellets are able to maintain ATRA serum concentrations in mice constant at the concentrations for Pin1 binding and inhibition as described previously22. We have packaged ATRA in slow-releasing microparticles, which alone shows more potent inhibitory activity both on Pin1 degradation in vitro and HCC tumor growth in vivo (data unpublished), indicating that developing novel slow-releasing method applicable to patients is worth further exploring. In addition, a combination of ATRA with current clinical drugs, such as sorafenib, paclitaxel, 5-Fu might have additive/synergistic therapeutic effects and represents a promising strategy for treating HCC.
In summary, our results have shown for the fist time that targeting Pin1 offers a promising therapeutic approach to simultaneously stop multiple cancer-driving pathways in HCC, further providing a rationale for developing longer half-life ATRA or more potent and specific Pin1-targeted ATRA derivatives to overcome drug resistance in treating aggressive cancers such as HCC.
Materials and Methods
Cell lines, antibodies and animals
Cell lines Huh-7 and PLC/PRF/5 were bought from Cell Bank of Chinese Academy of Sciences. Cells were maintained in DMEM media (#12800017; GIBCO) supplemented with 10% fetal bovine serum (#10437-028; GIBCO) and 1.5 g/L of NaHCO3. The protein samples were blotted with following antibodies: rabbit anti-Cyclin D1 (#2978 S; Cell Signaling Technology) 1:1000, mouse anti-CDK6 (#3136 S; Cell Signaling Technology) 1:1000, rabbit anti-Phospho-Akt(Thr308) (#9275 S; Cell Signaling Technology) 1:600, rabbit anti-Phospho-Akt(Ser473) (#9271 S; Cell Signaling Technology) 1:600, rabbit anti-c-Jun (60A8) (#9165 S; Cell Signaling Technology) 1:1000, rabbit anti-B-Raf (#sc-166; Santa Cruz Biotechnology) 1;400, rabbit anti-Cleaved Notch1 (Val1744) (#4147 S; Cell Signaling technology)1:300, rabbit anti-β-Catenin (#8480 S; Cell Signaling technology) 1:1000, rabbit anti-LC3B (#ab48394; abcam) 1:1000, mouse anti-GSTP1 (#3369 S; Cell Signaling technology) 1:1000, mouse anti-Actin (#HC201; TransGen Biotech) 1:3000. BALB/c nude mice were housed in laminar flow cabinets with free access to food and water in Laboratory Animal Center of Fujian Medical University. All of animal experiments were performed in accordance with the animal protocols and regulations approved by FJMU Experimental Animal Ethics Committee of Fujian Medical University.
Tumor implantation and drug treatment
5 × 105 PLC/PRF/5 cells together with matrigel (#356231; BD) or 2 × 106 Huh-7 cells were inoculated subcutaneously onto the left and right flank region of ~5-week old nude mice. Tumor volume was measured every week and calculated by the formula: length × width2/2. For Pin1 knockdown experiments, cells were infected by lentivirus expressing scrambled or Pin1 shRNA, selected by drug puromycin, expanded and inoculated onto nude mice at early passage (<6 passages).
For ATRA treatment, ATRA slow-releasing pellet (#V-111; Innovative Research of America) was embedded under the neck skin of mice 3 weeks after tumor cells inoculation (Huh-7) or when tumor volume reaches 60 mm3 (PLC/PRF/5) using Gauge Precision Trochar. It is normal that minor inflammation occurs around the drug pellet. It should be noted that anesthetic drug chloral hydrate together with ATRA pellet evokes very severe inflammation, which would complicate the results. Instead, anesthetic drug ketamine has no this problem.
Transwell assay
Hanging cell culture insert with pore size of 8.0 μm (#3442, Corning) were coated with 0.6 μg of collagen (#C4243; Sigma-Aldrich) and placed onto 24-well plate. 200 μl of 5 × 104 HCC cells starved O/N in media with 1% FBS were placed in the upper chamber and 500 ml of media with 10% PBS were placed in the lower chamber. The plate was placed in incubator to allow cell migration for 36 hrs. The remained cells in the upper chamber were removed using cotton swab and the inserts were fixed by methanol followed by staining with 0.1% crystal violet for 20 min. Migrated cells adhering to the underside of the inserts were photographed for 10–20 fields and counted under a light microscope at x200 magnification. For drug treatment experiments, cells were pretreated with ATRA, liarozole or combination of ATRA and liarozole for 24 hrs and were incubated with drugs during the whole transwell process.
