

Abstract

We present an atomistic understanding of the evolution of the size distribution with temperature and number of cycles in atomic layer deposition (ALD) of Pt nanoparticles (NPs). Atomistic modeling of our experiments teaches us that the NPs grow mostly via NP diffusion and coalescence rather than through single-atom processes such as precursor chemisorption, atom attachment, and Ostwald ripening. In particular, our analysis shows that the NP aggregation takes place during the oxygen half-reaction and that the NP mobility exhibits a size- and temperature-dependent scaling. Finally, we show that contrary to what has been widely reported, in general, one cannot simply control the NP size by the number of cycles alone. Instead, while the amount of Pt deposited can be precisely controlled over a wide range of temperatures, ALD-like precision over the NP size requires low deposition temperatures (e.g., T < 100 °C) when growth is dominated by atom attachment.
Supported nanoparticles (NPs) play a major role in a wide range of applications from catalysis to electronic, optical, and energy storage devices.1−5 Scaling materials down to the nanoscale not only maximizes the number of active surface sites but also brings about unique size-dependent functionalities.3,6−8 The latter, however, can only be understood and harnessed through the advent of synthesis routes that enable the deposition of NPs with narrow particle size distributions (PSDs), that is, size-selected NPs.9,10 Despite its potential, the scalable synthesis of size-selected NPs on high-surface-area supports, which are relevant to most practical applications, has so far proved elusive.10 Atomic layer deposition (ALD) is an established thin-film deposition technique that has recently seen use in the synthesis of supported NPs with very promising results in terms of control over the NP size.2,4,11−14 By relying on the cyclic repetition of self-limiting gas–solid reactions, ALD boasts digital control and atomic-level precision over the amount of material being deposited.4,11,15 While originally developed for flat substrates, ALD is readily scalable to high-surface-area substrates thanks to its solvent-free and surface-driven nature.4 Considerable research effort has thus been made to bring the unparalleled atomic-level precision of ALD to the synthesis of supported NPs. Nevertheless, the development of ALD routes for size-selected NPs is currently hampered by a lack of fundamental understanding.4,16,17 The majority of fundamental studies on ALD is, in fact, concerned with the surface chemistry behind the deposition of the atoms of choice on a given substrate, and far less research has been devoted to the understanding of the diffusion phenomena underlying the formation of NPs and the evolution of their PSD.4,16−21 As a result, ALD of NPs has been reported with different PSDs, and the growth mechanisms leading to these different PSDs are still under debate.1,2,4,12,14,17,20−23
The understanding of ALD growth is mostly framed in terms of sequential self-limiting chemisorption of ALD precursors, leading to a layer-by-layer deposition.21 However, the nucleation and growth of NPs is bound to be dictated by atomistic processes other than chemisorption such as surface diffusion of adatoms, NP nucleation, diffusion and coalescence, and atom attachment to and detachment from NPs.6,21,24−28 In a seminal work, Mackus et al.20 argued that during Pt ALD on oxides Pt NPs might grow not only due to ALD surface reactions but also through Ostwald ripening via volatile PtO2, that is, the growth of larger NPs at the expense of smaller ones through the exchange of single atoms driven by the minimization of the surface energy. Outside the field of ALD, the analysis of the PSD has revealed that apart from classical ripening, another mechanism is relevant to the growth of supported NPs: Smoluchowski aggregation,29−31 that is, NP diffusion and coalescence. We have recently shown that ALD of Pd NPs results in broad right-skewed PSDs and a reduction in the NPs density with the number of cycles that is consistent with NP sintering via NP diffusion and coalescence.14 Even if this suggests that Smoluchowski aggregation is a governing growth mechanism also in ALD of NPs, many questions remain, in particular: Can we understand NP growth in terms of elementary atomistic processes and thus identify the governing NP growth mechanisms among layer-by-layer growth due to direct precursor chemisorption, single atom deposition on the substrate followed by attachment to NPs, Ostwald ripening, and NP diffusion and coalescence? And, can we steer the growth so as to achieve size selection and control the size simply by the number of cycles?
We identify the governing mechanisms behind the nucleation and growth of Pt NPs during ALD. We do so by using the Pt(MeCp)Me3/O2 ALD process on bulk quantities of graphene nanoplatelets, which is, as we shall point out later, a model system relevant to several other Pt ALD processes. This platform allowed us to experimentally follow the evolution of the PSD and of the amount of metal deposited during the initial stages of NP growth, that is, the first 10 ALD cycles. In particular, to shed light on the role of the precursor chemisorption on the NPs nucleation and growth, we probed ex situ the morphology of the Pt/graphene composites after the first Pt(MeCp)Me3 and O2 half-cycles. To further deconvolute the effect of deposition and the growth due to NP sintering, we performed dedicated annealing experiments with the as-synthesized Pt/graphene composites. By modeling the relevant atomistic processes using a rate-equation approach, we could interpret, for the first time, the evolution of the shape of the PSD with the number of ALD cycles and the deposition temperature. Our analysis shows that NP growth is best described in terms of Smoluchowski aggregation kinetics over a wide range of temperatures, that is, 80–250 °C; that is, the NPs grow mostly through NP diffusion and coalescence rather than due to single-atom processes such as single-atom attachment/deposition and Ostwald ripening. The story is nuanced, though, because all depends on the cycle number and the deposition temperature: At lower temperatures, growth by single-atom attachment becomes relevant as NPs tend to pin on the substrate, such that the PSD remains narrow. Therefore, we show that while digital control over the amount of metal being deposited can be achieved over a wide range of temperatures, ALD precision over the NP size is only retained at low deposition temperatures. In other words, contrary to what has been widely claimed,4,12,22 in general, one cannot precisely control the NP size by simply varying the number of ALD cycles.
The
deposition experiments were carried out in a fluidized bed
reactor operated at atmospheric pressure, already described elsewhere,2,14 via a Pt(MeCp)Me3/O2 ALD process on gram-scale
batches of graphene nanoplatelets with a specific surface area of
150 m2 g–1. The latter translated into
cycle times of 19–24 min depending on the deposition temperature.
