

Abstract
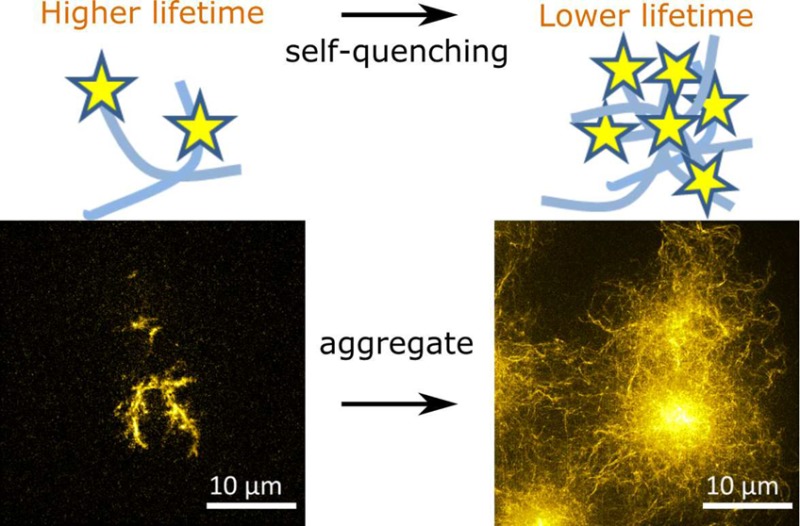
The characterization of the aggregation kinetics of protein amyloids and the structural properties of the ensuing aggregates are vital in the study of the pathogenesis of many neurodegenerative diseases and the discovery of therapeutic targets. In this article, we show that the fluorescence lifetime of synthetic dyes covalently attached to amyloid proteins informs on the structural properties of amyloid clusters formed both in vitro and in cells. We demonstrate that the mechanism behind such a “lifetime sensor” of protein aggregation is based on fluorescence self-quenching and that it offers a good dynamic range to report on various stages of aggregation without significantly perturbing the process under investigation. We show that the sensor informs on the structural density of amyloid clusters in a high-throughput and quantitative manner and in these aspects the sensor outperforms super-resolution imaging techniques. We demonstrate the power and speed of the method, offering capabilities, for example, in therapeutic screenings that monitor biological self-assembly. We investigate the mechanism and advantages of the lifetime sensor in studies of the K18 protein fragment of the Alzheimer’s disease related protein tau and its amyloid aggregates formed in vitro. Finally, we demonstrate the sensor in the study of aggregates of polyglutamine protein, a model used in studies related to Huntington’s disease, by performing correlative fluorescence lifetime imaging microscopy and structured-illumination microscopy experiments in cells.
Keywords: Self-quenching, FLIM, SIM, super-resolution, amyloid aggregation
Protein misfolding and aggregation has been linked to many neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD). A capability of monitoring amyloid protein aggregation both in vitro and in cells is vital for gaining further insights into the pathogenesis of, and to discover therapeutic strategies for, such disorders.1−6 Previous work has suggested that amyloid clusters formed under different environmental conditions are morphologically distinct and that the structural properties of aggregates are linked to disease pathology.7−12 A structural characterization of amyloid clusters is thus an important topic of research in the field. Various approaches have been employed to reveal the morphology of individual amyloid fibrils or clusters directly, for instance, electron microscopy (EM),2,5,8,12,13 atomic-force microscopy (AFM),11,12,14−16 and super-resolution fluorescence microscopy.17−22 However, such methods are slow to perform and can require elaborate sample preparation protocols, making them impractical to perform for screening applications or the analysis of large data sets. There are also limitations with these techniques in their applicability for studies in biological systems. For these reasons, a high-throughput method, which offers structural information on amyloid clusters and that is suitable for studies of protein aggregation both in vitro and in cells, is highly desirable.
Fluorescence imaging of proteins labeled covalently with synthetic organic fluorophores has proven to be a powerful method to study amyloid aggregation both in vitro and in cells.7,14,18,23,24 It indicates amyloid formation either by direct observation of the morphology of amyloid deposits via super-resolution microscopy or indirectly via changes in spectroscopic properties, such as the fluorescence intensity or lifetime. The fluorescence lifetime is particularly important in this respect, as it is an inherently ratiometric technique and thus less prone to concentration and intensity artifacts.25,26 Fluorescence lifetime imaging microscopy (FLIM) has previously been used on covalently labeled amyloid proteins to report on amyloid aggregation.7,18,23,24 One of these methods requires only a single color as dye label18,23,24 and holds great promise as a practical and powerful reporter for structural transformations of amyloid protein. However, to date the mechanism of lifetime changes in the reporter fluorophores upon aggregation has not been adequately elucidated. Furthermore, there have been no attempts to correlate lifetime values to underlying amyloid structure, and reported applications have so far been qualitative rather than quantitative in nature.