Additional Information
How to cite this article: Liao, X.-H. et al. Chemical or genetic Pin1 inhibition exerts potent anticancer activity against hepatocellular carcinoma by blocking multiple cancer-driving pathways. Sci. Rep. 7, 43639; doi: 10.1038/srep43639 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. U1205024), National Natural Science Foundation of China (No. 81573029), the Natural Science Foundation of Fujian Province (No. 2015J01312), the Collaborative Innovation Center for Stem Cells Translational Medicine (Fujian 2011 Program), the Fujian Province Natural Science Fund for Distinguished Young Scientist (No. 2015J06017) and National Institutes of Health grant R01CA167677.
Footnotes
Dr. Lu and Dr. Zhou are inventors of Pin1 technology, which was licensed by BIDMC to Pinteon Therapeutics. Both Dr. Lu and Dr. Zhou own equity in, and consult for, Pinteon. Their interests were reviewed and managed by BIDMC in accordance with its conflict of interest policy.
Author Contributions X.-H.L. developed the concepts and approaches, performed the experiments, analyzed the data and prepared the manuscript prior to submission. A.L.Z., M.Z., M.-Q.L., C.P.C., H.X., Q.-S.C., D.Y. and W.L. performed the experiments and analyzed the data. T.-F.T., H.L. helped to analyze the data. X.Z. and K.P.L. developed the concepts, prepared and revised the manuscript prior to submission. All authors approve the final manuscript.
References
- Mittal S. & El-Serag H. B. Epidemiology of hepatocellular carcinoma: consider the population. Journal of clinical gastroenterology 47 Suppl, S2–6, doi: 10.1097/MCG.0b013e3182872f29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo W. et al. A randomized phase III study of doxorubicin versus cisplatin/interferon alpha-2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy for unresectable hepatocellular carcinoma. Journal of the National Cancer Institute 97, 1532–1538, doi: 10.1093/jnci/dji315 (2005). [DOI] [PubMed] [Google Scholar]
- Llovet J. M. et al. Sorafenib in advanced hepatocellular carcinoma. The New England journal of medicine 359, 378–390, doi: 10.1056/NEJMoa0708857 (2008). [DOI] [PubMed] [Google Scholar]
- Stewart B. W. & Wild C. P. World Cancer Report 2014 (IARC Nonserial Publication) (2014). [Google Scholar]
- Mauro M. J., O’Dwyer M., Heinrich M. C. & Druker B. J. STI571: a paradigm of new agents for cancer therapeutics. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 20, 325–334 (2002). [DOI] [PubMed] [Google Scholar]
- Luo J., Solimini N. L. & Elledge S. J. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136, 823–837 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal S. Targeted cancer therapies. Nat Rev Drug Discov 9, 427–428 (2010). [DOI] [PubMed] [Google Scholar]
- Hanahan D. & Weinberg R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674, doi: 10.1016/j.cell.2011.02.013 (2011). [DOI] [PubMed] [Google Scholar]
- Sjoblom T. et al. The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274, doi: 10.1126/science.1133427 (2006). [DOI] [PubMed] [Google Scholar]
- Liou Y. C., Zhou X. Z. & Lu K. P. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends in biochemical sciences 36, 501–514, doi: 10.1016/j.tibs.2011.07.001 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K. P. & Zhou X. Z. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nature reviews. Molecular cell biology 8, 904–916, doi: 10.1038/nrm2261 (2007). [DOI] [PubMed] [Google Scholar]
- Zhou X. Z. & Lu K. P. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nature reviews. Cancer 16, 463–478, doi: 10.1038/nrc.2016.49 (2016). [DOI] [PubMed] [Google Scholar]
- Bao L. et al. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am J Pathol 164, 1727–1737, doi: 10.1016/S0002-9440(10)63731-5 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimori F., Takahashi K., Uchida C. & Uchida T. Mice lacking Pin1 develop normally, but are defective in entering cell cycle from G(0) arrest. Biochemical and biophysical research communications 265, 658–663, doi: 10.1006/bbrc.1999.1736 (1999). [DOI] [PubMed] [Google Scholar]
- Wulf G., Garg P., Liou Y. C., Iglehart D. & Lu K. P. Modeling breast cancer in vivo and ex vivo reveals an essential role of Pin1 in tumorigenesis. The EMBO journal 23, 3397–3407, doi: 10.1038/sj.emboj.7600323 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardini J. E. et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 20, 79–91, doi: 10.1016/j.ccr.2011.06.004 (2011). [DOI] [PubMed] [Google Scholar]
