

Abstract
GPR55 is a newly de-orphanized Class A GPCR that has been implicated in inflammatory pain, neuropathic pain, metabolic disorder, bone development, and cancer. Few potent GPR55 ligands have been identified to date. This is largely due to an absence of information about salient features of GPR55, such as residues important for signaling and residues implicated in the GPR55 signaling cascade. The goal of the work reported here was to identify residues that are key for the signaling of the GPR55 endogenous ligand, l-α-lysophosphatidylinositol (LPI), as well as the signaling of the GPR55 agonist, ML184, (CID 2440433, 3-[4-(2,3-dimethylphenyl)piperazine-1-carbonyl]-N,N-dimethyl-4-pyrrolidin-1-ylbenzenesulfonamide). Serum Response Element (SRE) and Serum Response Factor (SRF) luciferase assays were used as read-outs for studying LPI and ML184 signaling at the GPR55 mutants. A GPR55 R* model based on the recent delta-opioid receptor (DOR)crystal structure was used to interpret the resultant mutation data. Two residues were found to be crucial for agonist signaling at GPR55, K2.60 and E3.29, suggesting that these residues form the primary interaction site for ML184 and LPI at GPR55. Y3.32F, H(170)F and F6.55A/L mutation results suggested that these residues are part of the orthosteric binding site for ML184, while Y3.32F and H(170)F mutation results suggest that these two residues are part of the LPI binding pocket. Y3.32L, M3.36A and F6.48A mutation results suggest the importance of a Y3.32/M3.36/F6.48 cluster in the GPR55 signalling cascade. C(10)A and C(260)A mutations suggest that these residues form a second disulfide bridge in the extracellular domain of GPR55, occluding ligand extracellular entry in the TMH1-TMH7 region of GPR55. Taken together, these results provide the first set of discrete information on GPR55 residues important for LPI and ML184 signaling and for GPR55 activation. This information should aid in the rational design of next generation GPR55 ligands and hopefully the creation of the first high affinity GPR55 radioligand, a tool that is sorely needed in the field.
Introduction
GPR55 [GenBank accession number NM005683] is a Class A G-protein coupled receptor (GPCR) that recognizes a sub-set of cannabinoid CB1 and CB2 ligands, suggesting that GPR55 may be a cannabinoid receptor. 1Subsequently, l-α-lysophosphatidylinositol1-2 was reported to be the endogenous ligand of GPR55. GPR55 has been found to be implicated in inflammatory pain, neuropathic pain, metabolic disorder, bone development, and cancer,3 indicating the real potential of GPR55 ligands as therapeutics. In search of more potent GPR55 ligands and in collaboration with the Sanford-Burnham Institute, we performed high throughput screens for GPR55 agonists and antagonists using the NIH library of 300,000 compounds and identified several novel GPR55 chemotypes for each.4 One of these compounds from the agonist screen is ML184, (CID 2440433, 3-[4-(2,3-dimethylphenyl) piperazine-1-carbonyl]-N,N-dimethyl-4-pyrrol-idin-1-ylbenzenesulfonamide)5 (EC50=263nM). However, despite these ligand development efforts, the best GPR55 ligands (like ML184) remain active at sub-micromolar concentrations, concentrations not low enough for the development of a GPR55 radioligand.
To address this situation, we reasoned that knowledge of GPR55 structure, particularly binding pocket residues important for ligand signalling and those involved in the receptor activation would greatly aid rational GPR55 drug design approaches. To this end, we report here a GPR55 mutation study guided and analysed using a GPR55 R* model based on the recent delta opioid receptor (DOR) crystal structure. This study identifies multiple residues important for ML184 signaling and for GPR55 activation.
Results
Biological Evaluation
Serum Response Element (SRE) responses of mutant and wildtype GPR55 receptors
HEK293 cells transiently transfected with both pGL4.33 [luc2P/SRE/Hygro] and each of the mutants, as well as, WT were made to assess agonist-induced receptor activation of SRE. The SRE assay assesses the contribution of MAPK/ERK signalling pathway for GPR55 activity6. We found that the WT GPR55 has an EC50 value of 56 nM for the agonist ML184 (Fig 1 and Table 1). We determined that the EC50 value did not change regardless of the level of receptor expression (See S1 and S2 in Supporting Information). Mutants Y3.32F and Y3.32L showed right shifted SRE response curves with EC50's higher than WT (Fig. 1A). The mutant F6.55A and F6.55L induced a similar SRE response to ML184 with right shifted curves. The Q6.58M curve was almost identical to that of WT indicating this mutant is not important to GPR55 receptor function. The mutant C(10)A showed an increased EC50 value (636 nM). Very similarly, the mutant C(260)A induced an increased EC50 value (657 nM), suggesting the existence of a disulfide bridge (see below). Q7.36A and Q7.36N showed similar responses to WT indicating this amino acid is not critical for agonist induced GPR55 receptor activation. The response curve of H(170)F only showed a slight difference from that of WT. Mutant M3.36A, F6.48A, E3.29A and E3.29L all showed increased EC50 values. E3.29A and E3.29L showed very little responses. Among all the mutants, K2.60A did not show any response in the SRE assay indicating that K2.60A rendered the GPR55 receptor non-functional.
Figure 1.
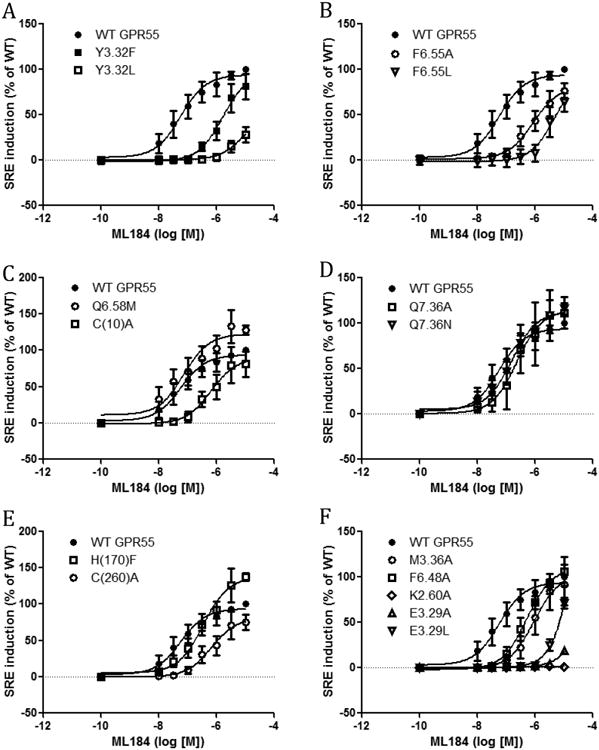
SRE responses induced by ML184 in GPR55 wild type and mutant transfected cells. (A) WT GPR55, Y3.32F and Y3.32L. (B) WT GPR55, F6.55A and F6.55L. (C) WT GPR55, Q6.58M and C(10)A. (D) WT GPR55, Q7.36A and Q7.36N. (E) WT GPR55, H(170)F and C(260)A. (F) WT GPR55, M3.36A, F6.48A, K2.60A, E3.29A and E3.29L.
Table 1.
ML184 induced SRE responses of wild-type and mutant transfected cells.
| ML184 | EC50 (nM) | CI (nM) | Fold/WT |
|---|---|---|---|
| WT | 56 | 24 to 133 | - |
| Y3.32F | 1659 | 670 to 4110 | 30 |
| Y3.32L | 9514 | 718 to 126100 | 170 |
| F6.55A | 840 | 345 to 2045 | 15 |
| F6.55L | 5131 | 1034 to 25460 | 92 |
| Q6.58M | 72 | 22 to 237 | 1 |
| C(10)A | 635 | 197 to 2044 | 11 |
| H(170)F | 341 | 158 to 734 | 6 |
| C(260)A | 657 | 222 to 1941 | 12 |
| Q7.36A | 230 | 57 to 920 | 4 |
| Q7.36N | 154 | 69 to 342 | 3 |
| M3.36A | 900 | 290 to 2790 | 16 |
| F6.48A | 514 | 266 to 993 | 9 |
| K2.60A | NO response | - | - |
| E3.29A | >30000 | very wide | - |
| E3.29L | >30000 | Very wide | - |
From n ≥ 3 experiments
In addition to the agonist ML184, the endogenous ligand (LPI) was used in the SRE assay for all the mutants (Fig 2). K2.60A did not show any LPI-induced responses compared with WT. E3.29A and E3.29L showed a slight response at high concentrations of LPI. F6.55A, F6.55L, Q6.58M, Q7.36A and Q7.36N all showed similar curves as that of WT. Y3.32F, Y3.32L, C(10)A, H(170)F, C(260)A, M3.36A and F6.48A showed right shifted curves compared with WT. The LPI-induced SRE response did not reach plateau for all the constructs including WT GPR55, thus no EC50 value was calculated.
Figure 2.