In particular, synthetic air (20 wt % O2) was used as the
oxygen source, and exposures of 5–10 min were employed to ensure
the complete removal of organic ligands at all temperatures (see Figure S11); the Pt(MeCp)Me3 exposure
time was of 4 min; the Pt(MeCp)Me3 and O2 exposures
were separated by purging steps of 5 min, where nitrogen was used
as the purging gas (N2, 99.999 vol %). Prior to deposition,
the graphene powders were treated with ozone for 30 min at 200 °C
to remove adventitious carbon and activate their inherently defective
surface by incorporating oxygen-containing functional groups for the
initial Pt(MeCp)Me3 chemisorption.14,22,32 Annealing experiments were performed ex
situ both in air and in argon atmospheres at 200 and 400 °C for
6, 12, 24, and 48 h. The Pt/graphene composites were analyzed ex situ
using a suite of characterization techniques including transmission
electron microscopy (TEM), high-resolution TEM (HRTEM), X-ray photoelectron
spectroscopy (XPS), X-ray diffraction (XRD), Raman spectroscopy, thermogravimetric
analysis (TGA), and instrumental neutron activation analysis (INAA).
The PSDs were obtained by determining the projected area (A) and thus the equivalent projected diameter (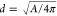 )
of more than 6000 NPs per sample by means
of semiautomatic image analysis of about 10–20 TEM pictures,
taken at different magnifications (e.g., 50 000× and 100 000×)
and sample locations. For more experimental details, the reader is
referred to the Supporting Information.
)
of more than 6000 NPs per sample by means
of semiautomatic image analysis of about 10–20 TEM pictures,
taken at different magnifications (e.g., 50 000× and 100 000×)
and sample locations. For more experimental details, the reader is
referred to the Supporting Information.
XPS, XRD, and HRTEM results are consistent with the deposition of metallic and crystalline Pt 3D NPs (see Figures S2, S3, and S8). According to the elemental analysis carried out via INAA, the platinum loading (i.e., Pt atoms per unit area of substrate) varies in an approximately linear fashion with the number of cycles while being virtually temperature-independent in the range of temperatures and ALD cycles considered here, that is, from 100 to 250 °C and from 1 to 10 cycles (Figure S10). Furthermore, the NP density was estimated to be fairly constant and ∼0.0025 nm–2 within the temperature range 100–200 °C and the cycle range 1–10 (Table S1). Several previous studies on thermal ALD of Pt have reported temperature-dependent growth rates.4,15,17,20,33 In particular, thermal ALD of Pt has never been reported for such low temperatures (i.e., T = 80 °C). This is due to the fact that while the Pt(MeCp)Me3 chemisorption can be active even at room temperature,17,34 the oxidation of carbon ligands on noncatalytic surfaces becomes relatively slow below ∼250 °C under typical ALD conditions, that is, low oxygen pressures (∼0.1 to 1 Torr) and oxygen exposure times on the order of a few seconds.4,15,17,20,33 Here, instead, we achieved ALD growth at temperatures as low as 80 °C by using oxygen partial pressures on the order of 0.2 bar (∼1500 Torr) and oxygen exposure times between 5 and 10 min (Figures S11 and S12). In fact, the combustion kinetics of adsorbed carbon is a function not only of temperature but also of the partial pressure of the oxidizer.4,20 This latter remark emphasizes that in all of those ALD processes leading to NP formation based on the combustion of organic ligands, the partial pressure of the oxidizer as well as the exposure time can be more important than the number of cycles, as far as the amount of metal deposited, and thus the average NP size, is concerned.
While the deposition temperature had little to no effect on the Pt loading, it did have a dramatic effect on the evolution of the shape of the PSD with the number of cycles (Figure 1, Figures S9 and S10). In particular, we observe two different growth regimes, one at low temperatures (i.e., T ≤ 100 °C) and one at high temperatures (i.e., T ≥ 150 °C). The high-temperature regime is characterized by broad right-skewed PSDs presenting a persistent peak at ∼1 nm, followed by a long tail, which shifts toward large sizes as the number of ALD cycles increases. On the contrary, at low temperatures, the PSD remains relatively narrow, and increasing the number of cycles results into the gradual disappearance of the peak on the small-size side and the appearance of a new peak toward large sizes, whose position gradually shifts to the right (Figures 1 and Figure S10). Surprisingly, low temperatures result in PSDs with larger average diameters (e.g., ⟨d⟩100 °C10 cycles = 2.6 nm and ⟨d⟩200 °C = 2.0 nm) that retain relatively low standard deviations (e.g., σ100 °C10 cycles = 1 nm and σ200 °C = 1.4 nm).
Figure 1.

Evolution of the experimental and simulated particle size distributions (PSDs) at 100 °C after 1 (a), 3 (b), 6 (c), and 10 (d) cycles and at 200 °C after 1 (e), 3 (f), 6 (g), and 10 (h) cycles, with a representative TEM micrograph in the respective inset. The PSDs are expressed in terms of probability density function and equivalent projected diameter. Each experimental point corresponds to a bin. The number of bins was calculated by using the Freedman–Diaconis method.
The ALD process used here is a representative model system for oxygen-based ALD of Pt NPs in that it reproduced the characteristics of the PSDs reported by different authors for various Pt ALD processes on different substrates and at different temperatures.1,2,17,22 For example, the features of the high-temperature PSDs can be found in the PSDs reported in the works of: Goulas et al.2 on the radical-enhanced and atmospheric pressure variant of Pt ALD (250 °C) on TiO2 nanopowders (1–5 ALD cycles); Sun et al.22 on the use of Pt ALD (250 °C) for single-atom catalysis (50–150 ALD cycles) on graphene powders; and Mackus et al.17 on Pt ALD (300 °C) on single-crystal alumina (20–100 ALD cycles). On the contrary, the PSD obtained at low temperature after 10 cycles (T ≤ 100 °C), being narrow and left-skewed, resembles the PSDs reported for plasma-enhanced Pt ALD carried out at room temperature on single-crystal alumina (20–100 ALD cycles).17 Furthermore, preliminary results on the application of the same ALD process used here to TiO2 Degussa P-25 nanopowders are also consistent with the above temperature regimes (Figure S9). Clearly, the different growth regimes reported here are an intrinsic characteristic of oxygen-based ALD of Pt NPs.