In this paper, we demonstrate that the fluorescence lifetime of synthetic dyes covalently attached to amyloid proteins informs on the density and morphological properties of amyloid clusters, and conclude that this is affected by fluorescence self-quenching of the dye molecules upon aggregation. The method requires only a single type of fluorescent dye to label the protein of interest. In what follows we refer to this concept as “lifetime sensor” for amyloid aggregation. We show that the lifetime sensor enables the probing of the structural density of amyloid clusters in a high-throughput manner with negligible influence on the kinetics of aggregation. We demonstrate the method both in vitro and in cells, via FLIM and optical sectioning structured-illumination microscopy (SROS-SIM) measurements of amyloid clusters and validated the method for different protein and dye label combinations. In particular, we studied the heparin-induced aggregation of labeled K18 tau protein in vitro. K18 tau is comprised of the aggregation prone four-repeat domain of the tau protein and is associated with AD and PD.4−6,13,27,28 Furthermore, we studied the aggregation of polyglutamine (polyQ) protein in human embryonic kidney (HEK) 293 cells, a protein related to HD,3,9 large aggregates of which interfere with mitosis.10 With the correlative FLIM/SIM experiments, we demonstrate that the lifetime sensor reports on the aggregation state of intracellular amyloid inclusions and that this offers a capability to enable high throughput screening applications.
Optical Sectioning with SIM Elucidates the Morphology of K18 Tau Aggregates Formed in Vitro with Subwavelength Resolution
We used the aggregation of labeled K18 tau as our in vitro test model for the characterization of the lifetime sensor. K18 tau monomer samples labeled with Atto 532 or Alexa Fluor 488 at different labeling ratios (defined as the percentage of monomer peptides that were labeled with the fluorescent dyes) were prepared, and their aggregation was induced by addition of heparin (see Supporting Information for details).
In order to provide reference data for the calibration of the lifetime sensor, we first characterized the morphology of K18 aggregates formed after various incubation times using SROS-SIM. SIM enables the imaging of amyloid clusters with sub-diffraction-limited resolution29,30 without any specific requirements on the labeling ratio or photochemical properties of the fluorescent reporter dyes. SROS-SIM is furthermore particularly capable of rejecting out-of-focus light, thus greatly enhancing image contrast. Example images to demonstrate these features are shown in Figure S1 of the Supporting Information for clusters of K18 tau. We used a home-built SIM system,31 and analyzed data using software developed in-house in Matlab (MathWorks) (see Supporting Information). The SROS-SIM software incorporates a number of previously proposed algorithms32−34 and is freely available online.35 Image stacks were acquired by performing SROS-SIM at different focal planes through the amyloid clusters.
Figure 1 displays maximum intensity projections of three-dimensional (3D) image stacks of representative K18-Atto532 amyloid clusters formed using varying incubation times and with labeling ratios of 5% and 30%, respectively. Clearly, fibrils were formed and assembled into loose clusters early on during the aggregation process. After about 30 h, clusters stopped growing bigger in size and instead grew into denser structures with time before forming dense spherulite structures that did not undergo further morphological transformation in time. We estimated the average radii (radii of the smallest enclosing spheres) of the amyloid clusters to be 6.6 ± 1.8 and 7.3 ± 2.2 μm after 29.3 h for the 5% and 30% labeled samples, respectively, and 7.5 ± 3.4 and 7.0 ± 2.1 μm for corresponding samples after 78.0 h. Sizes and structures of both the 5% and 30% labeled amyloid clusters appeared similar for comparable incubation times, indicating that the labels did not significantly influence K18 tau aggregation for labeling ratios up to 30%. A quantitative study of the influence of different labeling ratios on aggregation kinetics is the subject of subsequent sections. However, at very high labeling ratios the influence on aggregation may become significant.36 In Figure S2 in the Supporting Information, examples are shown for samples that were labeled at 100% and here clear differences can be observed in the morphology of aggregates compared to those obtained at labeling ratios lower than 50%.
Figure 1.

Maximum intensity projections of stacks of SROS-SIM super-resolution images obtained of heparin-induced K18-Atto532 amyloid clusters formed in vitro for different incubation times and labeling ratios. Scale bars: 10 μm.
Fluorescence Self-Quenching of Reporter Dyes Informs on K18 Aggregation in Vitro
Having characterized K18 aggregate morphology with optical super-resolution microscopy, we proceeded to validate the lifetime sensor with FLIM. To begin with, we measured the lifetime of fully aggregated K18 samples labeled with different dye ratios. Fully aggregated K18-Atto532 and K18-Alexa488 samples were obtained by incubating monomer and heparin mixtures for 9 days. The samples were then imaged on a home-built TCSPC (time-correlated single photon counting) -FLIM system15,37,38 (also see Supporting Information) and analyzed using the FLIMfit software.39 All data were fitted with monoexponential decay curves (see Supporting Information). FLIM data were segmented into regions containing individual amyloid clusters which were individually analyzed. We present fluorescence lifetime values as the average value of different K18 amyloid cluster lifetimes with error bars denoting the standard deviation. Figure 2a,b shows that for fully aggregated samples of K18 tau, the sensor lifetime decreases with increasing labeling ratio, irrespective of whether Alexa 488 or Atto 532 is used as label. For monomers, we measured lifetimes of 3.689 ± 0.005 ns for K18-Atto532 and 3.696 ± 0.002 ns for K18-Alexa488, which are higher than those corresponding to aggregates with any labeling ratios (Figure 2a,b).