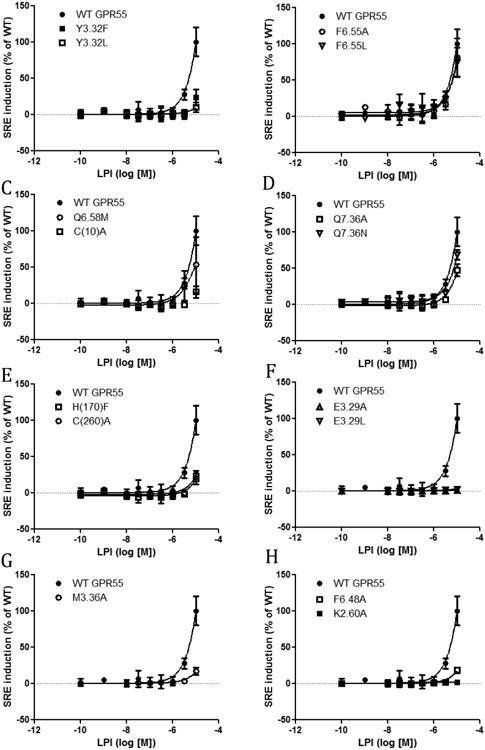
SRE responses induced by LPI in GPR55 wild type and mutant transfected cells. (A) WT GPR55, Y3.32F and Y3.32L. (B) WT GPR55, F6.55A and F6.55L. (C) WT GPR55, Q6.58M and C(10)A. (D) WT GPR55, Q7.36A and Q7.36N. (E) WT GPR55, H(170)F and C(260)A. (F) WT GPR55, E3.29A and E3.29L. (G) WT GPR55 and M3.36A. (H) WT GPR55, F6.48A and K2.60A,
Serum Response Factor (SRF) responses of mutant and wildtype GPR55 receptors
In addition to the SRE assay, the SRF assay was used to investigate another signaling pathway of GPR55. The SRF response is an indicator of G12-RhoA pathway activation, which GPR55 has been reported to induce.7 The study of two distinct signaling pathways of GPR55 broadens the understanding of the effects of these mutants on GPR55 function. Thus, the SRF assay was used to test GPR55 mutants in addition to the SRE assay (Fig 3 and Fig 4). Y3.32F and Y3.32L showed right shifted curves compared with WT. Y3.32L had a higher EC50 value (864μM (Fig 3). Similar to the SRE assay, the F6.55A and F6.55L had higher EC50 values than WT (Fig 3, Table 2). Both C(10)A and C(260)A showed less response than WT. M3.36A, F6.48A, E3.29A and E3.29L all behaved in the SRF assay similar to in the SRE assay with reduced responses. K2.60A was not able to induce any response in the SRF assay similar to its inactivity in the SRE assay.
Figure 3.
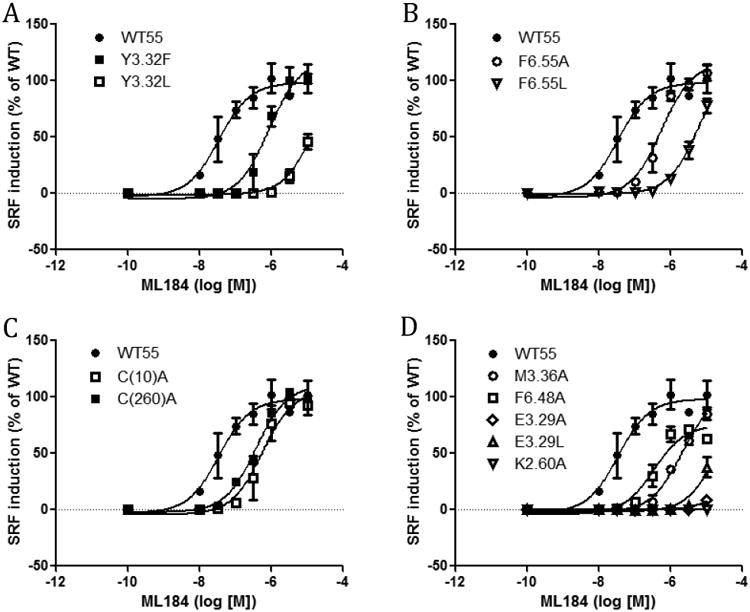
SRF responses induced by ML184 in GPR55 wild type and mutant transfected cells. (A) WT GPR55, Y3.32F and Y3.32L. (B) WT GPR55, F6.55A and F6.55L. (C) WT GPR55, C(10)A and C(260)A. (D) WT GPR55, M3.36A, F6.48A, K2.60A, E3.29A and E3.29L.
Figure 4.
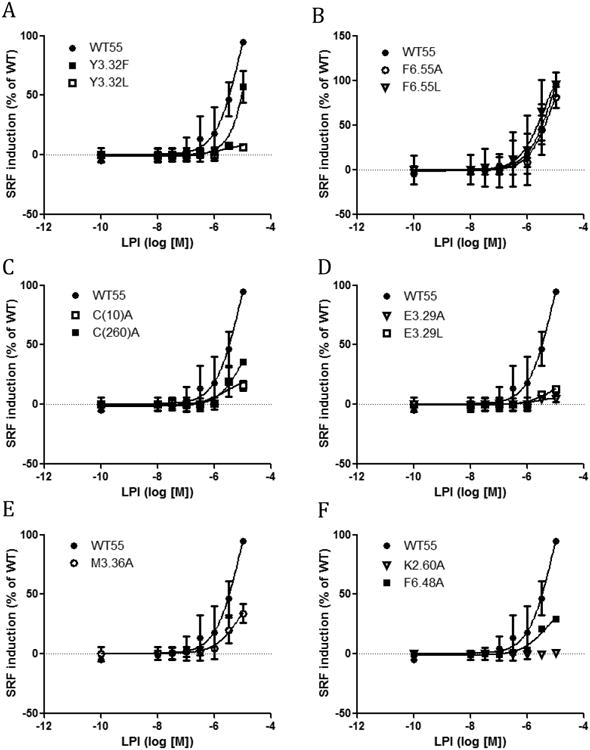
SRF responses induced by LPI in GPR55 wild type and mutant transfected cells. (A) WT GPR55, Y3.32F and Y3.32L. (B) WT GPR55, F6.55A and F6.55L. (C) WT GPR55, C(10)A and C(260)A. (D) WT GPR55, E3.29A and E3.29L. (E) WT GPR55 and M3.36A. (F) WT GPR55, F6.48A, K2.60A,
Table 2.
ML184 induced SRF responses of wild-type and mutant transfected cells.
| ML184 | EC50 (nM) | CI (nM) | Fold/WT |
|---|---|---|---|
| WT | 33 | 15 to 75 | - |
| Y3.32F | 851 | 449 to 1615 | 26 |
| Y3.32L | 864000 | very wide | 26182 |
| F6.55A | 550 | 338 to 893 | 17 |
| F6.55L | 10500 | 4663 to 23630 | 318 |
| C(10)A | 554 | 254 to 1206 | 17 |
| C(260)A | 363 | 201 to 656 | 11 |
| M3.36A | 2044 | 1415 to 2952 | 62 |
| F6.48A | 335 | 177 to 633 | 10 |
| K2.60A | NO response | - | - |
| E3.29A | > 30000 | very wide | - |
| E3.29L | > 30000 | very wide | - |
n ≥ 3 experiments.
Molecular Modeling
No x-ray crystal structure for GPR55 has been reported. For this reason, a GPR55 activated state (R*) model based upon the crystal structure of the delta-opioid receptor (DOR) was used to interpret the mutation results reported here.8 Glide docking studies in this model focused on ML184 and LPI. These studies were used to identify a docking site for each ligand in the GPR55 R* model that involves residues on TMHs2-3-5-6 and the EC-2 loop.
Primary Interactions for Agonist Signaling. ML184 Docked into the GPR55 R* Bundle
Figure 6 describes the ML184 binding site identified by Glide. Figure 6A shows all hydrogen bonding interactions for ML184 at GPR55. Here, the ML184 sulfonamide oxygen hydrogen bonds directly with K2.60 (H bond (N-O) distance, 2.8 Å; (N-H—O) angle, 171°). H(170) in the EC-2 loop also forms a hydrogen bond with an ML184 sulfonamide oxygen (H bond (N-O) distance, 2.8 Å; (N-H—O) angle, 163°). Y3.32 forms a hydrogen bond with the ML184 carboxamide oxygen (H bond (O-O) distance, 2.9 Å; (O-H—O) angle, 151°).
Figure 6.
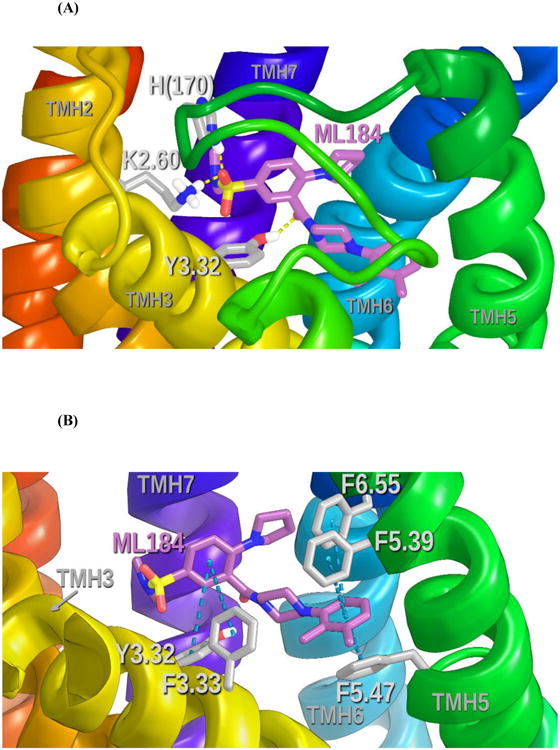
(A) This figure shows all hydrogen bonding interactions for ML184 at GPR55. The view is looking from TMHs 4 and 5 towards TMH7. Hydrogen bonding is shown with dotted yellow lines. (B) This figure shows all aromatic stacking interactions for ML184 at GPR55. The view is looking from TMH 4 towards TMH6. Aromatic stacking interactions are shown with light blue dotted lines between ring centroids. See Figure S-3 for a ligand interaction diagram for the ML184/GPR55 R* complex.