To resolve the NP formation and growth in finer detail, we studied the morphology of the Pt/graphene composites after the first half-cycle (i.e., Pt(MeCp)Me3/N2) as compared with the full cycle (Pt(MeCp)Me3/N2/O2/N2) and the stability of the as-synthesized Pt NPs against prolonged annealing periods at different temperatures. Ex situ TEM of the Pt/graphene after one-half-cycle and one full cycle at 100 °C revealed that the NPs form and grow mostly during the oxidation step (Figure 2a,b). This is direct evidence of the fact that in ALD NPs can grow via routes other than direct deposition, and thus layer-by-layer growth, at a temperature as low as 100 °C. Subsequent annealing periods in air up to 12 h at 200 °C resulted in virtually no further NP growth (Figure 2d), and only after 24 h did the PSD change d appreciably (Figure 2b–d). Annealing of the Pt/graphene obtained after 10 cycles at 100 °C revealed a similar picture; in particular, the composites were relatively stable at 200 °C even after 24 h in air (Figure S13). However, annealing periods of >6 h at 400 °C in air resulted in the formation of fractal aggregates of relatively large NPs (dp ≈ 5–10 nm), consistently with the onset of NP unpinning, diffusion, and aggregation above a certain thermal energy threshold.
Figure 2.
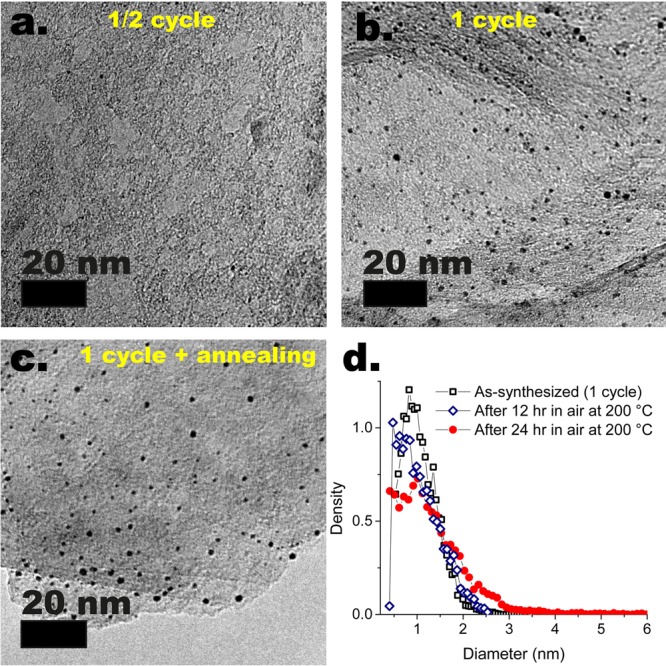
Ex situ TEM of the graphene nanoplatelets (a) after one half cycle (Pt(MeCp)Me3/N2) at 100 °C, (b) after one full cycle at 100 °C (Pt(MeCp)Me3/N2/O2/N2), and (c) after one cycle at 100 °C and annealing in air at 200 °C for 24 h. (d) Particle size distributions of the NPs deposited after one cycle at 100 °C and before and after annealing in air at 200 °C for 12 and 24 h.
The fact that the NPs can form and grow at 100 °C upon exposures to oxygen of only 5 min, while remaining virtually stable for 12 h at 200 °C in the same atmosphere, shows that the aggregation of Pt atoms and thus the NP growth during ALD is induced by the combustion of the organic ligands remaining on the surface after Pt(MeCp)Me3 chemisorption.35,36 Adsorbed organic materials are, in fact, known to suppress the aggregation of supported metals by mediating their mobility,10,25,37 especially on graphene surfaces. In addition, combustion reactions catalyzed by the metal itself can evolve heat and gas such as to generate local temperature and pressure fluctuations that can unpin and displace NPs as a whole, thus promoting sintering via NP diffusion and coalescence,38−40 even at temperatures that would not otherwise provide the thermal budget for the onset of mobility. Indeed, the burning of carbon coke adsorbed on commercial catalysts based on oxides-supported Pt is also known to induce metal sintering.38 Hence we propose the following NP formation and growth mechanism: The carbon remaining on the surface after the precursor chemisorption suppresses the Pt mobility by providing steric hindrance as well as preferential binding sites for both Pt atoms and NPs. As a result, the removal of carbon promotes the surface diffusion of Pt atoms and thus their aggregation, that is, NP formation. Once formed, NPs grow due to further capture of diffusing atoms as well as collision and subsequent coalescence with NPs diffusing as a whole. Transient NP mobility is induced by the heat and the gas locally evolved by the combustion reactions that unpin NPs from strong-binding sites.
To identify the governing mechanism behind the different evolution of the PSD at different temperatures, we devised two dynamic models: model (I) accounts for NP diffusion and coalescence, whereas model (II) accounts for Ostwald ripening. For a qualitative representation of the atomistic processes considered here and their effect on the morphology, see Figure 3. Furthermore, for a detailed description of the models and their parameters, the reader is referred to the Supporting Information. Both models describe the evolution in time of the population of single atoms and NPs with different sizes. Model (I) is based on Smoluchowski aggregation kinetics41−43 and allows for: cyclic generation of single atoms on both the substrate surface and the NPs (deposition); surface diffusion and irreversible aggregation of single atoms; atom attachment to NP (adatom capture); and NP diffusion and coalescence. The NP mobility was assumed to follow a power law of the kind Dk = D1(t) k–s,30,31,42,44−47 where k is the number of atoms comprising the NP, D1(t) is the diffusion rate of adatoms, and s > 0. The value of the exponent s typically ranges between 0 and 2 depending on the diffusion mechanism and is, in general, a poorly understood function of NP morphology, NP–substrate interaction, reacting atmosphere, and temperature.31,44,45 As such, here it is regarded as a fitting parameter.
Figure 3.

Schematic representation of the atomistic processes relevant to ALD of NPs and their effect on morphology, particularly on the shape of the PSD.