Figure 2.

Fluorescence lifetime data for K18-Atto532 and K18-Alexa488 tau samples reveal increasing degrees of fluorescence self-quenching during protein aggregation as the underlying mechanism for lifetime changes. (a,b) Fluorescence lifetime data and Stern–Volmer data fit for the fully aggregated K18-Alexa488 (a) and K18-Atto532 (b) samples with different labeling ratios. (c) Fluorescence lifetime of fully aggregated K18-Atto532 samples for different labeling ratios in PBS solution (yellow), dried state (red), and in PBS solution after rehydration of the dried state (blue). (d) Phasor plot of FLIM data of K18-Atto532 amyloid clusters. Circles denote phasors for aggregates formed after different incubation times with the specified labeling ratios. Solid diamonds denote phasors for monomers and fully aggregated samples with different labeling ratios, calculated from same data sets as panel b. As expected, the phasors for monomer, low-labeling ratio samples, and samples at early aggregation stages lie close to each other. A zoomed-in image (Figure S4) is available in the Supporting Information. (e) Illustration to show how cluster formation with different labeling ratios can lead to overlapping phasor trajectories. Phasors overlap in those samples where the local dye concentration, and thus self-quenching, is similar; for example, a more aggregated and thus denser sample with low labeling ratio (right panel) may have the same dye density as a less densely aggregated sample with high labeling ratio (left panel). Green, fluorescent dye labels; beige, proteins.
Potentially, the use of different labeling ratios could result in structural variations of forming amyloid clusters, which may lead to the dyes becoming more or less buried within aggregates, changing their solution exposure and potentially their lifetimes. To investigate potential effects of solvent quenching, we measured the sensor lifetimes of both fully aggregated and dried samples. Figure 2c shows the lifetime of fully aggregated samples contained in PBS with different Atto532 labeling ratios (yellow bars). The sample slides were then left to dry on a heated disk of a constant temperature of 37 °C. Figure 2c shows that the lifetimes of dried amyloid clusters displayed a similar decreasing trend as the dye labeling ratio increased, even in the absence of solvent (red bars). Finally, the samples were rehydrated again using Milli-Q water so that the original liquid volume was restored in the sample. The blue bars in Figure 2c show that the sensor lifetime of the recovered samples went back to values close to those measured before sample drying began. For all three conditions, lifetimes decreased with increasing labeling ratios, and hence solvent effects could be excluded as the (main) cause for the observed dependence of sensor lifetime on labeling ratio.
It had previously been speculated that the sensor lifetime decrease upon peptide aggregation may either be due to self-quenching23 or due to FRET-like energy transfer to energy states that are specific to amyloid structures.16,18,24,37 If the observed lifetime variation were solely due to FRET, then increasing the dye labeling ratio would correspond to an increase in donor concentration while the acceptor concentration (energy states of the aggregates) remains unchanged, and thus one would expect the fluorescence lifetime of the dye to increase accordingly or remain unchanged,40 which is the opposite of our observation (see Figure 2a,b). We thus hypothesized fluorescence self-quenching to be the underlying mechanism for the lifetime sensor: peptide aggregation brings the dye labels closer together, increases their local concentration and thus decreases their lifetimes. Self-quenching of a dye is often described by the Stern–Volmer equation26
| 1 |
Here, τ0 and τ represent the fluorescence lifetimes of unquenched or quenched dye, and I0 and I represent the fluorescence intensities of unquenched and quenched dyes, respectively. flabel is the labeling ratio of the sample, kq is the self-quenching rate coefficient of the dye, and ρ is the local concentration of K18 peptide in the aggregate. Equation 1 was fitted to the data in Figure 2a,b (red line) and excellent agreement was found with the experimental data. The monomer lifetimes τ0 recovered from the fitting are 3.68 ± 0.04 ns for K18-Atto532 and 3.61 ± 0.09 ns for K18-Alexa488, similar to the corresponding values 3.689 ± 0.005 and 3.696 ± 0.002 ns acquired experimentally. The sample corresponding to 100% dye labeling ratio was not taken into account in the fit, because these samples looked structurally different to aggregation formed using the lower labeling ratios (see Figure S2).