In addition to hydrogen bonding, ML184 also has aromatic stacking interactions in its binding site. Figure 6B shows all of these aromatic stacking interactions. Y3.32 and F3.33 form aromatic stacks with the ML184 benzene ring proximal to the ML184 sulfonamide moiety (Y3.32 ring centroid to centroid distance 5.5 Å; angle 77°); (F3.33 ring centroid to centroid distance 6.5 Å; angle 48°). F6.55 forms an aromatic stacking interaction with the distal, dimethyl-phenyl ring of ML184 (ring centroid to centroid distance 5.0 Å; angle 64°); while, F5.39 and F5.47 also form aromatic stacks with the central ML184 aromatic ring (F5.39 ring centroid to centroid distance 5.3 Å; angle 52°); (F5.47 ring centroid to centroid distance 4.5 Å; angle 45°).
M7.39 is engaged in a Met-aromatic ring interaction with the central benzene of ML184 (Distances to central benzene ring centroid from M7.39 sidechain atoms CG 5.0 Å, SD 5.3 Å, CE 4.4 Å) (see Figure 7A).9 In this orientation, the sulfur points up, with adjacent carbons pointing down towards phenyl ring carbons on ML184. ML184 also has an indirect interaction with E3.29, as this residue holds K2.60 (with which ML184 has a direct hydrogen bond) in a salt bridge that directs K2.60 towards the ML184 binding site (see Figure 7B). The ML184 conformer docked here is 1.14 kcal/mol above the ML184 global min. The Glide Dock score for the complex was -9.5. Table S-1 in Supplementary Information provides a summary of the residues with which ML184 interacts and the energy of each.
Figure 7.
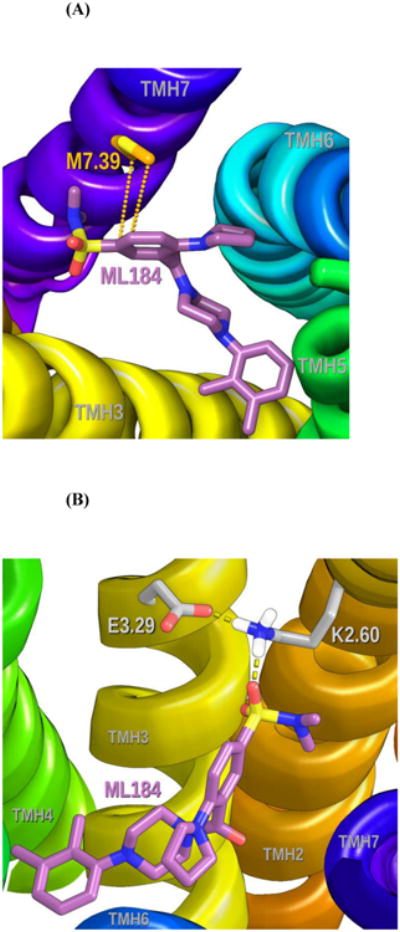
(A) M7.39 is engaged in a Met-aromatic ring interaction with the central benzene of ML184. 9 In this orientation, the sulfur points up, with adjacent carbons pointing down towards phenyl ring carbons on ML184.
(B) Although there is no direct interaction between ML184 and E3.29 in the current model, modeling suggests that E3.29 forms a salt bridge with K2.60. This salt bridge positions K2.60 for interaction with ML184. The view here is from TMH6 looking towards TMH2/3.
Toggle Switch Residues
GPR55 shares with the P2Y1210 and hPAR-1 11 receptors, a Y/F3.32-M3.36-F6.48 cluster of residues that likely acts as the toggle switch for activation of GPR55. Figure 8 illustrates these toggle switch residues contoured at their Van der Waals radii in the inactive (R) and the activated (R*) state.
Figure 8.
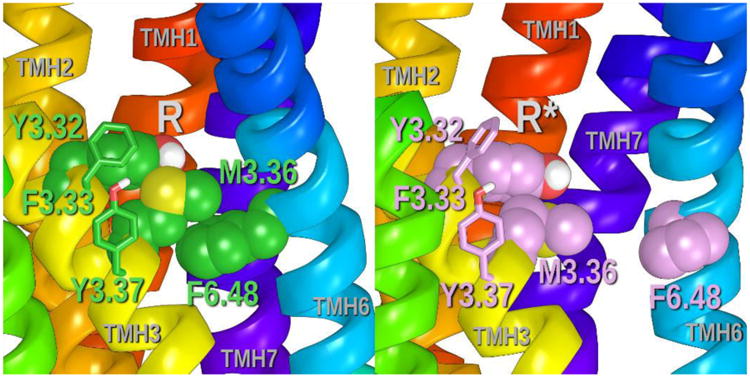
This figure illustrates the positions of the toggle switch residues, Y3.32, M3.36 and F6.48 (contoured at their Van der Waals radii) in the GPR55 R and R* models. Additional aromatic residues in the region of the toggle switch help stabilize the cluster of residues. These are F3.33 and Y3.37.
Inactive State
In the inactive state (Figure 8A), Y3.32 (χ1= 178°) sits extracellular to M3.36 (χ1= -170°), maintaining Van der Waals interactions with M3.36. M3.36 (χ1= -170°) is stacked over F6.48 (χ1= -90°). M3.36 also has a methione/aromatic ring interaction with F6.48 (Distances to F6.48 benzene ring centroid from M3.36 sidechain atoms CG 3.6 Å, SD 4.6 Å, CE 3.8 Å).9 Additional aromatic residues in the region of the toggle switch help stabilize the cluster of residues. These are F3.33 (χ1= -74°) and Y3.37 (χ1= -57°).
Agonist Activated State
ML184 binding causes Y3.32 to change conformations (χ1= 178°➔χ1= -157°), because Y3.32 is a direct ligand binding site residue. Figure 8B ilustrates that the movement of Y3.32 frees M3.36 to undergo a conformational change (χ1= -170°trans ➔χ1= -76° g+), which then allows F6.48 to undergo its (χ1= -90° g+➔χ1= -174° trans) conformational change. The F6.48 conformational change causes flexing in the SFXP hinge region of GPR55, straightening TMH6 and breaking the ionic lock (R3.50/Q6.30) to produce an opening at the intracellular end of GPR55 for G protein coupling. F3.33 (χ1= -79°) and Y3.37 (χ1= -59°) remain members of the extended cluster. Figure 9 illustrates the position of ML184 to the extended toggle switch residues in the GPR55 activated state.
Figure 9.
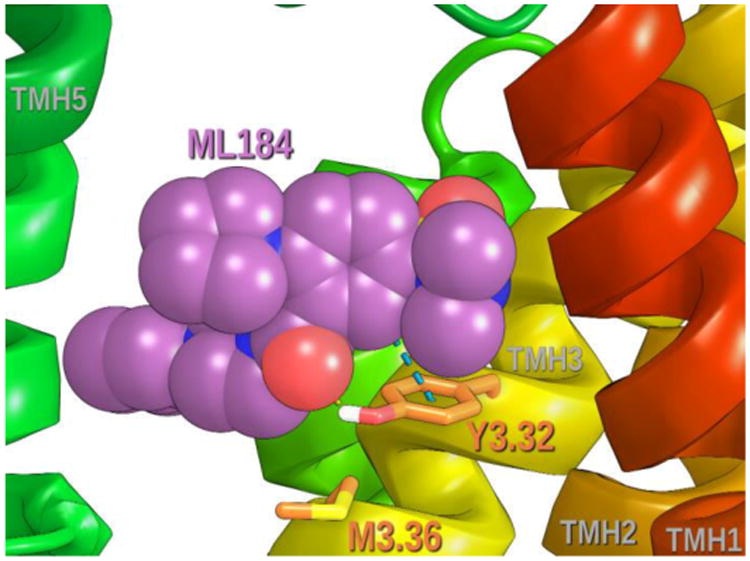
The position of ML184 relative to the extended toggle switch residues in the GPR55 activated state is illustrated here. ML184 (contoured at its Van der Waals radii) is positioned above Y3.32 and interacts directly with it. The movement of Y3.32 for this ML184 interaction, permits M3.36 (χ1 trans ➔ g+) and, in turn, F6.48 (χ1 g+➔ trans) to change to their R* conformations.
Disulfide Bridge Residues
As described in the Methods section, the high degree of sequence homology between GPR55 and the CXCR4 receptor12, particularly in the EC regions, dictated several modifications to our initial set of GPR55 models13. These were (1) the introduction of EC helical extensions on TMH5-7 of GPR55; (2) the introduction of a β sheet motif into the EC-2 loop in GPR55; and, (3) the introduction of a disulfide bridge between Cys(10) in the N-terminus and Cys(260) in the EC-3 loop near the top of TMH714. In the current paper, we tested the importance of a Cys(10)-Cys(260) disulfide bridge via single point mutations. Figure 10 illustrates the extracellular end of the receptor and the disulfide bridge between N-terminal C(10) and C(260) at the EC end of TMH7.
Figure 10.
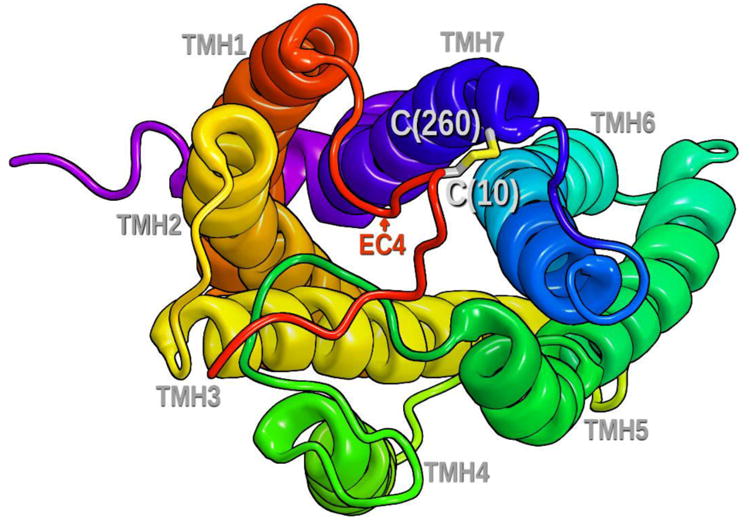
This figure illustrates the extracellular end of the receptor and the disulfide bridge between N-terminal C(10) and C(260) at the EC end of TMH7.
