

Abstract
The variable quality of histochemical and immunohistochemical staining of tissues may be attributed to pre-analytical and analytical variables. Both categories of variables frequently are undefined or inadequately controlled during specimen collection and preparation. Pre-analytical variables may alter the molecular composition of tissues, which results in variable staining; such variations may cause problems when different tissues are used as staining controls. We developed a standard tissue for use as a staining control. Our standard tissue contains five components: 1) nine combined human cell lines mixed with stroma from human spleen; 2) a squamous cancer cell line, A431; 3) fungus; 4) transverse sections of the mosquitofish and 5) normal human spleen. The first three components were embedded in HistoGel™ and all components were processed to paraffin and used to construct a single standard paraffin block. The muscles of mosquitofish and arteries of the spleen are positive controls for eosin staining, while other tissues are useful for assessing hematoxylin staining. The mosquitofish tissues also are excellent controls for the Masson trichrome stain and all mucin-related histochemical stains that we tested. The goblet cells of the intestine and skin stained strongly with Alcian blue, pH 2.5 (AB-2.5), mucicarmine, colloidal iron, periodic acid Schiff (PAS) or PAS-hematoxylin (PASH) and combination stains such as colloidal iron-PASH. Cell lines were not useful for evaluating histochemical stains except for PASH. The splenic stroma was a useful control for AB-2.5; however, eosin and mucin stains stained cell lines poorly, probably due to their rapid growth and associated loss of some differentiated characteristics such as production of mucins. Nevertheless, the cell lines were a critical control for immunohistochemical stains. Immunostaining of specific cell lines was consistent with the presence of markers, e.g., EGFr in DU145 cells. The cell lines expressed a wide range of markers, so they were useful controls for immunohistochemcial staining including EGFr, HER2, E-cadherin, cytokeratins, Ki67, PCNA, estrogen receptor, progesterone receptor, CD3, CD20 and CD45, activated (cleaved) caspase 3 and Bcl-2. The cell lines also were a control for the TUNEL stain.
Keywords: analytical variables, histochemistry, histology, immunohistochemistry, pre-analytical variables, standard tissue
Histology laboratory personnel sometimes must cope with variable quality of histochemical and immunohistochemical staining. The variability usually is due to pre-analytical and/or analytical variables, which have significant effects on staining, but often are unrecognized or inadequately controlled. Pre-analytical variables include patient age, race, sex, diet and existing diseases; medical interventions including chemotherapy, tissue biopsy and surgery; and tissue handling including collection, processing, storage and distribution (e.g., supplying tissues to investigators by biorepositories). Analytical variables include those that affect specific assays such as using different concentrations of dyes for histochemical staining or different concentrations, durations and temperatures for antibody incubation for immunohistochemistry (IHC). The problems associated with pre-analytical and analytical variables are exacerbated by the fact that different laboratories follow different protocols for fixation, tissue processing and staining (Goldstein et al. 2007). In addition, improper handling of tissues that may cause contamination with non-target tissues, organisms or extraneous substances also are problematic for histopathological analysis (Burke et al. 2009). If common and consistent standard tissues were readily available, some pre-analytical and most analytical variables could be controlled so that inter- and intra-laboratory reproducibility of all types of staining could be improved; this ultimately could lead to the development of more consistent quality control (QC) measures.
We attempted to identify, understand and control pre-analytical and analytical variables for histochemistry and IHC. We first developed a standard tissue that could facilitate evaluation and selection of different approaches to histochemistry and IHC including the choice of optimal concentrations of primary antibodies.
Our initial goal for developing a standard tissue was to standardize and optimize the hematoxylin and eosin (H & E) stain. Our original approach was to use a variety of human cell lines mixed with the stroma of normal spleen. Cell lines have the advantage that they can be cultured consistently and therefore avoid some pre-analytical variables such as those associated with removal of tissues from human subjects including warm ischemia during surgery and cold ischemia following surgery. We found, however, that rapidly growing cells in two-dimensional culture may lack some differentiated molecular features such as production of mucins. H & E staining of our original nine combined cell lines was not optimal; therefore, we developed an expanded standard tissue that could be used to improve it. We added cross sections of the thorax of the mosquitofish, Gambusia affins, an additional group of A431 cells without splenic stroma, fungus and a small portion of traumatic spleen. Each of these components was embedded separately in paraffin and used to construct a composite paraffin block (Taylor 1994). Our goal was to develop a standard tissue that contained components subject to minimal exposure to pre-analytical and analytical variables, which for a specific stain would yield consistent results for a number of histochemical and immunohistochemical stains used on paraffin embedded tissues. Our new standard tissue proved useful as a control for pre-analytical and analytical variables for histochemistry and IHC including the demonstration of mucins. We found that the cell lines were more useful for characterizing immunohistochemical staining than for standardizing the H & E stain.