Next, we performed FLIM imaging experiments for K18-Atto532 amyloid clusters, using different labeling ratios and incubation times, and performed a phasor plot analysis40−43 of the FLIM data (Figure 2d for phasor plot, corresponding lifetime data shown in Figure 3a). The phasors and error bars in Figure 2d are, respectively, the average values and standard deviations calculated from the phasors of different amyloid clusters. Phasors were obtained from Fourier transform of FLIM data as reported extensively in previous work.40,43 The methodology makes no model assumption and provides for a powerful tool to identify and differentiate contributions from different fluorescence decay components such as FRET or self-quenching. For example, if significant energy transfer between the dyes and energy states of aggregates took place, corresponding phasor trajectories (which means phasors for samples with different incubation times) of samples with different labeling ratios would be expected to be distinct without overlap occurring. We expect this to be true because aggregates with different structures (formed after different incubation times) are likely to feature different intrinsic energy states and hence the FRET efficiency between dye labels and different aggregate types should be distinct. Phasors for samples with different donor and acceptor stoichiometries and different FRET efficiencies should spread over an area on the phasor plot rather than lying on the same trajectory.40 However, as seen from Figure 2d, the phasor trajectories for different labeling ratios overlap, indicating that phasor positions are mainly related to the local dye concentrations rather than the specific type of aggregate structure forming. Figure 2e illustrates this idea schematically; the phasors for aggregates with different aggregate structure may overlap for samples with different labeling ratios if a lower aggregate density is compensated by a higher labeling ratio and vice versa, leaving local dye concentrations the same. For instance, the phasors highlighted by the dashed circle in Figure 2d correspond to 20% labeled sample that was fully aggregated (red phasor) and 30% labeled sample that was in the intermediate aggregation state (blue phasor). These phasors almost overlap because the local dye densities in the amyloid clusters are similar. Taken together, these results provide firm evidence that self-quenching is the main mechanism by which the lifetime sensor reports on aggregation. We also performed FLIM measurements for the Atto 532 maleimide dye that we used for K18 peptide labeling, dissolved in DMSO at different concentrations, and we verified that indeed the dye exhibits self-quenching at high concentration (see Figure S3 in Supporting Information).
Figure 3.

TCSPC-FLIM measurements of amyloid cluster formation kinetics of K18-Atto532 samples with different dye labeling ratios. (a) Fluorescence lifetimes of K18-Atto532 as a function of aggregation time for samples with 10%, 20%, and 30% labeling ratios. The sigmoidal curves are the fitted lines using the kinetic model described in the main text. (b) Same data represented with normalized fluorescence lifetimes, demonstrating that the cluster formation kinetics are not significantly affected by the presence of the dye labels. (c) Schematic to illustrate the K18 tau aggregation model used for FLIM data fitting. Green, fluorescent dye labels; beige, proteins.
Having established that fluorescence self-quenching is the underlying mechanism behind the lifetime sensor, we went on to explore how it could be used to report on the type and nature of amyloid clusters formed under different aggregation conditions. Figure 3a shows the fluorescence lifetime data from which the phasor plot shown in Figure 2d was generated. The sensor lifetime of amyloid clusters was shown to be approximately constant for all three labeling ratios for incubation times less than approximately 30 h. After about 30 h, the sensor lifetime began to drop and then reached a bottom plateau region after approximately 55 h of incubation. The top plateau in Figure 3a corresponds to lifetimes of 3.59 ± 0.04, 3.57 ± 0.02, and 3.56 ± 0.02 ns for the 10%, 20%, and 30% labeled samples, respectively, which is only about 100 ps lower than the monomer K18-Atto532 lifetime 3.689 ± 0.005 ns. We note that the SROS-SIM experiments revealed that for incubation times less than 30 h, aggregates consisted mostly of low density clusters of loosely assembled amyloid fibrils. This suggests that the lifetime sensor does not sensitively report on the conversion of labeled peptide into single amyloid fibrils at labeling ratios below 30% for K18. However, the sensor lifetime dropped significantly (by up to 19%, 34%, and 42%, respectively, for 10%, 20%, and 30% labeled samples) during the period when the aggregate density increased. Our lifetime sensor is thus highly sensitive to fibril packing density in the amyloid cluster, rather than the formation of single fibrils. This suggests that the distance between neighboring dyes on single fibrils is not sufficiently proximate to generate significant self-quenching, whereas in clusters on the other hand, this distance drops into a highly sensitive range for dye self-quenching.
The Fluorescence Lifetime Sensor Offers a Good Dynamic Range to Report on K18 Tau Aggregation without Significantly Influencing Its Kinetics
Figure 3a shows that it is advantageous to use a higher labeling ratio for a larger variation in lifetime to report on aggregation. However, as mentioned previously, the aggregation kinetics and structure of amyloid clusters may be affected by very high labeling ratios (see Figure S2). A good dynamic range was obtained at 30% labeling ratio for K18 tau with lifetimes dropping by 42% from monomeric to fully aggregated state. We thus investigated the effect of peptide labeling ratio up to 30% on aggregation kinetics. Structurally, the amyloid clusters formed with labeling ratios up to 30% were indistinguishable from one another, as verified by SROS-SIM calibration. In Figure 3b, we show that the kinetics of aggregation are similarly unaffected and the normalized lifetime profiles overlap very well for all labeling ratios, giving support to the notion that the labeling protocols with labeling ratios below 30% had minimal effects on the K18 aggregation processes investigated.