- D’Artista L. et al. Pin1 is required for sustained B cell proliferation upon oncogenic activation of Myc. Oncotarget, doi: 10.18632/oncotarget.7846 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franciosa G. et al. Prolyl-isomerase Pin1 controls Notch3 protein expression and regulates T-ALL progression. Oncogene, doi: 10.1038/onc.2016.5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore J. D. & Potter A. Pin1 inhibitors: Pitfalls, progress and cellular pharmacology. Bioorganic & medicinal chemistry letters 23, 4283–4291, doi: 10.1016/j.bmcl.2013.05.088 (2013). [DOI] [PubMed] [Google Scholar]
- Guo C. et al. Structure-based design of novel human Pin1 inhibitors (I). Bioorganic & medicinal chemistry letters 19, 5613–5616, doi: 10.1016/j.bmcl.2009.08.034 (2009). [DOI] [PubMed] [Google Scholar]
- Hennig L. et al. Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry 37, 5953–5960. (1998). [DOI] [PubMed] [Google Scholar]
- Wei S. et al. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nature medicine 21, 457–466, doi: 10.1038/nm.3839 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J. et al. Treatment of acute promyelocytic leukemia with ATRA and As2O3: a model of molecular target-based cancer therapy. Cancer biology & therapy 1, 614–620 (2002). [DOI] [PubMed] [Google Scholar]
- Huang M. E. et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 72, 567–572 (1988). [PubMed] [Google Scholar]
- de The H. & Chen Z. Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nature reviews. Cancer 10, 775–783 (2010). [DOI] [PubMed] [Google Scholar]
- Sanz M. A. & Lo-Coco F. Modern approaches to treating acute promyelocytic leukemia. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 29, 495–503 (2011). [DOI] [PubMed] [Google Scholar]
- Degos L. & Wang Z. Y. All trans retinoic acid in acute promyelocytic leukemia. Oncogene 20, 7140–7145, doi: 10.1038/sj.onc.1204763 (2001). [DOI] [PubMed] [Google Scholar]
- Nasr R. et al. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nature medicine 14, 1333–1342 (2008). [DOI] [PubMed] [Google Scholar]
- Ablain J. et al. Uncoupling RARA transcriptional activation and degradation clarifies the bases for APL response to therapies. J Exp Med 210, 647–653 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrieta O. et al. Randomized phase II trial of All-trans-retinoic acid with chemotherapy based on paclitaxel and cisplatin as first-line treatment in patients with advanced non-small-cell lung cancer. J Clin Oncol 28, 3463–3471 (2010). [DOI] [PubMed] [Google Scholar]
- Budd G. T. et al. Phase I/II trial of all-trans retinoic acid and tamoxifen in patients with advanced breast cancer. Clin Cancer Res 4, 635–642 (1998). [PubMed] [Google Scholar]
- Bryan M. et al. A pilot phase II trial of all-trans retinoic acid (Vesanoid) and paclitaxel (Taxol) in patients with recurrent or metastatic breast cancer. Investigational new drugs 29, 1482–1487, doi: 10.1007/s10637-010-9478-3 (2011). [DOI] [PubMed] [Google Scholar]
- Muindi J. et al. Continuous treatment with all-trans retinoic acid causes a progressive reduction in plasma drug concentrations: implications for relapse and retinoid “resistance” in patients with acute promyelocytic leukemia. Blood 79, 299–303 (1992). [PubMed] [Google Scholar]
- Marusyk A., Almendro V. & Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nature reviews. Cancer 12, 323–334, doi: 10.1038/nrc3261 (2012). [DOI] [PubMed] [Google Scholar]
- Lu L. C., Hsu C. H., Hsu C. & Cheng A. L. Tumor Heterogeneity in Hepatocellular Carcinoma: Facing the Challenges. Liver cancer 5, 128–138, doi: 10.1159/000367754 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nault J. C. & Villanueva A. Intratumor molecular and phenotypic diversity in hepatocellular carcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research 21, 1786–1788, doi: 10.1158/1078-0432.CCR-14-2602 (2015). [DOI] [PubMed] [Google Scholar]
- Shi J. Y. et al. Inferring the progression of multifocal liver cancer from spatial and temporal genomic heterogeneity. Oncotarget 7, 2867–2877, doi: 10.18632/oncotarget.6558 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang R. et al. PIN1 overexpression and beta-catenin gene mutations are distinct oncogenic events in human hepatocellular carcinoma. Oncogene 23, 4182–4186, doi: 10.1038/sj.onc.1207493 (2004). [DOI] [PubMed] [Google Scholar]