The use of the standard tissue permitted experimental studies such as identifying the effects of varying approaches to fixation and/or decalcification on histochemical (e.g., mucin stains) and immunohistochemical stains. In addition, the standard tissue could be used to test new antibodies or new lots of antibodies, new reagents, methods of antigen retrieval, initial antibody titrations, new secondary detection systems or new lots of reagents for the secondary detection system.
Material and methods
One of our goals for developing the standard tissue was to evaluate histochemical and immunohistochemical procedures and reagents. For example, for IHC, use of the nine cell lines combined with stroma and the A431 cell line without stroma could identify problems in immunostaining. Specifically, each cell line expresses a set of biomarkers and it is known that some of the cells do not express these markers, e.g., the cell line, MiaPaca 2, does not stain with EGFr. One result of using the nine combined cell lines for many specific immunohistochemical assays was that the combined cells could serve as both positive and negative controls for several immunohistochemical stains.
By immunostaining separately each of the cell lines grown without splenic stroma, we identified which antibodies or assays each of the nine combined cell lines plus the A431 cell line would stain with specific antibodies. Therefore if an antibody did not stain any of the nine combined cell lines or the individual A431 cell line, and therefore did not stain with an antibody (or assay) known to stain at least one of these cell lines, then it would be clear that the immunostaining or assay method used did not work. By contrast, by deliberate choice of the cell lines, not all nine combined cell lines should stain with all the antibodies tested, e.g., lymphoma cell lines do not stain with cytokeratins, so the nine combined cell lines serve as both positive and negative controls for immunohistochemical stains. If during the same immunostaining run, staining for some antibodies worked, but one did not, the problem would be limited to either an operator error or to problems with the specific primary antibody.
In a similar way, transverse sections of the mosquitofish, sections of traumatic spleen and sections of the selected cell lines serve as positive and negative controls for some histochemical stains, e.g., PAS, PASH. In addition to serving as controls, the standard tissue permits evaluation modifications of immunohistochemical and histochemical techniques including titration of antibodies, staining of more than one antigen per cell, effects of decalcification solutions, optimization of antigen retrieval methods and modifications of histochemical techniques.
Standard tissue
Our standard tissue was consisted of five components: 1) nine combined human cell lines with stroma from traumatic spleen, 2) the squamous cancer cell line, A431 without splenic stroma, 3) a small portion of normal human spleen, 4) fungus, and 5) transverse sections of the mosquitofish, G. affins. Each component was embedded separately in paraffin, then combined into a single standard paraffin block (Taylor 1994); details concerning the components and their embedment are given below.
Splenic stroma
Pieces of traumatic human spleen from splenectomies that had been stored in liquid nitrogen vapor phase were obtained from the University of Alabama at Birmingham Tissue Procurement Shared Facility of the Comprehensive Cancer Center. Before thawing completely, a 100 mg sample of frozen spleen was minced into < 1 mm pieces in lysis buffer (Margolis et al. 1989). The lysis buffer contained 50 mM pH 7.5 HEPES, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 200 μM sodium orthovanadate, 10 mM sodium pyrophosphate, 100 mM sodium fluoride, 10% glycerol and 1% Triton X-100. After thawing, the minced spleen was sieved through 250 μm pore size precision woven nylon mesh (TETKO Inc., Kansas City, MO) and washed with room temperature lysis buffer. The preparations were considered satisfactory when red blood cells were lysed and stroma appeared yellow, typically after five or six washes with extraction buffer. The stroma then was mixed with the combined six adenocarcinoma and three lymphoma cell lines, transferred to 6 ml 10% NBF (Richard-Allan Scientific, Kalamazoo, MI) and incubated for 2 h at room temperature. After centrifugation at 150 × g for 5 min, the supernatant was discarded and the resulting pellet of stroma and combined cell lines was mixed with a pre-melted matrix of either HistoGel™ (Richard-Allan Scientific) or 1.2% Agar (Sigma-Aldrich, St. Louis, MO) and poured into a plastic a 30 × 24 mm base mold (Surgipath Medical Industries, Inc., McGaw Park, IL), which is a plastic mold for embedding tissues in paraffin. The objective of using the two matrices was to test for differences in the quality of gel formation, histology (e.g., sectioning) and staining characteristics between the commercially available HistoGel™ and agar prepared in our laboratory. Agar is a widely used, relatively inexpensive laboratory reagent with a defined composition that therefore might be a more cost effective matrix for some research laboratories. The matrices were allowed to solidify at 4° C for 30 min, then they were removed from the mold and placed in labeled cassettes for tissue processing. The pellet with cell lines and stroma in either HistoGel™ or 1.2% agar was used as a component of the standard tissue.