We developed a kinetic model to describe the in vitro aggregation process of K18 tau based on the SROS-SIM results presented above and used it to quantify the effect of labeling on aggregation kinetics. Figure 3c shows the model schematically. Early amyloid clusters were modeled to consist of loose assemblies of K18 tau fibrils. The amyloid clusters grow until they reach a certain size and become “mature”. At this point, clusters stop growing larger and instead act as “sinks” that absorb the remaining amyloid species (monomer/oligomer/fibrils) in their vicinity and thus increase in density. We assume that the number of “sinks” does not change after the amyloid clusters are “mature” and we describe the densification process of such “sinks” using the Smoluchowski sink absorption model.44−46 Combining the Smoluchowski model and Stern–Volmer eq (eq 1), we derived the following equation to describe K18 tau aggregation kinetics (for details of our model and derivation of this equation see Supporting Information)
| 2 |
In eq 2, τ0 is the fluorescence lifetime
of monomeric sample, and τ(t) is the average
lifetime of amyloid clusters at time t. flabel is the labeling ratio, kq is the self-quenching rate coefficient. ρsink(t) is the average local concentration of K18 monomers inside
each sink, at time t. V is the average
volume of the sinks. 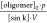 is a constant.
4πDR[sink] represents the K18 aggregation rate
constant. θ is the average sink formation time, and s is the standard deviation of sink formation time. We perform
a global fit of the aggregation kinetic data in Figure 3a using eq 2. Results are presented in Table S1 in the Supporting Information, where a discussion of
the model and demonstration of its robustness are also presented.
Using global fitting for all other parameters (kqρsink (∞),
is a constant.
4πDR[sink] represents the K18 aggregation rate
constant. θ is the average sink formation time, and s is the standard deviation of sink formation time. We perform
a global fit of the aggregation kinetic data in Figure 3a using eq 2. Results are presented in Table S1 in the Supporting Information, where a discussion of
the model and demonstration of its robustness are also presented.
Using global fitting for all other parameters (kqρsink (∞), 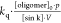 , θ
and s), we were able to recover aggregation rate
constants 4πDR[sink] for the 10%, 20%, and
30% labeled samples as, respectively, 0.034 ± 0.006, 0.046 ±
0.010, and 0.057 ± 0.014/h. The average sink formation time θ
can be recovered with a similar approach and was found to be 43.0
± 1.1, 44.6 ± 1.0, and 42.3 ± 0.9 h for the 3 samples,
respectively. Both aggregation rate constant and the average sink
formation time did not vary significantly for different labeling ratios,
providing firm evidence that labeling ratios up to 30% do not significantly
influence the aggregation kinetics of the K18 peptide.
, θ
and s), we were able to recover aggregation rate
constants 4πDR[sink] for the 10%, 20%, and
30% labeled samples as, respectively, 0.034 ± 0.006, 0.046 ±
0.010, and 0.057 ± 0.014/h. The average sink formation time θ
can be recovered with a similar approach and was found to be 43.0
± 1.1, 44.6 ± 1.0, and 42.3 ± 0.9 h for the 3 samples,
respectively. Both aggregation rate constant and the average sink
formation time did not vary significantly for different labeling ratios,
providing firm evidence that labeling ratios up to 30% do not significantly
influence the aggregation kinetics of the K18 peptide.
The Fluorescence Lifetime Reports on the Structural Density of PolyQ Aggresomes Formed in Cells
To demonstrate the lifetime sensor concept for measurement of amyloid cluster formation in cell models, we performed correlated FLIM/SIM imaging of PolyQ aggresomes formed under physiological conditions inside cells. SNAP-HDQ72-expressing HEK 293T cells were labeled with SNAP-Cell 505-Star and fixed on gridded slides9,10 (see Supporting Information); they were then imaged using both TCSPC-FLIM and SROS-SIM as described previously. Note that we fixed the cells only for the purpose of enabling correlative FLIM/SIM measurements; in principle, the lifetime sensor can equally be applied in living cells. Figure 4d–f shows the intensity-merged FLIM images of the polyQ aggresomes, and Figure 4a–c shows the corresponding SROS-SIM images of the same aggresomes (animated volume renderings of the data are available in the Supporting Information). Comparing both data sets, it is seen that aggresomes with a lower lifetime display a denser structure via SIM and vice versa.
Figure 4.