Binding Pocket Position
Figure 11 illustrates the positions of Q6.58 and Q7.36 relative to the ML184 binding site. These residues are positioned relatively high on the EC end of TMHs 6 and 7 and above the ML184 binding pocket in our current GPR55 R* model. In our first GPR55 model, Q6.58 was near the nitrogen in the pendant five membered ring of ML184 and able to form a hydrogen bonding interaction with this nitrogen.15 As discussed in the next section, mutation results suggest that Q6.58 and Q7.36 do not interact with ML184. This necessitated a re-orientation of ML184 in the binding pocket in the current model.
Figure 11.
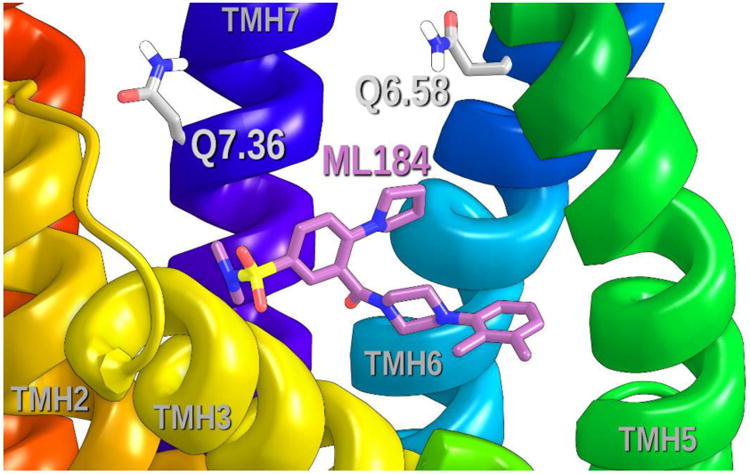
This figure illustrates the positions of Q6.58 and Q7.36 relative to the ML184 binding site. Both residues are clearly above the ML184 binding site.
LPI in GPR55 R* Model
Figure 12 illustrates the dock of the GPR55 endogenous ligand, LPI in the GPR55 R* model. In general, Glide docked LPI in the same region of GPR55 R* as ML184, i.e., TMHs2-3-5-6 and the EC-2 loop region, except that the LPI binding site is slightly higher in the binding pocket. LPI appears designed to take advantage of both E3.29 and K2.60, with K2.60 being the primary interaction site. Here a phosphate group oxygen can accept a hydrogen bond from K2.60 (H bond (N-O) distance, 2.6 Å; (N-H—O) angle, 173°), while the C-2 hydroxyl group can donate a hydrogen bond to E3.29 (H bond (O-O) distance 2.7 Å; (O-H—O) angle, 176°). The LPI-K2.60 hydrogen bond is the largest contributor to the Interaction Energy in the LPI/GPR55 R* complex, -16.60 kcal/mol. The energy of interaction for LPI with E3.29 is -4.79 kcal/mol.
Figure 12.
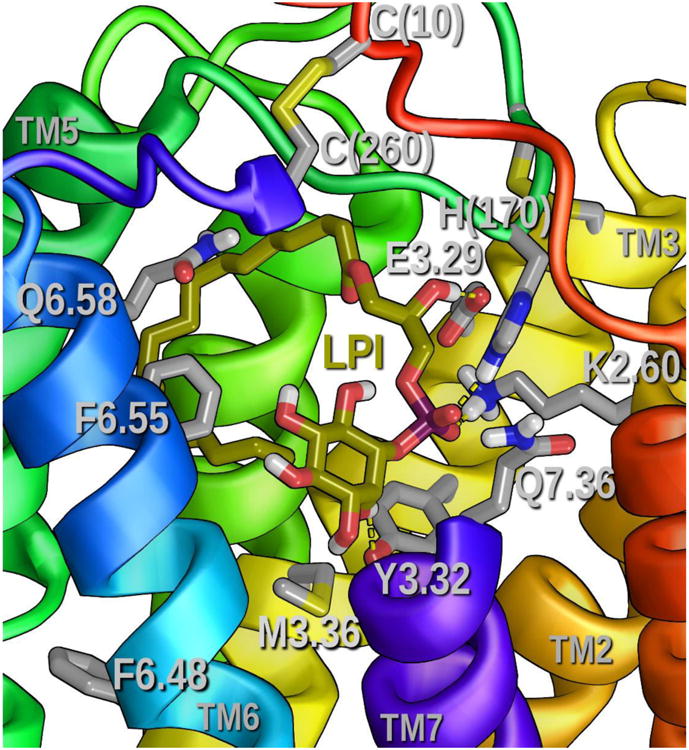
This figure shows LPI docked in the GPR55 R* model. All hydrogen bonding interactions for LPI are indicated by yellow dashed lines. The view is looking from TMHs 7 towards TMH4. The extracellular portion of TMH7 has been removed from the view. See Table S-2 in Supplementary Information for the list of residues with which LPI has predominantly Van der Waals interactions. A ligand interaction diagram is also available for the LPI/GPR55 R* complex (see Figure S-4.
The second phosphate group oxygen can accept a hydrogen bond from H(170) (H bond (N-O) distance, 3.0 Å; (N-H—O) angle, 167°). The energy of interaction for LPI with H(170) is the second highest energy in the complex, -8.74 kcal/mol. In addition, the inositol ring C-2′ hydroxyl can donate a hydrogen bond to Y3.32 (H bond (O-O) distance, 3.1 Å; (O-H—O) angle, 121°). In addition, as one might expect, the LPI alkyl chain has principally Van der Waals interactions with other residues in the binding pocket (see Table S-2- in Supplementary Information for the energy breakdown). The conformational energy cost for LPI to assume the docked conformation was 8.09 kcal/mol. The Glide score for the dock was -11.7 and the total Interaction Energy for the LPI/GPR55 R* complex was -75.16 kcal/mol.
Other GPR55 Ligands
In Supplemental Information, we docked several other GPR55 agonists in the GPR55 R* model reported here. These include anandamide and GSK494581 (R and S enantiomers)16. We also address the reported species differences between human and rat/mouse GPR55 that has been discovered for some GPR55 ligands.16
Discussion
Ml184 Docked into the GPR55R* Bundle
When one begins the characterization of the ligand binding pocket in a newly de-orphanized receptor, like GPR55, mutation studies are crucial to understanding the binding pocket. Our original GPR55 models (R and R*) were based on a beta-2-adrenergic receptor template.13-14 In this model, we docked ML184 so that the ligand piperazine and di-methyl phenyl ring segment was vertical in the binding pocket, while the rest of the ligand occupied a horizontal space near the extracellular loops. Mutation results reported here clearly necessitate that a reorientation of ML184 occur in the binding pocket. In the refined model reported here, we also took advantage of crystal structures that were unavailable at the time of the original model creation and found that the delta-opioid receptor (DOR) crystal structure 8 was a more appropriate template for the next generation model. The DOR, for example has a PRO at position 4.59, as does GPR55. The beta-2-adrenergic receptor, on the other hand, has a PRO shifted by one residue to 4.60. In addition, the DOR has the same toggle switch partner as GPR55, M3.36.
Ligand Interactions within the Binding Crevice
As predicted by previous studies conducted on the GPR55 receptor in the Reggio lab, the current model has a crucial hydrogen bonding interaction between ML184 and K2.60 on TMH 2 (see Figure 6A or 7B) that results in the strongest ligand Interaction Energy (-9.3 kcal/mol) of any residue in the GPR55 R* bundle (See Table S-1). A strong hydrogen bond between the electronegative sulfonamide and the positive lysine is consistent with the experimental results that show a complete loss of signalling upon the K2.60 mutation to an alanine (see Tables 1 and 2).
Mutation of E3.29 to either an alanine or a leucine resulted in a profound reduction of receptor signalling as well. Though there is no direct interaction between ML184 and E3.29 in the current model, modelling suggests that E3.29 forms a salt bridge with K2.60 (see Figure 7B). This salt bridge positions K2.60 for interaction with ML184. The loss of this residue's directing capability for K2.60 via the E3.29/A/L mutations effectively make K2.60 less available in the binding pocket, leading to a considerable loss of function.
At the ML184 binding site, the EC-2 loop residue, H(170), serves as a hydrogen bond donor to the ML184 sulfonamide oxygen (see Figure 6A). The Interaction Energy for ML184 with H(170) is -3.7 kcal/mol (see Table S-1). Simultaneously, the sulfonamide oxygen also receives a hydrogen bond from K2.60. Because hydrogen bonding is stronger when the donating residue is charged, the H170 hydrogen bond with ML184 is weaker (-3.7 kcal/mol) than that with K2.60 (-9.3 kcal/mol) (see Table S-1). The pKa of histidine is 6.0. At physiological pH, about 10% of HIS residues are protonated. We assume here that H(170) is uncharged. An H(170)F mutation removes the hydrogen bonding ability of residue 170, but not its ability to form aromatic interactions. Mutation of His(170) to a phenylalanine resulted in a 6-fold reduction in SRE responses (see Table 1). The magnitude of the effect upon mutation is consistent with the loss of a hydrogen bond. In addition, the fact that H(170) mutation affected ML184's signalling suggests indirectly that there is also a C(168)/C3.25 disulfide bond in GPR55. This bond requires that the EC-2 loop extend over to TMH3 to link with C3.25 and therefore is responsible for the H(170) location in the GPR55 R* model. In addition, inspection of GPCR crystal structures that contain the analogous disulfide bridges reveal that the second residue after the disulfide bridge typically points down into the binding crevice. H(170) is the second residue after the C(168) / C3.25 disulfide bridge.