Cell lines and cell culture
Initially, we evaluated 12 cell lines to develop the standard tissue from which 10 were selected for the cell line components of the standard tissue, i.e., nine cell lines combined and mixed with splenic stroma and a separate area of A431 cells without splenic stroma. Two cell lines, CRL5928 and HTB182, were evaluated but not used in the standard tissue, because they did not add enough different information from the other cell lines; however, we include here some results from these two cell lines as additional information. Nine of these cell lines included the six adenocarcinoma cell lines, MDA-MB-231 and MCF7 (both breast), SKOV3 (ovary), MiaPaca 2 (pancreas), DU145 and LNCaP (both prostate), and three squamous cancer cell lines, A431 (skin/epidermis), and CRL5928 and HTB182 (both lung). These nine cell lines were obtained from the American Type Culture Collection (ATCC) (Manassas, VA) and maintained according to ATCC recommended guidelines as shown in Table 1. The diffuse B-cell lymphoma cell lines, SU-DHL-4 and SU-DHL-6, were obtained from Deutsche Sammlung von Microorganismen und Zellkulturen (Braunschweig, Germany) (Drexler et al. 2014) and OCI-Ly3, a diffuse large B-cell lymphoma cell line from Ontario Cancer Institute (OCI, Toronto, Canada). These lymphoma cell lines grow in suspension. The adherent cells, i.e., the adenocarcinoma and squamous cell lines, were detached from the culture flasks using 0.25% trypsin-EDTA (Gibco, Grand Island, NY) when 70–90% confluency of was reached. Approximately the same number of cells of each of the cell lines were combined and mixed with stroma from spleen.
Table 1.
Selected cell lines, sources and media
| Cell line | Source | Medium | Medium source | Medium additives | Molecule expressed |
|---|---|---|---|---|---|
| MCF7 (breast) | ATCC, Manassas, VA | DMEM | Mediatech, Inc. Manassas, VA | 10% FCS, 0.01 μg/ml | Estrogen receptor |
| MDA-MB-231 (breast) | ATCC, | DMEM/F12 (1:1) | Life Technologies Corp., Grand Island, NY | 10% FCS | EGF receptor |
| CRL5928 (lung) | ATCC, | RPMI 1640 | Mediatech, Inc. | 10% FCS | E-cadherin |
| HTB182 (lung) | ATCC, | RPMI 1640 | Mediatech, Inc. | 10% FCS | Cytokeratins AE1/AE3 |
| Ly3 (lymphoma) | OCI, Toronto, Canada | IMDM | Life Technologies Corp. | 10% FCS | Bcl-2 |
| SU-DHL-4 (lymphoma) | DSMZ, Braunschweig, Germany | RPMI 1640 | Mediatech, Inc. | 10% FCS | CD20 |
| SU-DHL-6 (lymphoma) | DSMZ, | RPMI 1640 | Mediatech, Inc. | 10% FCS | CD45 |
| SKOV3 (ovary) | ATCC, | DMEM | Mediatech, Inc. | 10% FCS | HER2 |
| MiaPaca 2 (pancreas) | ATCC, | DMEM | Mediatech, Inc. | 12.5% FCS | E-cadherin |
| DU145 (prostate) | ATCC, | RPMI 1640 | Mediatech, Inc. | 10% FCS | EGF receptor |
| LNCaP (prostate) | ATCC, | RPMI 1640 | Mediatech, Inc. | 10% FCS | Androgen resistant |
| A431 (skin/epidermis) | ATCC, | DMEM | Mediatech, Inc. | 10% FCS | EGF receptor |
RPMI, Roswell Park Memorial Institute; DMEM, Dulbecco’s modification Eagle’s medium; IMDM, Iscove’s modified Dulbecco’s medium; FCS, fetal calf serum.
All 12 cell lines were maintained in an incubator with at 37° C and 5% CO2.
Individual cell lines
To determine the immunohistochemical phenotype of each cell line, each cell line was cultured individually and processed without splenic stroma to paraffin using the same protocol as for the combined cells. This permitted characterization of the antibody expression of each cell line to facilitate interpretation of staining of the pooled cells. A fungus was included to increase the types of stains that could be tested using the standard tissue.
Fungus
Approximately 50 mg of fungus obtained from visible colonies growing on food was minced in room temperature 10% NBF, fixed for 6 h, then a pellet was prepared using the protocol for cell lines described above. The species of the fungus was not determined; however, microscopically, it is a branching filamentous structure.
Gambusia affins
Mosquitofish, Gambusia affins, eastern subspecies, were harvested from a local domestic pond and transferred to 4° C water. The fish were immobilized, decapitated, then the 4° C water was replaced with 4° C 10% NBF, which gradually was allowed to warm for 3 h to room temperature. Two adjacent transverse sections 1–2 mm thick were cut immediately caudal to the gill plates. These provided a clear representation of internal organs (e.g., liver, intestines gonad). The cross sections were fixed in 10% NBF for 6–8 h at room temperature, then processed to paraffin for addition to the standard tissue block.