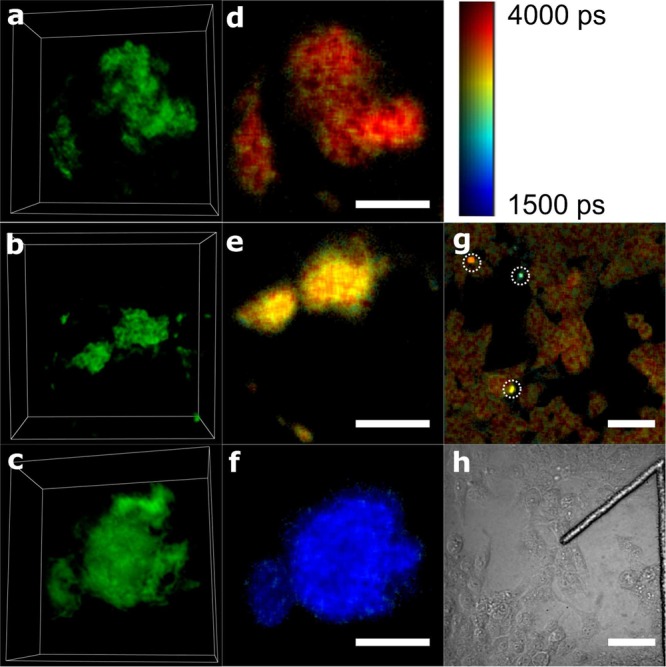
Correlated FLIM/SIM images of polyQ aggresomes labeled with SNAP-Cell 505-Star dye in fixed HEK cells. (a–c) Three-dimensional (3D) images of polyQ aggresomes reconstructed from the SROS-SIM images, presented using Icy software.49 Size for boxes in panels a–c is 15 μm × 15 μm × 7.1 μm. Animated 3D renderings are available in the Supporting Information. (d–f) FLIM images of the same polyQ aggresomes corresponding to panels a–c. Scale bar: 5 μm. (g) FLIM image of polyQ-expressing HEK cells stained with SNAP-Cell 505-Star dye. Scale bar: 50 μm. (h) Transmitted light image of the same field of view as the FLIM image g. The shape of “1” is the number label on the gridded dish. The colorbar indicates the fluorescence lifetime values for panels d–f.
Figure 4g displays a zoomed-out FLIM image with several aggresomes appearing as bright spots (highlighted by white circles) in the same field of view. This image clearly demonstrates the high-throughput capability of the lifetime sensor. It took only about 1 min to acquire the image on a TCSPC-FLIM system, which could be sped up further if time-gated FLIM were used instead of TCSPC.26 The lifetime sensor reveals structural density variations between different aggresomes in a single FLIM image containing a large number of cells (see Figure 4g), providing a fast read out tool that informs on amyloid structure without requirement for imaging each cluster individually with high-resolution optical microscopy or EM techniques. At the same time, the FLIM image keeps enough spatial resolution to locate the position of the aggresomes within cells and to distinguish between aggresomes in different cells. Moreover, the lifetime provides an indication of the structural density and morphology of different amyloid clusters, whereas a similar global parameter to quantify structural density variation does not exist for high-resolution methods. The lifetime sensor is thus a powerful tool for the identification of large numbers of amyloid clusters in bulk, for instance, for the screening of aggresomes with different structural densities in cells or for the acquisition of statistical data of the aggresome structural densities, and such data could be further correlated with cellular phenotypes, for example, a capability for cells to undergo mitosis.10
A drawback of the lifetime sensor comes from the fact that fluorescence self-quenching is not only related to the structural properties of the amyloid clusters, but also to the labeling ratio of the sample (as shown in Figure 3a). This requires control over the labeling ratio of the sample. This can easily be achieved for in vitro experiments where monomer labeling ratios can be controlled, such as our K18 tau experiments shown earlier. In cell experiments, covalent bio-orthogonal labeling techniques47,48 such as SNAP-tag and CLIP-tag can be used to control the labeling ratios, as we have employed here for polyQ experiments in HEK cells.
In summary, we present a lifetime sensor for high-throughput structural characterization of amyloid clusters both in vitro and in cells. Performing SROS-SIM and FLIM experiments on in vitro aggregated K18 tau labeled with Alexa 488 or Atto 532, we show that the lifetime sensor reports on the amyloid structural density according to the fluorescence lifetime variation of synthetic dyes covalently labeled on peptides. We demonstrate that the mechanism of the sensor is via fluorescence self-quenching and that the sensor offers a good dynamic range of fluorescence lifetime variation without significantly influencing the aggregation formation. The power of lifetime sensor concept was demonstrated for cell experiments via correlative FLIM/SIM experiments in HEK cells on aggresomes of polyQ labeled with SNAP-Cell 505-Star. This high-throughput method allows for the screening or statistical analysis for aggresomes with different structural density formed in cells, and it can potentially be applied on studies in more physiological conditions such as brain slices and so forth.
Acknowledgments
This work was supported by grants from the Leverhulme Trust; the Engineering and Physical Sciences Research Council, UK [EP/H018301/1]; the Medical Research Council [MR/K015850/1, and MR/K02292X/1]; the Wellcome Trust [089703/Z/09/Z]; and the Alzheimer’s Research UK [ARUK-EG2012A-1]. WeiYue Chen is funded by China Scholarship Council; and Cambridge Commonwealth, European and International Trust for her PhD.
Supporting Information Available
Details of the sample preparation, imaging parameters, data analysis, the fitting results and discussions of the K18 aggregation kinetics data, and the supporting figures are available in the .pdf file. Animated 3D renderings corresponding to Figure 4a-c are presented in the .avi files. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nanolett.6b03686.