Y3.32 serves two functions in GPR55. It is a binding site residue (see Figure 6 A and B) and also part of the extended toggle switch for GPR55 activation (see Figure 8). A stepwise loss of function is seen when Y3.32 is first mutated to a phenylalanine (30-fold loss, see Table 1) and then a leucine (170-fold loss, see Table 1). At the ML184 binding site, Y3.32 donates a hydrogen bond to the ML184 carboxamide oxygen (Figure 6A) and forms an aromatic stacking interaction with the ML184 central benzene ring (Figure 6B) proximal to the ML184 sulfonamide moiety. The first mutation, Y3.32F, removes the residue's potential to donate a hydrogen bond. The magnitude of the effect (30-fold loss) is consistent with Y3.32 serving as the lone hydrogen bond donor to ML184 in this region. The second mutation, Y3.32L, removes both hydrogen bonding and aromatic stacking interactions from the ML184 binding site at this position. The 170-fold reduction in EC50 (see Table 1) is consistent with the loss of two important interactions for ML184. Y3.32F mutation results are also consistent with Y3.32's participation in the extended toggle switch region in GPR55, as loss of aromaticity at 3.32 in the Y3.32L mutant should have a profound effect on signalling (170-fold reduction in EC50 (see Table 1)).
The current model of GPR55 places F6.55 facing inward towards the binding crevice. This residue forms an aromatic stacking interaction with the distal, dimethyl-phenyl ring of ML184 (Figure 6B; Interaction Energy= -4.3 kcal/mol, see Table S-1). The F6.55A mutation resulted in a 15-fold loss in EC50 for ML184, consistent with the loss of this aromatic stacking interaction. The F6.55L resulted in an even larger loss in EC50 (92-fold). This mutation not only removes an aromatic stacking interaction, but also causes crowding in the ML-84 binding pocket. This crowding causes ML184 to shift position and the net result would be reduced binding site interactions. The 92-fold loss in EC50 is consistent with such an alteration.
The x-ray crystal structure of the beta-1-adrenergic receptor complexed with Gs protein shows that TMH6 has straightened by flexing its proline kink at P6.50.17 This conformational change also impacts binding pocket residues. W6.48 has been shown to change its conformational state within the binding pocket upon receptor activation (χ1 g+ → trans). In the inactive state, W6.48 is typically held in its χ1=g+ conformation by another binding pocket residue. Together, this pair of residues is known as the “toggle switch”, 18 In the GPR55 activated state model reported here, Y3.32, M3.36 and F6.48 form an extended toggle switch (see Figure 8), with additional interactions from F3.33 and Y3.37. The mutation effects of Y3.32 are discussed above in the context of their effects on the ML184 binding pocket interactions and upon signalling. For both the SRE and SRF readouts used here, mutation of M3.36 to alanine had a significant impact on signalling (16-fold, 62-fold). Mutation of the toggle switch residues M3.36 and F6.48 resulted in increased EC50 values, but these mutants were able to ultimately reach SRE induction comparable to wild-type. As ML184 is a GPR55 agonist, it is expected that its position in the bundle would not cause it to interfere with the receptors ability to activate. This is verified by the experimental data and is what is observed in the current ligand receptor complex.
Class A GPCRs typically have a disulfide bridge between a residue in the EC-2 loop and Cys3.25 near the top of TMH3. The sequence of GPR55 suggests that it likely has this disulfide bridge as well. Mutation studies of these Cys residues typically result in loss of function as the EC-2 – C3.25 disulfide bridge is important for binding pocket structure.19 The CXCR4 12, 20 and CCR5 21 crystal structures reveal the existence of a second disulfide bridge that links the N terminus of the receptor to the EC end of TMH7, forming what has been called the fourth EC loop.21 This loop has been proposed to shape the entrance of the ligand-binding pocket and to add rigidity to the overall surface of the receptor.21 The sequences of ∼30% of Class A GPCRs contain such Cys residues, including the lysophospholipid (LPA), bradykinin (B1-2), endothelin (ETA-B), melanocortin (MC1-5), serotonin (5-HT), purinergic (P2Y), and orphan receptors, such as GPR55. The structure of one of the latest resolved rhodopsin-like receptors, P2Y12, revealed the presence of such an EC-4 loop; 10 however, the conservation of these residues does not necessarily imply the formation of a EC-4 loop (see crystal structures of dopamine D322 and serotonin, 5HT1B23 receptors). Because the GPR55 sequence suggests the presence of an EC-4 loop, this second disulfide bridge was incorporated into our GPR55 model and was tested via C(10)A and C(260)A mutations here. Results reported here show that each mutation impacts ML184 activation of GPR55, although these mutations are not devastating (11-fold for C(10)A; 12-fold for C(260)A). We have taken these results as evidence that this disulfide bridge is present in GPR55 and have retained this bridge in our model (see Figure 10). It should be noted that the EC-4 loop may have larger effects on GPR55 agonists other than ML184, as it has been proposed that this fourth loop shapes the entrance of the ligand-binding pocket.21
Q6.58 and Q7.36 are located at the extreme EC ends of TMH6 and TMH7 (see Figure 11). In our first GPR55 model, Q6.58 was near the nitrogen in the pendant five membered ring of ML184 and able to form a hydrogen bonding interaction with this nitrogen.13 A Q6.58M mutation should have resulted in the loss of this hydrogen bond and a significant reduction in ML184 EC50. However, at the Q6.58M/A mutations, ML184 retained WT signalling. This result is a key result from a modelling perspective because it clearly suggested that a different binding mode for ML184 should be sought.
Also in our previous GPR55 R model, Q7.36 interacted simultaneously with K2.60 and with the GPR55 antagonist, ML192. 14 The re-orientation of ML184 dictated by the Q6.58M/A mutations dictated that Q7.36 should have no ligand interactions. Consistent with this result, experimental data reported here shows that at both the Q7.36A and Q7.36N mutant, ML184 retains WT signalling.
Correlation Between LPI dock and Mutation Results for LPI
The dock of LPI at GPR55 R* illustrated in Figure 12 is consistent with the functional effects of mutations shown in Figures 2 (SRE responses to LPI) and 4 (SRF responses to LPI). As discussed above, LPI has a very strong interaction with K2.60 (Interaction E= -16.60 kcal/mol). For this reason, K2.60 appears to be the primary interaction site for LPI. In Figure 2, the K2.60A mutation results (panel H) show a loss of function for LPI. This is consistent with loss of the primary interaction site. In Figure 2, the E3.29A mutation (Panel F) results show a loss of function for LPI here as well. This result is similar to that seen for ML184 with E3.29. In the GPR55 R* complex, E3.29 forms a salt bridge with K2.60 (see Figure 7B). This salt bridge positions K2.60 for interaction with LPI. The loss E3.29's directing capability for K2.60 via the E3.29A/L mutations likely makes K2.60 less available in the binding pocket, leading to the considerable loss of function for LPI.
The H(170)F mutation (Figure 2, Panel E) shows reduced signaling for LPI at this mutant. This is consistent with the loss of a hydrogen bond for LPI with this residue. A similar result was obtained for Y3.32 (Figure 2, Panel A), where reduced signaling for LPI occurs as the result of Y3.32F and Y3.32L mutations. These mutations cause the loss of the hydrogen bond between Y3.32 and the inositol ring hydroxyl.
In Figure 2, the Q6.58M (Panel C), Q7.36A/N(Panel D) and F6.55A/L (Panel B) mutations have little to no effect on LPI signaling. Figure 12 shows that Q6.58 and Q7.36 are not part of the LPI binding site, consistent with the lack of effect on signaling in these mutants. It is clear in Figure 12 that F6.55 is in the binding pocket for LPI. The Interaction Energy Table for LPI (see Table S-2) shows that LPI has a small, predominantly Van der Waals interaction with F6.55 (-3.13 kcal/mol). This interaction is between the LPI alkyl chain (near end) and F6.55. Even when F6.55 is mutated to an Ala or Leu, the terminal portion of the chain can expend very little energy to establish a Van der Waals interaction with residue 6.55. For this reason, we would expect little if any change in LPI's ability to activate GPR55 at these mutants.
Since M3.36 and F6.48 are part of the GPR55 toggle switch, mutation of either residue (M3.36A; Panel G and F6.48A; Panel H) reduces LPI's ability to signal. This would be expected for any agonist of GPR55. Figure 4 results for SRF responses triggered by LPI are consistent with results in Figure 2 and therefore are consistent with the dock of LPI as discussed above.
LPI Docked into the GPR55R* Bundle
As discussed above, the mutation data reported here prompted us to re-orient ML184 in the binding pocket from our initial dock of ML184.13 We report here also an LPI dock in the refined GPR55 R* model that is altered in orientation from our original dock. 13 The original dock had the LPI inositol group high enough in the binding pocket for an inositol hydroxyl to interact with Q6.58. Mutation results in Figure 2, show essentially no effect on LPI signalling in the Q6.58M mutant. Instead residues deeper in the binding pocket, such as Y3.32, showed an effect on LPI signalling when mutated. These results clearly suggested that while K2.60 remained a primary interaction site for LPI in the early paper and in the current manuscript, the LPI headgroup had to penetrate deeper in the binding site crevice than initially thought.
Quite recently, additional synthetic ligands, as well as another endogenous agonist for GPR55 have been described.24 The synthetic ligands employed our previous model as a guide for ligand design. The mutation results reported here will be important for the design of more potent and efficacious agonists for GPR55.