Model (II) is a variation of model (I) and allows for: deposition, surface diffusion and aggregation of single atoms, atom attachment to NP, and Ostwald ripening. The thermodynamic driving force for the latter lies in the size-dependent stability of NPs (Gibbs–Thomson effect).48,49 However, for Ostwald ripening to take place, several kinetic steps have to be realized: atom detachment from small NPs, surface diffusion or transport through the vapor phase to larger NPs, and subsequent attachment.27,50,51 Each of these steps can be kinetically limited due to large energy barriers. In the case of platinum, the high Pt–Pt bond energy makes the detachment of an atom from Pt NPs an unlikely event,27,48,50,51 especially when Pt does not enjoy a very strong adsorption on the support. For example, Zhou et al.48 argued that Pt NPs adsorbed on TiO2 consisting of only few tens of atoms (∼0.5 to 1 nm) are kinetically stable against surface-mediated Ostwald ripening under ALD conditions (at vacuum). A more favorable pathway for Ostwald ripening is the exchange of single atoms in the form of volatile PtO2 through the vapor phase in the presence of oxidizing atmospheres.20,27,50,51 Plessow and Abild-Pedersen50 have recently shown through simulations that gas-phase-mediated ripening can account for the temperature and oxygen pressure dependence of the sintering of Pt NPs supported on alumina and silica at high temperatures (600 °C). On the contrary, they also argued that hardly any sintering via Ostwald ripening is expected to occur up to temperatures of ∼400 °C, even at relatively high oxygen partial pressures (0.1 bar) and for annealing times on the order of several hours. Nonetheless, given that during ALD of Pt, combustion reactions can lead to local temperatures that are considerably higher than the actual deposition temperature, we still consider Ostwald ripening among the possible growth mechanisms. Because gas-phase-mediated ripening has more favorable and less substrate-dependent energetics than surface-mediated ripening, the treatment of Ostwald ripening in model (II) is based on the work of Plessow and Abild-Pedersen.50 At any rate, this allows us to study the hypothetical effect of Ostwald ripening on the shape of the PSD during ALD of NPs.
The dynamic models allowed us to rule out nucleation and growth scenarios that are typically invoked in the literature to describe the growth of NPs in ALD:17,18,20,21 (1) NP nucleation in the first cycle followed by layer-by-layer growth on preexisting nuclei; (2) deposition on both the substrate and the NPs, single atom diffusion, aggregation, and attachment, that is, Model (I) letting s → + ∞; and (3) deposition on both the substrate and the NPs, single atom diffusion, aggregation, and attachment, and gas-phase-mediated Ostwald ripening, that is, Model (II). None of these scenarios described the observed PSD evolution (Figure 4 and Figures S17–S19). In particular, scenario (1) fails to describe the evolution of both the shape of the PSD and the Pt loading (Figure S17). Scenario (2) is characterized by left-skewed PSDs with a very narrow peak and a clear-cutoff on the large-size side. In this case, increasing the number of cycles results in a shift of the peak toward larges sizes and increasingly longer tails on the small-size side. Furthermore, the cyclic generation of single atoms on the substrate coupled to the absence of an annealing mechanism (e.g., NP diffusion and coalescence, and Ostwald ripening) gives rise to a series of nucleation peaks on the small-size side, whose number correlates with the number of cycles (Figure S15). Interestingly, such feature is observable in the left-skewed and narrow PSDs reported for plasma-enhanced Pt ALD at room temperature.17 This points to the fact that at such low temperatures the NPs are stable and effectively pinned on the substrate surface and that ALD growth proceeds, indeed, via single-atom attachment/deposition. Finally, it is worth noting that in scenario (II) each cycle results in an increase in the number of NPs per unit area, which, in the limit of low coverages, eventually saturates to a critical value depending on the ratio between single atoms surface diffusion and generation rates.26
Figure 4.

Experimental and simulated PSDs rescaled with respect to their respective average diameter and then normalized with respect to their integral. The black solid lines are representative PSDs obtained with model (I), the dark gray dashed line is the classical Lifshitz–Slyozov–Wagner (LSW) PSD for diffusion-limited Ostwald ripening, and the blue dotted lines are representative PSDs obtained with model (II). (a) Experimental PSDs obtained after one cycle in the temperature range 80–250 °C; the sum of squared errors (SSE) for model (I), model (II), and the LSW PSD is equal to 3, 131, and 49, respectively. (b) Experimental PSDs obtained in the temperature range 80–100 °C and in the cycle range 3–10; the SSE for model (I), model (II), and the LSW PSD is equal to 9, 182, and 65, respectively. (c) Experimental PSDs obtained in the temperature range 150–250 °C and in the cycle range 3–10;the SSE for model (I), model (II), and the LSW PSD is equal to 3, 157, and 72, respectively.
We also ruled out Ostwald ripening as the dominant growth mechanism because model (II) fails to give a qualitative description of the evolution of the PSD (Figures S19 and S20 and Figure 4) regardless of the temperature used for the simulations. In brief, below 200 °C, the addition of Ostwald ripening has little to no effect on the PSDs, which thus overlap with the ones of scenario (2). As expected, the onset of Ostwald ripening at higher temperatures results in the disappearance of the smaller NPs and thus of the long tails on the small-sizes side of scenario (2). Less trivially, the disappearance of smaller NPs in each cycle is also accompanied by the progressive narrowing (“focusing”) of the PSD, which remains nearly symmetric (Figures S19 and S20 and Figure 4c). On the contrary, simulated annealing periods after deposition on the order of tens to hundreds of hours result in the gradual broadening of the PSD (“defocusing”), which eventually approaches its the stationary self-similar form (Figures S20 and S21). The latter is slightly more symmetric than the left-skewed PSD predicted by the Lifshitz–Slyozov–Wagner (LSW) theory for diffusion-limited Ostwald ripening.29,49 Nevertheless the LSW PSD is a fair approximation, and, as such, it is plotted in Figure 4 as a reference for the asymptotic PSD in the case of Ostwald ripening. The transient focusing of the PSD discussed here is analogous to the behavior of an ensemble of NPs in a colloidal solution undergoing Ostwald ripening in the presence of an excess of monomers that was described by Talapin et al.49 via Monte Carlo simulations. Johnson et al.52 exploited the same phenomenon to grow core–shell NPs with very narrow PSDs in a layer-by-layer fashion by employing successive injections of small sacrificial NPs of the shell material to a colloidal solution containing the larger core NPs. Although the focusing of the PSD by Ostwald ripening could inspire the development of analogous NP synthesis routes based on ALD, it clearly cannot account for the PSDs observed here. Finally, the fact that model (II) cannot describe the PSD evolution reinforces the notion that Ostwald ripening of supported noble metals NPs is a slow process that dominates the late stages of growth of large and immobile NPs (i.e., dp> 5 nm) at high temperatures (∼400–1000 °C) and on long time scales (several hours to years).25,27,30,51,53−58
We could describe the shape of the experimental PSDs and their evolution with the number of cycles and temperature only when allowing for NP diffusion and coalescence, that is, through model (I) (Figure 4). Analysis of the full evolution of the PSD shape (Figure 1) revealed that at low deposition temperatures the NP mobility drops with increasing NP–substrate contact area (Dk ≈ k–2/3), whereas at high temperatures the NP mobility is virtually size-independent (s ≈ 0). In other words, at low temperatures, upon reaching a critical size, the NPs are effectively pinned on the substrate surface, thus acting as sinks for the mobile NPs that nucleate in their neighborhood, which explains the gradual disappearance of the peak on the small-size side (i.e., the nucleation peak). On the contrary, at high temperatures, because all of the diffusing species enjoy the same mobility and approximately the same capture efficiency: New nuclei can form and persist in each cycle, such that the nucleation peak remains in every cycle and the PSD gradually broadens. This picture is also in agreement with a qualitative assessment of the space-correlation of the NPs in the two different temperature regimes (see Figure 1 and Figures S4–S9): At low temperatures the NPs tend to be well-spaced, whereas at high temperatures small NPs can coexist next to large ones, and the distribution of the interparticle distance is qualitatively broad. Finally, we ascribe the temperature-dependent scaling of the NP mobility to the temperature dependence of the combustion rate of carbon ligands. At high temperatures, high combustion rates result in local heat and gas evolution that induces the unpinning of small NPs as well as large ones, whereas at low temperatures, slower combustion kinetics result in temperature and pressure fluctuations of lesser magnitude that can drive the diffusion of only relatively small NPs.