Details of the sample preparation, imaging parameters, data analysis, the fitting results and discussions of the K18 aggregation kinetics data, and the supporting figures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ciryam P.; Tartaglia G. G.; Morimoto R. I.; Dobson C. M.; Vendruscolo M. Cell Rep. 2013, 5, 781–790. 10.1016/j.celrep.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe J. D.; Cohen A. S. J. Struct. Biol. 2000, 130, 88–98. 10.1006/jsbi.2000.4221. [DOI] [PubMed] [Google Scholar]
- Bates G. Lancet 2003, 361, 1642–1644. 10.1016/S0140-6736(03)13304-1. [DOI] [PubMed] [Google Scholar]
- Aguzzi A.; O’Connor T. Nat. Rev. Drug Discovery 2010, 9, 237–248. 10.1038/nrd3050. [DOI] [PubMed] [Google Scholar]
- Stefani M.; Dobson C. M. J. Mol. Med. 2003, 81, 678–699. 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. Nat. Cell Biol. 2004, 6, 1054–1061. 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- Bacskai B. J.; Skoch J.; Hickey G. A.; Allen R.; Hyman B. T. J. Biomed. Opt. 2003, 8, 368–375. 10.1117/1.1584442. [DOI] [PubMed] [Google Scholar]
- Kollmer M.; et al. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 5604–5609. 10.1073/pnas.1523496113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M.; Williamson N.; Boschetti C.; Ellis T.; Yoshimi T.; Tunnacliffe A. Biotechnol. Bioeng. 2015, 112, 1883–1892. 10.1002/bit.25606. [DOI] [PubMed] [Google Scholar]
- Lu M.; Boschetti C.; Tunnacliffe A. J. Biol. Chem. 2015, 290, 27986–28000. 10.1074/jbc.M115.676437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S.; Akhremitchev B. B. Biomacromolecules 2006, 7, 1630–1636. 10.1021/bm0600724. [DOI] [PubMed] [Google Scholar]
- Stine W. Jr; Snyder S.; Ladror U.; Wade W.; Miller M.; Perun T.; Holzman T.; Krafft G. J. Protein Chem. 1996, 15, 193–203. 10.1007/BF01887400. [DOI] [PubMed] [Google Scholar]
- Mukrasch M. D.; Biernat J.; von Bergen M.; Griesinger C.; Mandelkow E.; Zweckstetter M. J. Biol. Chem. 2005, 280, 24978–24986. 10.1074/jbc.M501565200. [DOI] [PubMed] [Google Scholar]
- Roberti M. J.; Bertoncini C. W.; Klement R.; Jares-Erijman E. A.; Jovin T. M. Nat. Methods 2007, 4, 345–351. 10.1038/nmeth1026. [DOI] [PubMed] [Google Scholar]
- Chan F. T.; Kaminski Schierle G. S.; Kumita J. R.; Bertoncini C. W.; Dobson C. M.; Kaminski C. F. Analyst 2013, 138, 2156–2162. 10.1039/c3an36798c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinotsi D.; Grisanti L.; Mahou P.; Gebauer R.; Kaminski C. F.; Hassanali A.; Kaminski Schierle G. S. J. Am. Chem. Soc. 2016, 138, 3046–3057. 10.1021/jacs.5b11012. [DOI] [PubMed] [Google Scholar]
- Ries J.; Udayar V.; Soragni A.; Hornemann S.; Nilsson K. P. R.; Riek R.; Hock C.; Ewers H.; Aguzzi A. A.; Rajendran L. ACS Chem. Neurosci. 2013, 4, 1057–1061. 10.1021/cn400091m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esbjörner E. K.; Chan F.; Rees E.; Erdelyi M.; Luheshi L. M.; Bertoncini C. W.; Kaminski C. F.; Dobson C. M.; Kaminski Schierle G. S. Chem. Biol. 2014, 21, 732–742. 10.1016/j.chembiol.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski Schierle G. S.; Van De Linde S.; Erdelyi M.; Esbjörner E. K.; Klein T.; Rees E.; Bertoncini C. W.; Dobson C. M.; Sauer M.; Kaminski C. F. J. Am. Chem. Soc. 2011, 133, 12902–12905. 10.1021/ja201651w. [DOI] [PubMed] [Google Scholar]
- Pinotsi D.; Buell A. K.; Galvagnion C.; Dobson C. M.; Kaminski Schierle G. S.; Kaminski C. F. Nano Lett. 2014, 14, 339–345. 10.1021/nl4041093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine R. F.; Kaminski Schierle G. S.; van de Linde S.; Kaminski C. F. Methods Appl. Fluoresc. 2016, 4, 022004. 10.1088/2050-6120/4/2/022004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski C. F.; Kaminski Schierle G. S. Neurophotonics 2016, 3, 041807. 10.1117/1.NPh.3.4.041807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn S. D.; Dalgarno P. A.; Cameron R. T.; Hedley G. J.; Hacker C.; Lucocq J. M.; Baillie G. S.; Samuel I. D.; Penedo J. C. Mol. BioSyst. 2014, 10, 34–44. 10.1039/C3MB70272C. [DOI] [PubMed] [Google Scholar]