Conclusions
Results reported here identify key GPR55 residues that are important for agonist signaling, as well as residues implicated in the agonist activated signaling cascade. Two residues crucial for ML184 and LPI signaling at GPR55 are K2.60 and E3.29. Two additional residues, Y3.32 and H(170), are important for both ML184 and LPI signaling. A third residue, F6.55, was found to be important only for ML184 signaling. Further, results suggest that a cluster of residues, F3.32/M3.36/F6.48 serves as the toggle switch for activation of GPR55. GPR55 also likely possesses a second disulfide bridge that links the N terminus of the receptor to the EC end of TMH7. This loop has been called the fourth EC loop. 21 All of these results provide, for the first time, structural information that should aid in the rational design of next generation GPR55 ligands. It is hoped that this will lead to a high affinity GPR55 radioligand, a tool that is sorely needed in the field.
Experimental Section
Material and Reagents
Soy LPI (purchased from Avanti Polar Lipids), and ML184 (MolPort) were dissolved in dimethyl sulfoxide to a concentration of 10 mM. The SRE reporter, pGL4.33[luc2P/SRE/Hygro] and the SRF-RE reporter, pGL4.34[luc2P/SRF-RE/Hygro] were from Promega.
Mutagenesis and Cell Culture
The M3.36A, F6.48A, K2.60A, E3.29A, E3.29L, Y3.32F, Y3.32L, F6.55A, F6.55L, Q6.58M, C(10)A, H(170)F, C(260)A, Q7.36A and Q7.36N mutants of the HA tagged human GPR55 in the vector pcDNA3 were constructed using the QuikChange site-directed mutagenesis kit (Stratagene). DNA sequencing subsequently confirmed the presence of the desired mutation only.
Quantification of receptor surface expression
On-Cell Western (Licor) was used to quantify the receptor surface expression levels 25. Briefly, HEK293 cells were transfected with WT or mutant GPR55 receptor and grown in 96 well plates overnight. HA primary antibody and subsequent near-infrared fluorescent secondary antibodies were used to detect the surface receptor. Plates were scanned on LI-COR Odyssey IR Imager. Data were analyzed using Excel and Prism 5.01 software.
Serum Response Element (SRE) and Serum Response Factor (SRF) assay
HEK293 cells were transiently transfected with GPR55 and pGL4.33 [luc2P/SRE/Hygro] or pGL4.34 [luc2P/SRF-RE/Hygro] vector reporter plasmids using Lipofectamine 2000 as described by the manufacturer (Invitrogen). Transfected HEK293 cells were seeded (60,000 cells per well) in 96-well plates. Five hours later, medium was changed to 1% FBS/DMEM. Cells were incubated overnight. The next day cells were treated with ligands for 5 h in serum-free DMEM medium at 37°C. After treatment, cells were lysed by 1× lysis buffer for 10 min at room temperature. Plates were read to record bioluminescent light immediately after the injection of 40μl Luciferin (≥250μM) per well. Luminescence was measured in an Envision 2104 multilabel Reader (PerkinElmer). Luminescence values are given as relative light units. Concentration-effect curves for agonist-mediated receptor activation were analyzed by nonlinear regression techniques using GraphPad Prism 5.01 software (GraphPad) and data were fitted to sigmoidal dose-response curves to obtain EC50 values.
Modeling
Amino Acid Numbering System
The amino acid numbering system used here is the Ballesteros-Weinstein numbering system 26 in which the most highly conserved residue across Class A GPCRs in each TMH is assigned a number .50. This number is preceded by the TMH number and can be followed by the absolute sequence number in parentheses. For example, the most highly conserved residue in TMH4 is W4.50. For GPR55, this residue is W4.50(146). The residue preceding this residue is I4.49(145) and the residue following it is V4.51(147). Loop residues in this system are identified by their absolute sequence numbers only.
Modeling of hGPR55 active-state bundle (GPR55 R*) using GPCR x-ray crystallography data
The model of the activated form of GPR55 (GPR55 R*) described in the current work was created using the 1.8 Å crystal structure of the human delta opioid receptor (hDOR, PDB id: 4N6H) as a template. 8 Transmembrane regions of GPR55 vs. hDOR in which the placement of prolines differed (GPR55 transmembrane helices (TMHs) 1, 5, 6 and 7) were explored using the Conformational Memories (CM) method described below.27
Conformational Memories (CM) Method for Calculating TMH Conformation
The CM method uses multiple Monte Carlo/simulated annealing random walks employing the CHARMM force field. Backbone φ and ψ torsions in regions of interest (i to i-4 of a proline) were allowed to vary +/- 50°, while all other backbone torsion angles were allowed to vary +/- 10°. Side chain torsions were allowed to vary +/- 180°. All bond angles were allowed to vary +/- 8° except for C-S-C angles that were allowed to vary +/- 15°. A minimum set of 108 conformers was generated for each GPR55 helix, independently, in a distance dependent dielectric at 310 K.
TMH1 in GPR55 has a Pro at position 1.41 not found in DOR. An ideal helix (φ= -62.9°, and ψ = -41.6°) with the GPR55 sequence was built, and the region containing the proline and 4 residues prior to the Pro (i to i-4, A1.37 - P1.41) was varied using CM.
TMH5 in GPR55 has two prolines, the highly conserved P5.50 and an additional one at position 5.41. TMH5 of the DOR crystal structure was mutated to the GPR55 sequence and the backbone dihedrals from P5.41 to K5.37 were varied to explore the possible conformations caused by this second, proline.
TMH6 in GPR55 has a conservative SFXP substitution in place of the highly conserved Class A CWXP motif. Biophysical studies have indicated that there is a salt bridge, or ionic lock (between R3.50 near the intracellular (IC) end of TMH3 and D/E6.30 at the IC end of TMH6), common to all Class A GPCRs that is broken upon activation. The breaking of this ionic lock allows TMH6 to straighten, moving its intracellular (IC) end away from the TMH bundle.28 For this reason, it was crucial to explore the conformational space of GPR55 TMH6 which has a glutamine (Q) at position 6.30 in place of a typical aspartic (D) or glutamic (E) acid. The GPR55 TMH6 sequence was built, using the ideal helix values (φ= -62.9°, and ψ = -41.6°) and the i to i-4 region around the Pro (P6.50-V6.46) was varied using CM.
TMH7 in GPR55 lacks the highly conserved Class A GPCR NPXXY motif having instead a DVXXY sequence. Traditionally the NPXXY motif influences the conformation of TMH7 and places Y7.53 in the correct position to interact with F7.60 on Helix 8 (Hx8, a short intracellular extension of TMH7 that lies usually parallel to the cell membrane). For receptors that possess an NPXXY motif, we have typically designated the backbone region of P7.50- X7.46 region as variable. For GPR55 TMH7, an ideal helix (φ= -62.9°, and ψ = -41.6°) was built and the i to i-4 region in which there is normally a Pro (V7.50 - C7.46) was varied using CM.
Construction of the GPR55 Active-State Bundle
Transmembrane helices 2, 3 and 4 of the DOR were mutated to the corresponding GPR55 residues, while helices chosen from the CM output that would fit in the bundle were substituted in the bundle. This included a straightened TMH6 conformer for which the TMH3-TMH6 ionic lock was broken, reflecting the R* state. Incorporated into this new bundle were updates from previous homology models of GPR55 14-15 created in this lab: The high degree of sequence homology between GPR55 and the CXCR4 receptor, 12 particularly in the EC regions, dictated several modifications to the model. These were (1) the introduction of EC helical extensions on TMH5-7 of GPR55; (2) the introduction of a β sheet motif into the EC-2 loop in GPR55; and, (3) the introduction of a disulfide bridge between Cys(10) in the N-terminus and Cys(260) in the EC-3 loop near the top of TMH7.
The resulting homology model was then optimized using the following protocol 14: The energy of the GPR55 R* bundle was minimized using the OPLS 2005 force field in Macro-model 9.9 (Schrödinger Inc., Portland, OR). An 8.0-Å extended non-bonded cutoff (updated every 10 steps), a 20.0-Å electrostatic cutoff, and a 4.0-Å hydrogen bond cutoff were used in each stage of the calculation. The minimization was performed in three stages. Each stage consisted of a Polak- Ribier conjugate gradient minimization in 1000-step increments until the bundle reached a 0.05 kJ/mol gradient. In the first stage of the calculation, the TMH region of the receptor was held stationary and the loops were allowed to relax using the generalized born/surface area continuum solvation model for water (Macro-model). In the second stage, the loops were frozen and the side chains of the TMHs were allowed to adjust. A distance dependent dielectric was used for this minimization. In the third stage, the N and C termini were minimized using the protocol described, for the loops, above. In this stage, only the termini were minimized.
Conformational Analysis of ML184 and LPI
ML184
A complete conformational analysis of the GPR55 agonist ML184 (CID2440433) was performed using ab initio Hartree-Fock (HF) calculations at the 6–31G* basis set level as encoded in Jaguar (version 9.3, Schrodinger, LLC, New York, NY). Hartree-Fock 6–31G* six-fold conformer searches were performed for the rotatable bonds, N1-S2, S2-C3, C4′-N5′, C4-C5, C5-N6, N7-C8 (see numbering system in Fig 5), of ML184 as follows: In each conformer search, local energy minima were identified by rotation of a subject torsion angle through 360° in 60° increments (6-fold search), followed by optimization at the HF 6-31G* level. To account for dispersion forces that might play a role in ML184, all unique conformers were re-optimized with Density Functional Theory (DFT) B3LYP-D3 at the 6–31G** level.29 To calculate the energy difference between the global minimum energy conformer of ML184 and its final docked conformation, rotatable bonds in the global minimum energy conformer were driven to their corresponding value in the final docked conformation and the single point energy of the resultant structure was calculated at the DFT B3LYP-D3 6–31G** level. This difference was calculated to be 1.14 kcal/mol.