Although our analysis shows that NP diffusion and coalescence is certainly relevant to ALD of Pt NPs, one might expect that the exact size and temperature dependence of the NP mobility depend on the nature of the substrate and on the choice of coreactant (e.g., O2 and H2).25,27,36,38 For example, defects and functional groups on the substrate surface can act as strong binding sites or anchoring points for the Pt NPs and are expected to reduce the NP mobility30 and thus the metal aggregation.36 In particular, the nature of the defect determines its trapping energy and thus the temperature at which an NP of a given size has enough thermal energy to unpin.59 From this perspective, given the autocatalytic nature of combustion reactions, we speculate that there exists a well-defined critical deposition temperature for a given substrate and a given oxygen partial pressure at which combustion reactions locally evolve enough heat and gas to unpin small and large NPs alike. In other words, the exact temperature discriminating between growth regimes might differ between substrates, yet two temperature regimes are expected to arise on substrates with different nature, for example, TiO2 and graphene nanoplatelets. The considerations put forward so far for oxygen might hold for other coreactants, as they also inherently result in the removal of organic ligands and thus in local evolution of heat and gas. For example, Lubers et al.36 have shown that using H2 instead of O2 as a coreactant leads to smaller NPs and a less effective ligand removal. In this case, the latter might have mitigated the NP aggregation due to the mediating effect of the remaining ligands on NP diffusion and a less vigorous heat and gas evolution. However, using H2 also resulted in significantly lower deposition rates, which alone might account for the smaller NPs. Therefore, the effect of the coreactant on the evolution of the PSD certainly merits further study.
The inclusion of NP diffusion and coalescence among the NP-growth mechanisms relevant to ALD has important implications for its potential use in the synthesis of supported NPs. One implication is that the deposition temperature can be used to tailor the shape of the PSD and the NP space-correlation depending on the target application: narrow PSDs and well-spaced NPs at low temperatures and broad PSDs and particle-distance distributions at high temperatures. A corollary to this is that in general one cannot precisely control the NP size by simply varying the number of cycles, as in most instances this would result not only in an increase in the average diameter but also in a broadening of the PSD. In particular, control over the NP size with ALD-like atomic-level resolution can only be retained when the growth is dominated by single-atom attachment/deposition, that is, when the operating conditions during removal of organic ligands are such that the NPs are virtually immobile. The second important implication regards the understanding of selective ALD of NPs on surface defects. For instance, on the basis of the observation of the morphology of graphene after 500–1000 Pt ALD cycles, Kim et al.60 concluded that Pt is predominately deposited on line defects as opposed to basal planes. In light of our results, however, one can explain the same observation by invoking uniform deposition over the entire substrate surface, followed by the formation of mobile NPs that, over time, selectively pin onto the line defects, thus depleting the basal planes from ALD-grown material.
In summary, we studied the evolution of the PSD of Pt NPs in oxygen-based Pt ALD on bulk quantities of graphene nanoplatelets via a joint experimental and modeling approach. While different deposition temperatures resulted in virtually the same amount of deposited metal, the temperature had a dramatic effect on the PSD shape and its evolution with the number of cycles. Low deposition temperatures resulted in narrow PSDs and a mode that gradually shifts toward the large-size side with increasing number of cycles. On the contrary, high deposition temperatures resulted in PSDs presenting a persistent peak in the small-size side and a gradual broadening with increasing number of cycles. Ex situ TEM of the ALD half-cycles and dedicated annealing experiments show that the NPs form and grow mostly during the oxidation of the carbon ligands, after which they are relatively stable. Both analytical and numerical modeling show that the shape of the PSD and its evolution with temperature and number of cycles are best described in terms of sintering via NP diffusion and coalescence. In particular, the NP mobility was found to exhibit a temperature- and size-dependent scaling. At low temperatures, the NP mobility scales as the inverse of the contact area with the substrate, whereas at high temperatures it is virtually size-independent. As a result, at low temperatures, the NPs effectively pin onto the susbtrate surface once they reach a certain size, which then act as sinks for the small mobile NPs nucleating in their proximity. This explains the gradual disappearance of the peak on the small-size side with increasing number of cycles. At high temperatures, instead, the NP mobility is size-independent; that is, both nucleating NPs and preexisting large NPs enjoy approximately the same capture efficiency, hence the persistent nucleation peak in the PSD and its broadening with the number of cycles. We attribute the temperature-dependent scaling of the NP mobility to the different magnitudes of the local temperature and pressure fluctuations brought about by the combustion of carbon ligands at different temperatures. The insights presented here are relevant not only to ALD of Pt NPs but also to ALD of noble-metal NPs, in general, because these processes are typically based on the cyclic combustion of organic ligands and NP formation through metal aggregation. Finally, the fundamental insights presented in this Letter open up an avenue for the development of ALD routes for the scalable synthesis of size-selected NPs on high-surface-area substrates.