- Michel C. H.; Kumar S.; Pinotsi D.; Tunnacliffe A.; George-Hyslop P. S.; Mandelkow E.; Mandelkow E.-M.; Kaminski C. F.; Kaminski Schierle G. S. K. J. Biol. Chem. 2014, 289, 956–967. 10.1074/jbc.M113.515445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty J.; Taylor H. B.; Chen L.; Bugeon L.; Lamb J. R.; Dallman M. J.; French P. M. Biomed. Opt. Express 2011, 2, 1340–1350. 10.1364/BOE.2.001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz J. R.Principles of fluorescence spectroscopy; Springer: New York, 2013. [Google Scholar]
- Shammas S. L.; Garcia G. A.; Kumar S.; Kjaergaard M.; Horrocks M. H.; Shivji N.; Mandelkow E.; Knowles T. P.; Mandelkow E.; Klenerman D. Nat. Commun. 2015, 6, 7025. 10.1038/ncomms8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C.; Lee V. M.-Y.; Trojanowski J. Q. Nat. Rev. Neurosci. 2007, 8, 663–672. 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Ströhl F.; Kaminski C. F. Optica 2016, 3, 667–677. 10.1364/OPTICA.3.000667. [DOI] [Google Scholar]
- Gustafsson M. G. J. Microsc. 2000, 198, 82–87. 10.1046/j.1365-2818.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- Young L. J.; Ströhl F.; Kaminski C. F. J. Visualized Exp. 2016, 111, e53988. 10.3791/53988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson M. G.; Shao L.; Carlton P. M.; Wang C. R.; Golubovskaya I. N.; Cande W. Z.; Agard D. A.; Sedat J. W. Biophys. J. 2008, 94, 4957–4970. 10.1529/biophysj.107.120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker K. Opt. Express 2013, 21, 24692–24701. 10.1364/OE.21.024692. [DOI] [PubMed] [Google Scholar]
- Shaw M.; Zajiczek L.; O’Holleran K. Methods 2015, 88, 11–19. 10.1016/j.ymeth.2015.03.020. [DOI] [PubMed] [Google Scholar]
- The SROS-SIM software is freely available online via laser.ceb.cam.ac.uk.
- Amaro M.; Wellbrock T.; Birch D. J.; Rolinski O. J. Appl. Phys. Lett. 2014, 104, 063704. 10.1063/1.4865197. [DOI] [Google Scholar]
- Kaminski Schierle G. S.; Bertoncini C. W.; Chan F. T. S.; van der Goot A. T.; Schwedler S.; Skepper J.; Schlachter S.; van Ham T.; Esposito A.; Kumita J. R.; Nollen E. A. A.; Dobson C. M.; Kaminski C. F. ChemPhysChem 2011, 12, 673–680. 10.1002/cphc.201000996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J.; Elder A.; Swartling J.; Venkitaraman A.; Jeyasekharan A.; Kaminski C. J. Microsc. 2007, 227, 203–215. 10.1111/j.1365-2818.2007.01803.x. [DOI] [PubMed] [Google Scholar]
- Warren S. C.; Margineanu A.; Alibhai D.; Kelly D. J.; Talbot C.; Alexandrov Y.; Munro I.; Katan M.; Dunsby C.; French P. M. PLoS One 2013, 8, e70687. 10.1371/journal.pone.0070687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.; Avezov E.; Schlachter S. C.; Gielen F.; Laine R. F.; Harding H. P.; Hollfelder F.; Ron D.; Kaminski C. F. Biophys. J. 2015, 108, 999–1002. 10.1016/j.bpj.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digman M. A.; Caiolfa V. R.; Zamai M.; Gratton E. Biophys. J. 2008, 94, L14–L16. 10.1529/biophysj.107.120154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redford G. I.; Clegg R. M. J. Fluoresc. 2005, 15, 805–815. 10.1007/s10895-005-2990-8. [DOI] [PubMed] [Google Scholar]
- Clayton A.; Hanley Q.; Verveer P. J. Microsc. 2004, 213, 1–5. 10.1111/j.1365-2818.2004.01265.x. [DOI] [PubMed] [Google Scholar]
- Von Smoluchowski M. Zeitschrift für physikalische Chemie 1917, 92, 129–168. [Google Scholar]
- Rice S. A.Diffusion-limited reactions; Elsevier: New York, 1985; Vol. 25. [Google Scholar]
- Dorsaz N.; De Michele C.; Piazza F.; De Los Rios P.; Foffi G. Phys. Rev. Lett. 2010, 105, 120601. 10.1103/PhysRevLett.105.120601. [DOI] [PubMed] [Google Scholar]
- Dean K. M.; Palmer A. E. Nat. Chem. Biol. 2014, 10, 512–523. 10.1038/nchembio.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay C. S.; Finn M. Chem. Biol. 2014, 21, 1075–1101. 10.1016/j.chembiol.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Chaumont F.; Dallongeville S.; Chenouard N.; Hervé N.; Pop S.; Provoost T.; Meas-Yedid V.; Pankajakshan P.; Lecomte T.; Le Montagner Y.; Lagache T.; Dufour A.; Olivo-Marin J.-C. Nat. Methods 2012, 9, 690–696. 10.1038/nmeth.2075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.