Figure 5.

The structures of GPR55 agonists, ML184 and LPI, are illustrated here. The ML184 structure is numbered for reference in the text to torsion angles varied for conformational analysis.
LPI
To account for the conformational cost for L-α-lysophosphatidylinositol (LPI) to complex with the GPR55 R* model, a low free energy extended conformation of LPI with a tilted headgroup and internal hydrogen bonding was used for comparison to the docked conformation of LPI.30 The low free energy structure was relaxed in the OPLS3 all atom force field, as encoded in Macromodel (version 11.3, Schrödinger, LLC, NY 2016), using an 8.0 Å nonbonded cutoff (updated every 10 steps), a 20.0 Å electrostatic cutoff, and a 4.0 Å hydrogen bond cutoff. A distance dependent dielectric set to a base constant of 2 was applied as the structure was relaxed to a gradient of 0.01 kcal/mol·A. To obtain the energy difference for the docked conformation of LPI, the single point energy was calculated in OPLS3 with identical non-bonded cutoffs and dieletric treatment. The difference was calculated to be 8.09 kcal/mol.
Glide docking of ML184 and LPI
A low free-energy conformer of ML184 and LPI was used as input for docking into the minimized receptor model. ML184 and LPI were initially docked manually in the binding site of the GPR55 R* model, after which the automatic docking program, Glide (version 7.2, Schrödinger, LLC, NY 2016), was used to explore other possible binding conformations and receptor site interactions. Extra precision (XP) and flexible docking with ring sampling were selected for ML184's docking setup. No ring sampling was selected to avoid changing chiral centers in the inositol ring of LPI, and standard precision (SP) mode was used in the docking setup. Glide rigid redocking with XP mode was performed for LPI from the output of the SP mode dock. Lysine 2.60(80), a previously identified ligand interaction site,13 was defined as a hydrogen bond donor and was used as a constraint for the automatic docking of the ligands. The energy of the ligand/GPR55 R* complex was minimized using the OPLS3 force field in Macromodel 11.3 (Schrödinger, LLC, NY 2016). An 8.0-Å extended non-bonded cutoff (updated every 10 steps), a 20.0-Å electrostatic cutoff, and a 4.0-Å hydrogen bond cutoff were used in each stage of the calculation as described previously. The docks with the best Glide scores were: -9.5 kcal/mol for the ML184/GPR55 R* model, and -11.7 kcal/mol for the LPI/GPR55 R* model.
Assessment of Pair-wise Interaction Energies
After defining the atoms of ML184 or LPI as one group (Group 1) and the atoms corresponding to a residue that lines the binding site in the final ligand/GPR55 R* complexes as another group (Group 2), Macromodel was used to output the pair-wise interaction energy (coulombic and Van der Waals) for a given pair of atoms. The pairs corresponding to Group 1 (ligand) and Group 2 (residue of interest) were then summed to yield the interaction energy between the ligand and that residue. The ML184/GPR55 R* complex was found to have interaction energies totaling to -57.85 kcal/mol. Taking the conformational energy cost for ML184 (1.14 kcal/mol) into account, the final total energy for the ML184/GPR55 R* complex was found to be -56.71 kcal/mol. A breakdown of interaction energies is provided in Table S-1. Subsequently the LPI/GPR55 R* complex was found to have interaction energies totaling to -83.25 kcal/mol. Taking the conformational energy cost for LPI (8.09 kcal/mol) into account, the final total energy for the LPI/GPR55 R* complex was found to be -75.16 kcal/mol. A breakdown of interaction energies is provided in Table S-2.
Supplementary Material
Acknowledgments
This research was supported by grants from the National Institute on Drug Abuse R01DA023204, R01DA035926, P30DA013429, and K05DA021358 and from the National Institute of Neurological Disorders and Stroke R21NS077347. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We would like to thank Lawrence Barak of the NIDA sponsored Duke P30 Center of Excellence for technical support.
Abbreviations
- A2-AR
A2-adenosine receptor
- βarr2-GFP
β-arrestin2-green fluorescent protein
- β2-AR
β2-adrenergic receptor
- CID2745687
methyl-5-[(tert-butylcarbamothioylhydrazinylidene)methyl]-1-(2,4-difluorophenyl)pyrazole-4-carboxylate
- CM
Conformational Memories
- DMEM
Dulbecco's modified Eagle's Medium
- DOR
delta opioid receptor
- ERK
extracellular signal related kinase
- FBS
fetal bovine serum
- GPCR
G-protein-coupled receptor
- HBSS
Hanks Balanced Salt Solution
- LPA
lysophosphatidic acid
- Rho
rhodopsin
- SRE
Serum Response Element
- SRF
Serum Response Factor
- TMH
transmembrane helix
Footnotes
Author Contributions: MAL and PZ also contributed to writing the manuscript and contributed equally to this work. MAL and DPH constructed the molecular model in consultation with PHR. PZ designed and constructed vectors for expression of mutant proteins and analyzed the mutant phenotypes in vitro. HPS designed and constructed vectors for expression of mutant proteins. All authors reviewed the results and approved the final version of the manuscript.
Model Coordinates: The coordinates of the GPR55 R* model described here are available upon request. Please e-mail requests to Dow Hurst at dphurst@uncg.edu
Literature Cited
- 1.Kapur A, Zhao P, Sharir H, Bai Y, Caron MG, Barak LS, Abood ME. Atypical responsiveness of the orphan receptor GPR55 to cannabinoid ligands. The Journal of biological chemistry. 2009;284(43):29817–27. doi: 10.1074/jbc.M109.050187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Henstridge CM, Balenga NA, Ford LA, Ross RA, Waldhoer M, Irving AJ. The GPR55 ligand L-alpha-lysophosphatidylinositol promotes RhoA-dependent Ca2+ signaling and NFAT activation. Faseb J. 2009;23(1):183–93. doi: 10.1096/fj.08-108670. [DOI] [PubMed] [Google Scholar]; (b) Oka S, Toshida T, Maruyama K, Nakajima K, Yamashita A, Sugiura T. 2-Arachidonoyl-sn-glycero-3-phosphoinositol: A Possible Natural Ligand for GPR55. Journal of biochemistry. 2009;145(1):13–20. doi: 10.1093/jb/mvn136. [DOI] [PubMed] [Google Scholar]; (c) Oka S, Nakajima K, Yamashita A, Kishimoto S, Sugiura T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem Biophys Res Commun. 2007;362(4):928–34. doi: 10.1016/j.bbrc.2007.08.078. [DOI] [PubMed] [Google Scholar]
- 3.(a) Sawzdargo M, Nguyen T, Lee DK, Lynch KR, Cheng R, Heng HH, George SR, O'Dowd BF. Identification and cloning of three novel human G protein-coupled receptor genes GPR52, PsiGPR53 and GPR55: GPR55 is extensively expressed in human brain. Brain Res Mol Brain Res. 1999;64(2):193–8. doi: 10.1016/s0169-328x(98)00277-0. [DOI] [PubMed] [Google Scholar]; (b) Staton PC, Hatcher JP, Walker DJ, Morrison AD, Shapland EM, Hughes JP, Chong E, Mander PK, Green PJ, Billinton A, Fulleylove M, Lancaster HC, Smith JC, Bailey LT, Wise A, Brown AJ, Richardson JC, Chessell IP. The putative cannabinoid receptor GPR55 plays a role in mechanical hyperalgesia associated with inflammatory and neuropathic pain. Pain. 2008;139(1):225–36. doi: 10.1016/j.pain.2008.04.006. [DOI] [PubMed] [Google Scholar]; (c) Whyte LS, Ryberg E, Sims NA, Ridge SA, Mackie K, Greasley PJ, Ross RA, Rogers MJ. The putative cannabinoid receptor GPR55 affects osteoclast function in vitro and bone mass in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(38):16511–6. doi: 10.1073/pnas.0902743106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ford LA, Roelofs AJ, Anavi-Goffer S, Mowat L, Simpson DG, Irving AJ, Rogers MJ, Rajnicek AM, Ross RA. A role for L-alpha-lysophosphatidylinositol and GPR55 in the modulation of migration, orientation and polarization of human breast cancer cells. Br J Pharmacol. 2010;160(3):762–71. doi: 10.1111/j.1476-5381.2010.00743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Andradas C, Caffarel MM, Perez-Gomez E, Salazar M, Lorente M, Velasco G, Guzman M, Sanchez C. The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene. 2010;30:245–252. doi: 10.1038/onc.2010.402. [DOI] [PubMed] [Google Scholar]; (f) Pineiro R, Maffucci T, Falasca M. The putative cannabinoid receptor GPR55 defines a novel autocrine loop in cancer cell proliferation. Oncogene. 2010;30:142–152. doi: 10.1038/onc.2010.417. [DOI] [PubMed] [Google Scholar]
- 4.(a) Heynen-Genel S, Dahl R, Shi S, Milan L, Hariharan S, Sergienko E, Hedrick M, Dad S, Stonich D, Su Y, Vicchiarelli M, Mangravita-Novo A, Smith LH, Chung TDY, Sharir H, Caron MG, Barak LS, Abood ME. Screening for Selective Ligands for GPR55 - Antagonists. Probe Reports NIH Molecular Libraries Program. 2010 [PubMed] [Google Scholar]; (b) Heynen-Genel S, Dahl R, Shi S, Milan L, Hariharan S, Bravo Y, Sergienko E, Hedrick M, Dad S, Stonich D, Su Y, Vicchiarelli M, Mangravita-Novo A, Smith LH, Chung TDY, Sharir H, Barak LS, Abood ME. Screening for Selective Ligands for GPR55 - Agonists. Probe Reports NIH Molecular Libraries Program. 2010 [PubMed] [Google Scholar]