Acknowledgments
We acknowledge the support and the help provided by Damiano La Zara and Aris Goulas during the preparation of the manuscript. We also thank Máté Erdős for carrying out some preliminary work regarding the study of Ostwald ripening at the very beginning of this project and David Valdesueiro Gonzalez for the acquisition of the XPS spectra. The research leading to these results has received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC Grant, Agreement No. 279632.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpclett.6b02978.
Experimental details; comprehensive materials characterization including: XPS, Raman spectroscopy, XRD, INAA, TGA, TEM, and HRTEM; supplementary experimental results including TEMs and PSDs of ALD of Pt NPs on TiO2 nanopowders; detailed description of the dynamic model including NP diffusion and coalescence and of the one allowing for Ostwald ripening; LSW PSD; and overview of the simulations results including case studies and parameter sensitivity. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- King J. S.; Wittstock A.; Biener J.; Kucheyev S. O.; Wang Y. M.; Baumann T. F.; Giri S. K.; Hamza A. V.; Baeumer M.; Bent S. F. Ultralow Loading Pt Nanocatalysts Prepared by Atomic Layer Deposition on Carbon Aerogels. Nano Lett. 2008, 8, 2405–2409. 10.1021/nl801299z. [DOI] [PubMed] [Google Scholar]
- Goulas A.; van Ommen J. R. Atomic Layer Deposition of Platinum Clusters on Titania Nanoparticles at Atmospheric Pressure. J. Mater. Chem. A 2013, 1, 4647–4650. 10.1039/c3ta01665j. [DOI] [Google Scholar]
- Shao M.; Peles A.; Shoemaker K. Electrocatalysis on Platinum Nanoparticles: Particle Size Effect on Oxygen Reduction Reaction Activity. Nano Lett. 2011, 11, 3714–3719. 10.1021/nl2017459. [DOI] [PubMed] [Google Scholar]
- Van Bui H.; Grillo F.; van Ommen J. R. Atomic and Molecular Layer Deposition: off the Beaten Track. Chem. Commun. 2017, 53, 45–71. 10.1039/C6CC05568K. [DOI] [PubMed] [Google Scholar]
- Gür F. N.; Schwarz F. W.; Ye J.; Diez S.; Schmidt T. L. Toward Self-Assembled Plasmonic Devices: High-Yield Arrangement of Gold Nanoparticles on DNA Origami Templates. ACS Nano 2016, 10, 5374–5382. 10.1021/acsnano.6b01537. [DOI] [PubMed] [Google Scholar]
- Li J.; Wang Z.; Chen C.; Huang S. Atomic-Scale Observation of Migration and Coalescence of Au Nanoclusters on YSZ Surface by Aberration-Corrected STEM. Sci. Rep. 2014, 4, 5521. 10.1038/srep05521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K.; Imaoka T.; Chun W.-J.; Enoki O.; Katoh H.; Takenaga M.; Sonoi A. Size-Specific Catalytic Activity of Platinum Clusters Enhances Oxygen Reduction Reactions. Nat. Chem. 2009, 1, 397–402. 10.1038/nchem.288. [DOI] [PubMed] [Google Scholar]
- Eberhardt W.; Fayet P.; Cox D. M.; Fu Z.; Kaldor A.; Sherwood R.; Sondericker D. Photoemission from Mass-Selected Monodispersed Pt Clusters. Phys. Rev. Lett. 1990, 64, 780–783. 10.1103/PhysRevLett.64.780. [DOI] [PubMed] [Google Scholar]
- Wettergren K.; Schweinberger F. F.; Deiana D.; Ridge C. J.; Crampton A. S.; Rötzer M. D.; Hansen T. W.; Zhdanov V. P.; Heiz U.; Langhammer C. High Sintering Resistance of Size-Selected Platinum Cluster Catalysts by Suppressed Ostwald Ripening. Nano Lett. 2014, 14, 5803–5809. 10.1021/nl502686u. [DOI] [PubMed] [Google Scholar]
- Cuenya B. R.; Behafarid F. Nanocatalysis: Size- and Shape-Dependent Chemisorption and Catalytic Reactivity. Surf. Sci. Rep. 2015, 70, 135–187. 10.1016/j.surfrep.2015.01.001. [DOI] [Google Scholar]
- George S. M. Atomic Layer Deposition: An Overview. Chem. Rev. 2010, 110, 111–131. 10.1021/cr900056b. [DOI] [PubMed] [Google Scholar]
- Lu J.; Elam J. W.; Stair P. C. Atomic Layer Deposition–Sequential Self-limiting Surface Reactions for Advanced Catalyst “Bottom-up” Synthesis. Surf. Sci. Rep. 2016, 71, 410–472. 10.1016/j.surfrep.2016.03.003. [DOI] [Google Scholar]
- O’Neill B. J.; Jackson D. H. K.; Lee J.; Canlas C.; Stair P. C.; Marshall C. L.; Elam J. W.; Kuech T. F.; Dumesic J. A.; Huber G. W. Catalyst Design with Atomic Layer Deposition. ACS Catal. 2015, 5, 1804–1825. 10.1021/cs501862h. [DOI] [Google Scholar]
- Van Bui H.; Grillo F.; Helmer R.; Goulas A.; van Ommen J. R. Controlled Growth of Palladium Nanoparticles on Graphene Nanoplatelets via Scalable Atmospheric Pressure Atomic Layer Deposition. J. Phys. Chem. C 2016, 120, 8832–8840. 10.1021/acs.jpcc.6b02285. [DOI] [Google Scholar]
- Puurunen R. L. Surface Chemistry of Atomic Layer Deposition: A Case Study for the Trimethylaluminum/Water Process. J. Appl. Phys. 2005, 97, 121301. 10.1063/1.1940727. [DOI] [Google Scholar]
- Christensen S. T.; Elam J. W.; Lee B.; Feng Z.; Bedzyk M. J.; Hersam M. C. Nanoscale Structure and Morphology of Atomic Layer Deposition Platinum on SrTiO 3 (001). Chem. Mater. 2009, 21, 516–521. 10.1021/cm8026863. [DOI] [Google Scholar]