- Pang R. W. et al. PIN1 expression contributes to hepatic carcinogenesis. The Journal of pathology 210, 19–25, doi: 10.1002/path.2024 (2006). [DOI] [PubMed] [Google Scholar]
- Ao R., Zhang D. R., Du Y. Q. & Wang Y. Expression and significance of Pin1, beta-catenin and cyclin D1 in hepatocellular carcinoma. Molecular medicine reports 10, 1893–1898, doi: 10.3892/mmr.2014.2456 (2014). [DOI] [PubMed] [Google Scholar]
- Wang H. et al. PIN1 gene overexpression and beta-catenin gene mutation/expression in hepatocellular carcinoma and their significance. Journal of Huazhong University of Science and Technology. Medical sciences = Hua zhong ke ji da xue xue bao. Yi xue Ying De wen ban = Huazhong keji daxue xuebao. Yixue Yingdewen ban 27, 54–57, doi: 10.1007/s11596-007-0116-z (2007). [DOI] [PubMed] [Google Scholar]
- Bae J. S. et al. PIN1 in hepatocellular carcinoma is associated with TP53 gene status. Oncology reports 36, 2405–2411, doi: 10.3892/or.2016.5001 (2016). [DOI] [PubMed] [Google Scholar]
- Huang L. et al. PIN1 genetic polymorphisms and the susceptibility of HBV-related hepatocellular carcinoma in a Guangxi population. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine 37, 6599–6606, doi: 10.1007/s13277-015-4539-z (2016). [DOI] [PubMed] [Google Scholar]
- Pang R. et al. Pin1 interacts with a specific serine-proline motif of hepatitis B virus X-protein to enhance hepatocarcinogenesis. Gastroenterology 132, 1088–1103, doi: 10.1053/j.gastro.2006.12.030 (2007). [DOI] [PubMed] [Google Scholar]
- Luo M. L. et al. Prolyl isomerase Pin1 acts downstream of miR200c to promote cancer stem-like cell traits in breast cancer. Cancer research 74, 3603–3616, doi: 10.1158/0008-5472.CAN-13-2785 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wauwe J. et al. Liarozole, an inhibitor of retinoic acid metabolism, exerts retinoid-mimetic effects in vivo. The Journal of pharmacology and experimental therapeutics 261, 773–779 (1992). [PubMed] [Google Scholar]
- Liou Y. C. et al. Loss of Pin1 function in the mouse causes phenotypes resembling cyclin D1-null phenotypes. Proc. Natl. Acad. Sci. USA 99, 1335–1340 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y. et al. Peptidyl-prolyl cis/trans isomerase Pin1 is critical for the regulation of PKB/Akt stability and activation phosphorylation. Oncogene 28, 2436–2445, doi: 10.1038/onc.2009.98 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulf G. M. et al. Pin1 is overexpressed in breast cancer and potentiates the transcriptional activity of phosphorylated c-Jun towards the cyclin D1 gene. EMBO J. 20, 3459–3472 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustighi A. et al. The prolyl-isomerase Pin1 is a Notch1 target that enhances Notch1 activation in cancer. Nature cell biology 11, 133–142, doi: 10.1038/ncb1822 (2009). [DOI] [PubMed] [Google Scholar]
- Ryo A., Nakamura M., Wulf G., Liou Y. C. & Lu K. P. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nature cell biology 3, 793–801, doi: 10.1038/ncb0901-793 (2001). [DOI] [PubMed] [Google Scholar]
- Smith M. A. et al. Phase I and pharmacokinetic evaluation of all-trans-retinoic acid in pediatric patients with cancer. J Clin Oncol 10, 1666–1673 (1992). [DOI] [PubMed] [Google Scholar]
- Lefebvre P. et al. Pharmacokinetics of oral all-trans retinoic acid in patients with acute promyelocytic leukemia. Leukemia 5, 1054–1058 (1991). [PubMed] [Google Scholar]
- Villanueva A. & Llovet J. M. Liver cancer in 2013: Mutational landscape of HCC--the end of the beginning. Nature reviews. Clinical oncology 11, 73–74, doi: 10.1038/nrclinonc.2013.243 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramlau R. et al. Randomized phase III trial comparing bexarotene (L1069-49)/cisplatin/vinorelbine with cisplatin/vinorelbine in chemotherapy-naive patients with advanced or metastatic non-small-cell lung cancer: SPIRIT I. J Clin Oncol 26, 1886–1892, doi: 10.1200/JCO.2007.12.2614 (2008). [DOI] [PubMed] [Google Scholar]
- Decensi A. et al. Randomized double-blind 2 × 2 trial of low-dose tamoxifen and fenretinide for breast cancer prevention in high-risk premenopausal women. J Clin Oncol 27, 3749–3756, doi: 10.1200/JCO.2008.19.3797 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly R. M., Nguyen N. K. & Sukumar S. Molecular pathways: current role and future directions of the retinoic acid pathway in cancer prevention and treatment. Clin Cancer Res 19, 1651–1659 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]