- 5.Heynen-Genel S, Dahl R, Shi S, Milan L, Hariharan S, Bravo Y, Sergienko E, Hedrick M, Dad S, Stonich D, Su Y, Vicchiarelli M, Mangravita-Novo A, Smith LH, Chung TDY, Sharir H, Barak LS, Abood ME. Screening for Selective Ligands for GPR55. Probe Reports NIH Molecular Libraries Program. 2010 [Google Scholar]
- 6.Cheng Z, Garvin D, Paguio A, Stecha P, Wood K, Fan F. Luciferase Reporter Assay System for Deciphering GPCR Pathways. Current chemical genomics. 2010;4:84–91. doi: 10.2174/1875397301004010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lauckner JE, Jensen JB, Chen HY, Lu HC, Hille B, Mackie K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc Natl Acad Sci U S A. 2008;105(7):2699–704. doi: 10.1073/pnas.0711278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Structure of the delta-opioid receptor bound to naltrindole. Nature. 2012;485(7398):400–4. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valley CC, Cembran A, Perlmutter JD, Lewis AK, Labello NP, Gao J, Sachs JN. The methionine-aromatic motif plays a unique role in stabilizing protein structure. J Biol Chem. 2012;287(42):34979–91. doi: 10.1074/jbc.M112.374504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang K, Zhang J, Gao ZG, Zhang D, Zhu L, Han GW, Moss SM, Paoletta S, Kiselev E, Lu W, Fenalti G, Zhang W, Muller CE, Yang H, Jiang H, Cherezov V, Katritch V, Jacobson KA, Stevens RC, Wu B, Zhao Q. Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature. 2014;509(7498):115–8. doi: 10.1038/nature13083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang C, Srinivasan Y, Arlow DH, Fung JJ, Palmer D, Zheng Y, Green HF, Pandey A, Dror RO, Shaw DE, Weis WI, Coughlin SR, Kobilka BK. High-resolution crystal structure of human protease-activated receptor 1. Nature. 2012;492(7429):387–92. doi: 10.1038/nature11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330(6007):1066–71. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kotsikorou E, Madrigal KE, Hurst DP, Sharir H, Lynch DL, Heynen-Genel S, Milan LB, Chung TD, Seltzman HH, Bai Y, Caron MG, Barak L, Abood ME, Reggio PH. Identification of the GPR55 Agonist Binding Site Using a Novel Set of High-Potency GPR55 Selective Ligands. Biochemistry. 2011;50:5633–5647. doi: 10.1021/bi200010k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kotsikorou E, Sharir H, Shore DM, Hurst DP, Lynch DL, Madrigal KE, Heynen-Genel S, Milan LB, Chung TD, Seltzman HH, Bai Y, Caron MG, Barak LS, Croatt MP, Abood ME, Reggio PH. Identification of the GPR55 antagonist binding site using a novel set of high-potency GPR55 selective ligands. Biochemistry. 2013;52(52):9456–69. doi: 10.1021/bi4008885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kotsikorou E, Madrigal KE, Hurst DP, Sharir H, Lynch DL, Heynen-Genel S, Milan LB, Chung TD, Seltzman HH, Bai Y, Caron MG, Barak L, Abood ME, Reggio PH. Identification of the GPR55 agonist binding site using a novel set of high-potency GPR55 selective ligands. Biochemistry. 2011;50(25):5633–5647. doi: 10.1021/bi200010k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown AJ, Daniels DA, Kassim M, Brown S, Haslam CP, Terrell VR, Brown J, Nichols PL, Staton PC, Wise A, Dowell SJ. Pharmacology of GPR55 in yeast and identification of GSK494581A as a mixed-activity glycine transporter subtype 1 inhibitor and GPR55 agonist. J Pharmacol Exp Ther. 2011;337(1):236–46. doi: 10.1124/jpet.110.172650. [DOI] [PubMed] [Google Scholar]
- 17.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477(7366):549–55. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McAllister SD, Hurst DP, Barnett-Norris J, Lynch D, Reggio PH, Abood ME. Structural mimicry in class A G protein-coupled receptor rotamer toggle switches: the importance of the F3.36(201)/W6.48(357) interaction in cannabinoid CB1 receptor activation. The Journal of biological chemistry. 2004;279(46):48024–37. doi: 10.1074/jbc.M406648200. [DOI] [PubMed] [Google Scholar]
- 19.(a) Perlman JH, Wang W, Nussenzveig DR, Gershengorn MC. A disulfide bond between conserved extracellular cysteines in the thyrotropin-releasing hormone receptor is critical for binding. J Biol Chem. 1995;270(42):24682–5. doi: 10.1074/jbc.270.42.24682. [DOI] [PubMed] [Google Scholar]; (b) Hwa J, Reeves PJ, Klein-Seetharaman J, Davidson F, Khorana HG. Structure and function in rhodopsin: further elucidation of the role of the intradiscal cysteines, Cys-110, -185, and -187, in rhodopsin folding and function. Proc Natl Acad Sci U S A. 1999;96(5):1932–5. doi: 10.1073/pnas.96.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qin L, Kufareva I, Holden LG, Wang C, Zheng Y, Zhao C, Fenalti G, Wu H, Han GW, Cherezov V, Abagyan R, Stevens RC, Handel TM. Structural biology. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science. 2015;347(6226):1117–22. doi: 10.1126/science.1261064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szpakowska M, Perez Bercoff D, Chevigne A. Closing the ring: a fourth extracellular loop in chemokine receptors. Science signaling. 2014;7(341):pe21. doi: 10.1126/scisignal.2005664. [DOI] [PubMed] [Google Scholar]
- 22.Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330(6007):1091–5. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C, Jiang Y, Ma J, Wu H, Wacker D, Katritch V, Han GW, Liu W, Huang XP, Vardy E, McCorvy JD, Gao X, Zhou XE, Melcher K, Zhang C, Bai F, Yang H, Yang L, Jiang H, Roth BL, Cherezov V, Stevens RC, Xu HE. Structural basis for molecular recognition at serotonin receptors. Science. 2013;340(6132):610–4. doi: 10.1126/science.1232807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Morales P, Whyte L, Chicharro R, Gomez-Canas M, Pazos R, Goya P, Irving AJ, Fernandez-Ruiz J, Ross R, Jagerovic N. Identification of Novel GPR55 Modulators Using Cell-Impedance-Based Label-Free Technology. Journal of medicinal chemistry. 2016 doi: 10.1021/acs.jmedchem.5b01331. [DOI] [PubMed] [Google Scholar]; (b) Yrjola S, Parkkari T, Navia-Paldanius D, Laitinen T, Kaczor AA, Kokkola T, Adusei-Mensah F, Savinainen JR, Laitinen JT, Poso A, Alexander A, Penman J, Stott L, Anskat M, Irving AJ, Nevalainen TJ. Potent and selective N-(4-sulfamoylphenyl)thiourea-based GPR55 agonists. European journal of medicinal chemistry. 2016;107:119–32. doi: 10.1016/j.ejmech.2015.10.050. [DOI] [PubMed] [Google Scholar]; (c) Guy AT, Nagatsuka Y, Ooashi N, Inoue M, Nakata A, Greimel P, Inoue A, Nabetani T, Murayama A, Ohta K, Ito Y, Aoki J, Hirabayashi Y, Kamiguchi H. Neuronal Development. Glycerophospholipid regulation of modality-specific sensory axon guidance in the spinal cord. Science. 2015;349(6251):974–7. doi: 10.1126/science.aab3516. [DOI] [PubMed] [Google Scholar]
- 25.Zhao P, Sharir H, Kapur A, Cowan A, Geller EB, Adler MW, Seltzman HH, Reggio PH, Heynen-Genel S, Sauer M, Chung TD, Bai Y, Chen W, Caron MG, Barak LS, Abood ME. Targeting of the orphan receptor GPR35 by pamoic acid: a potent activator of extracellular signal-regulated kinase and beta-arrestin2 with antinociceptive activity. Mol Pharmacol. 2010;78(4):560–8. doi: 10.1124/mol.110.066746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ballesteros J, Weinstein H. Integrated methods for the construction of three-dimensional models and computational modeling of structure-function relations in G-protein-coupled receptors. Methods in Neurosciences. 1995;25:366–428. [Google Scholar]
- 27.(a) Guarnieri F, Weinstein H. Conformational Memories and the Exploration of Biologically Relevant Peptide Conformations: An Illustration for the Gonadotropin-Releasing Hormone. J Amer Chem Soc. 1996;118:5580–5589. [Google Scholar]; (b) Whitnell RM, Hurst DP, Reggio PH, Guarnieri F. Conformational memories with variable bond angles. J Comput Chem. 2008;29:741–752. doi: 10.1002/jcc.20822. [DOI] [PubMed] [Google Scholar]
- 28.Jensen AD, Guarnieri F, Rasmussen SG, Asmar F, Ballesteros JA, Gether U. Agonist-induced conformational changes at the cytoplasmic side of transmembrane segment 6 in the beta 2 adrenergic receptor mapped by site-selective fluorescent labeling. J Biol Chem. 2001;276(12):9279–90. doi: 10.1074/jbc.M004871200. [DOI] [PubMed] [Google Scholar]
- 29.Grimme S, Antony J, Ehrlich S, Krieg H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. Journal of Chemical Physics. 2010;132:154104–19. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 30.Kotsikorou E, Lynch DL, Abood ME, Reggio PH. Lipid bilayer molecular dynamics study of lipid-derived agonists of the putative cannabinoid receptor, GPR55. Chemistry and Physics of Lipids. 2011;164(2):131–143. doi: 10.1016/j.chemphyslip.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.