- Mackus A. J. M.; Weber M. J.; Thissen N. F. W.; Garcia-Alonso D.; Vervuurt R. H. J.; Assali S.; Bol A. A.; Verheijen M. A.; Kessels W. M. M. Atomic Layer Deposition of Pd and Pt Nanoparticles for Catalysis: on the Mechanisms of Nanoparticle Formation. Nanotechnology 2016, 27, 034001. 10.1088/0957-4484/27/3/034001. [DOI] [PubMed] [Google Scholar]
- Miikkulainen V.; Leskelä M.; Ritala M.; Puurunen R. L. Crystallinity of Inorganic Films Grown by Atomic Layer Deposition: Overview and General Trends. J. Appl. Phys. 2013, 113, 021301. 10.1063/1.4757907. [DOI] [Google Scholar]
- Knez M. Diffusion Phenomena in Atomic Layer Deposition. Semicond. Sci. Technol. 2012, 27, 074001. 10.1088/0268-1242/27/7/074001. [DOI] [Google Scholar]
- Mackus A. J. M.; Verheijen M. A.; Leick N.; Bol A. A.; Kessels W. M. M. Influence of Oxygen Exposure on the Nucleation of Platinum Atomic Layer Deposition: Consequences for Film Growth, Nanopatterning, and Nanoparticle Synthesis. Chem. Mater. 2013, 25, 1905–1911. 10.1021/cm400562u. [DOI] [Google Scholar]
- Shi J.; Li Z.; Kvit A.; Krylyuk S.; Davydov A. V.; Wang X. Electron Microscopy Observation of TiO 2 Nanocrystal Evolution in High-Temperature Atomic Layer Deposition. Nano Lett. 2013, 13, 5727–5734. 10.1021/nl403566u. [DOI] [PubMed] [Google Scholar]
- Sun S.; Zhang G.; Gauquelin N.; Chen N.; Zhou J.; Yang S.; Chen W.; Meng X.; Geng D.; Banis M. N.; et al. Single-atom Catalysis Using Pt/Graphene Achieved through Atomic Layer Deposition. Sci. Rep. 2013, 3, 1775. 10.1038/srep01775. [DOI] [Google Scholar]
- Griffiths M. B. E.; Pallister P. J.; Mandia D. J.; Barry S. T. Atomic Layer Deposition of Gold Metal. Chem. Mater. 2016, 28, 44–46. 10.1021/acs.chemmater.5b04562. [DOI] [Google Scholar]
- Zhang Z. Atomistic Processes in the Early Stages of Thin-Film Growth. Science 1997, 276, 377–383. 10.1126/science.276.5311.377. [DOI] [PubMed] [Google Scholar]
- Zinke-Allmang M.; Feldman L. C.; Grabow M. H. Clustering on Surfaces. Surf. Sci. Rep. 1992, 16, 377–463. 10.1016/0167-5729(92)90006-W. [DOI] [Google Scholar]
- Venables J. A.; Spiller G. D. T.; Hanbucken M. Nucleation and Growth of Thin Films. Rep. Prog. Phys. 1984, 47, 399–459. 10.1088/0034-4885/47/4/002. [DOI] [Google Scholar]
- Wynblatt P.; Gjostein N. Supported metal crystallites. Prog. Solid State Chem. 1975, 9, 21–58. 10.1016/0079-6786(75)90013-8. [DOI] [Google Scholar]
- La Torre A.; del Carmen Giménez-López M.; Fay M. W.; Rance G. A.; Solomonsz W. A.; Chamberlain T. W.; Brown P. D.; Khlobystov A. N. Assembly, Growth, and Catalytic Activity of Gold Nanoparticles in Hollow Carbon Nanofibers. ACS Nano 2012, 6, 2000–2007. 10.1021/nn300400z. [DOI] [PubMed] [Google Scholar]
- Woehl T. J.; Park C.; Evans J. E.; Arslan I.; Ristenpart W. D.; Browning N. D. Direct Observation of Aggregative Nanoparticle Growth: Kinetic Modeling of the Size Distribution and Growth Rate. Nano Lett. 2014, 14, 373–378. 10.1021/nl4043328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jak M.; Konstapel C.; van Kreuningen A.; Verhoeven J.; Frenken J. Scanning Tunnelling Microscopy Study of the Growth of Small Palladium Particles on TiO2(110). Surf. Sci. 2000, 457, 295–310. 10.1016/S0039-6028(00)00431-3. [DOI] [Google Scholar]
- Bell G. R.; Dawson P. M.; Pandey P. A.; Wilson N. R.; Mulheran P. A. Sizedependent Mobility of Gold Nano-Clusters During Growth on Chemically Modified Graphene. APL Mater. 2014, 2, 012109. 10.1063/1.4862696. [DOI] [Google Scholar]
- Karasulu B.; Vervuurt R.; Kessels E.; Bol A. A. Continuous and Ultrathin Platinum Films on Graphene using Atomic Layer Deposition: A Combined Computational and Experimental Study. Nanoscale 2016, 8, 19829–19845. 10.1039/C6NR07483A. [DOI] [PubMed] [Google Scholar]
- Aaltonen T.; Ritala M.; Sajavaara T.; Keinonen J.; Leskelä M. Atomic Layer Deposition of Platinum Thin Films. Chem. Mater. 2003, 15, 1924–1928. 10.1021/cm021333t. [DOI] [Google Scholar]
- Takakusagi S.; Fukui K.-i.; Tero R.; Nariyuki F.; Iwasawa Y. Self-Limiting Growth of Pt Nanoparticles from MeCpPtMe3 Adsorbed on TiO2(110) Studied by Scanning Tunneling Microscopy. Phys. Rev. Lett. 2003, 91, 066102. 10.1103/PhysRevLett.91.066102. [DOI] [PubMed] [Google Scholar]
- Mackus A. J. M.; Leick N.; Baker L.; Kessels W. M. M. Catalytic Combustion and Dehydrogenation Reactions during Atomic Layer Deposition of Platinum. Chem. Mater. 2012, 24, 1752–1761. 10.1021/cm203812v. [DOI] [Google Scholar]
- Lubers A. M.; Muhich C. L.; Anderson K. M.; Weimer A. W. Mechanistic Studies for Depositing Highly Dispersed Pt Nanoparticles on Carbon by use of Trimethyl(methylcyclopentadienyl)platinum(IV) Reactions with O2 and H2. J. Nanopart. Res. 2015, 17, 179. 10.1007/s11051-015-2982-9. [DOI] [Google Scholar]
- Zan R.; Bangert U.; Ramasse Q.; Novoselov K. S. Interaction of Metals with Suspended Graphene Observed by Transmission Electron Microscopy. J. Phys. Chem. Lett. 2012, 3, 953–958. 10.1021/jz201653g. [DOI] [PubMed] [Google Scholar]
- Chu Y.; Ruckenstein E. Behavior of Platinum Crystallites on Carbon Substrates. Surf. Sci. 1977, 67, 517–540. 10.1016/0039-6028(77)90012-7. [DOI] [Google Scholar]
- Rubets V.; Kukushkin S. Determination of Migration Mechanisms and Their Influence on the Structure of Films. Thin Solid Films 1992, 221, 267–270. 10.1016/0040-6090(92)90825-V. [DOI] [Google Scholar]
- Kukushkin S.; Osipov A. Soliton Model of Island Migration in Thin Films. Surf. Sci. 1995, 329, 135–140. 10.1016/0039-6028(95)00020-8. [DOI] [Google Scholar]
- von Smoluchowski M. Drei Vortrage Uber Diffusion. Brownsche Bewegung und Koagulation von Kolloidteilchen. Z. Phys. 1916, 17, 557–585. [Google Scholar]
- Vicanek M.; Ghoniem N. The Effects of Mobility Coalescence on the Evolution of Surface Atomic Clusters. Thin Solid Films 1992, 207, 90–97. 10.1016/0040-6090(92)90107-M. [DOI] [Google Scholar]
- Sholl D. S.; Skodje R. T. Late-stage Coarsening of Adlayers by Dynamic Cluster Coalescence. Phys. A 1996, 231, 631–647. 10.1016/0378-4371(96)00112-4. [DOI] [Google Scholar]
- Jensen P. Growth of Nanostructures by Cluster Deposition: Experiments and Simple Models. Rev. Mod. Phys. 1999, 71, 1695–1735. 10.1103/RevModPhys.71.1695. [DOI] [Google Scholar]
- Winkler K.; Wojciechowski T.; Liszewska M.; Górecka E.; Fiałkowski M. Morphological Changes of Gold Nanoparticles due to Adsorption onto Silicon Substrate and Oxygen Plasma Treatment. RSC Adv. 2014, 4, 12729–12736. 10.1039/C4RA00507D. [DOI] [Google Scholar]
- Kryukov Y. A.; Amar J. G. Effects of Cluster Diffusion on the Island Density and Size Distribution in Submonolayer Island Growth. Phys. Rev. E 2011, 83, 041611. 10.1103/PhysRevE.83.041611. [DOI] [PubMed] [Google Scholar]
- Hubartt B. C.; Kryukov Y. A.; Amar J. G. Rate-Equation Approach to Irreversible Island Growth with Cluster Diffusion. Phys. Rev. E 2011, 84, 59901. 10.1103/PhysRevE.84.059901. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Muhich C. L.; Neltner B. T.; Weimer A. W.; Musgrave C. B. Growth of Pt Particles on the Anatase TiO2 (101) Surface. J. Phys. Chem. C 2012, 116, 12114–12123. 10.1021/jp302273m. [DOI] [Google Scholar]
- Talapin D. V.; Rogach A. L.; Haase M.; Weller H. Evolution of an Ensemble of Nanoparticles in a Colloidal Solution: Theoretical Study. J. Phys. Chem. B 2001, 105, 12278–12285. 10.1021/jp012229m. [DOI] [Google Scholar]
- Plessow P. N.; Abild-Pedersen F. Sintering of Pt Nanoparticles via Volatile PtO2: Simulation and Comparison with Experiments. ACS Catal. 2016, 6, 7098–7108. 10.1021/acscatal.6b01646. [DOI] [Google Scholar]
- Harris P. J. F. Growth and Structure of Supported Metal Catalyst Particles. Int. Mater. Rev. 1995, 40, 97–115. 10.1179/imr.1995.40.3.97. [DOI] [Google Scholar]
- Johnson N. J. J.; Korinek A.; Dong C.; van Veggel F. C. J. M. Self-Focusing by Ostwald Ripening: A Strategy for Layer-by-Layer Epitaxial Growth on Upconverting Nanocrystals. J. Am. Chem. Soc. 2012, 134, 11068–11071. 10.1021/ja302717u. [DOI] [PubMed] [Google Scholar]; PMID: 22734596
- Simonsen S. B.; Chorkendorff I.; Dahl S.; Skoglundh M.; Sehested J.; Helveg S. Direct Observations of Oxygen-induced Platinum Nanoparticle Ripening Studied by In Situ TEM. J. Am. Chem. Soc. 2010, 132, 7968–7975. 10.1021/ja910094r. [DOI] [PubMed] [Google Scholar]
- Granqvist C. Size Distributions for Supported Metal Catalysts: Coalescence Growth Versus Ostwald Ripening. J. Catal. 1976, 42, 477–479. 10.1016/0021-9517(76)90125-1. [DOI] [Google Scholar]
- Heinemann K.; Poppa H. Direct Observation of Small Cluster Mobility and Ripening. Thin Solid Films 1976, 33, 237–251. 10.1016/0040-6090(76)90084-5. [DOI] [Google Scholar]
- Prestat E.; Popescu R.; Blank H.; Schneider R.; Gerthsen D. Coarsening of Pt Nanoparticles on Amorphous Carbon Film. Surf. Sci. 2013, 609, 195–202. 10.1016/j.susc.2012.12.014. [DOI] [Google Scholar]
- Ruckenstein E. Growth Kinetics and the Size Distributions of Supported Metal Crystallites. J. Catal. 1973, 29, 224–245. 10.1016/0021-9517(73)90226-1. [DOI] [Google Scholar]
- Wang F.; Richards V. N.; Shields S. P.; Buhro W. E. Kinetics and Mechanisms of Aggregative Nanocrystal Growth. Chem. Mater. 2014, 26, 5–21. 10.1021/cm402139r. [DOI] [Google Scholar]
- Carrey J.; Maurice J.-L.; Petroff F.; Vaurès A. Growth of Au Clusters on Amorphous Al2O3: are Small Clusters More Mobile than Atoms?. Surf. Sci. 2002, 504, 75–82. 10.1016/S0039-6028(01)01796-4. [DOI] [Google Scholar]
- Kim K.; Lee H.-B.-R.; Johnson R. W.; Tanskanen J. T.; Liu N.; Kim M.-G.; Pang C.; Ahn C.; Bent S. F.; Bao Z. Selective Metal Deposition at Graphene Line Defects by Atomic Layer Deposition. Nat. Commun. 2014, 5, 4781. 10.1038/ncomms5781. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.