The goldfish, Carassius auratus, rosy red, Pimephales promelas and feeder guppies, Poecillidae reticulate, were purchased from a pet store and processed using the mosquitofish protocol. We evaluated these fish to evaluate them as possible alternatives to mosquitofish. For example, the guppies might be more available to some laboratories or histology facilities than mosquitofish.
Decalcification
Although the method for processing mosquitofish to paraffin blocks produced good histological sections, occasionally sections were torn by the bones of the fish. To reduce tearing, some transverse sections of the mosquitofish thorax were fixed in 10% NBF overnight. The 10% NBF was replaced with 10% formic acid (Fisher Scientific, Fair Lawn, NJ) for 8 h at room temperature under continuous agitation using a magnetic stirrer (Sigma-Aldrich). Formic acid was used for decalcification, because it can be used after fixation by formalin and does not contain proprietary reagents. Formic acid also has been reported to act as a formalin related fixative with less distortion of nuclear staining (Callis 2008).
Tissue processing
All specimens in labeled cassettes were processed in a tissue processor (Tissue-Tek® VIP™ 5, Sakura Finetek Inc., Torrance, CA) using the animal tissue processing protocol for our laboratory: 80% ethanol for 45 min, 95% ethanol for 45 min, three changes of absolute ethanol for 45 min each, two changes of xylene for 45 min each and three changes of 61° C paraffin wax for 30 min each.
Paraffin embedding
A standard tissue paraffin block contained separate areas for A431 cells (1 mm diameter × 3 mm), the nine combined cell lines mixed with splenic stroma (1 mm diameter × 3 mm), two cross sections of male and female mosquitofish (each approximately 1–2 mm diameter × 3 mm), fungus (1 mm diameter × 3 mm) and the portion of a traumatic human spleen (approximately 4 × 4 × 2 mm) obtained from routinely processed paraffin blocks. After tissue processing, each component of the artificial tissue was assembled in an array on a 30 × 24 × 5 mm base mold (Surgipath Medical Industries, Inc., McGaw Park, IL) that contained molten paraffin.
Histochemistry
The standard tissue paraffin block was sectioned at 4 μm. Sections mounted on Bond-Rite® microscope slides (Richard-Allan Scientific, Kalamazoo, MI) and heated at 60° C for 1 h to attach the sections to the slide. Prior to staining, sections were deparaffinized in three changes of xylene and rehydrated through graded concentrations of ethanol. The histological stains evaluated were: H & E (Gill et al. 1974), periodic acid Schiff (PAS) (McManus 1946), periodic acid Schiff-hematoxylin (PASH) (Mowry 1958), colloidal iron (Muller 1955), mucicarmine (Southgate 1927), Alcian blue pH 1.0 (AB-1.0) (Lev and Spicer 1964, Mowry 1956), Alcian blue pH 2.5 (AB-2.5) (Mowry 1956), and combination stains including colloidal iron-PASH (McManus 1948, Mowry 1958) and Alcian blue 2.5-PASH (Mowry 1956, 1963). We also evaluated the Masson trichrome stain (Polysciences, Inc., Warrington, PA) (Masson 1929).
Antigen recovery/retrieval (AR)
IHC was performed with and without AR according to the standard protocol of the laboratory. Initial titrations when testing new antibodies in specific tissues, e.g., standard tissue, are performed under varying conditions of AR. The conditions typically tested are: 1) no AR, 2) boiling in a pressure cooker at 15 psi in 0.01 M citric acid monohydrate, pH 6, or 3) boiling in a pressure cooker at 15 psi in 10 mM Tris/1 mM EDTA/l, pH 9. In general, we have found that 10 mM Tris/1 mM EDTA/l, pH 9, is the most sensitive retrieval method for a wide range of antibody-antibody conditions, i.e., many primary antibody-antigen combinations with a defined secondary detection exhibit stronger staining at pH 9. Therefore, most experiments were performed using 10 mM Tris/1 mM EDTA/l, pH 9. The standard tissue and all 12 individual cells lines (without splenic stroma) also were immunostained using 14 antibodies that included markers of proliferation, targets used in therapy and markers used to evaluate the characteristics of tumors (Table 2). The standard tissue was immunostained in parallel with appropriate formalin fixed and paraffin embedded tissues, e.g., breast carcinoma with uninvolved tissue adjacent to the area with cancer that was positive for ER/PR/HER2, a lymphoma, and a tonsil, both positive for CD3, CD20 and CD45.
Table 2.
Clones, vendors and dilution/concentration of monoclonal antibodies
| Antibody | Clone (species) | Vendor | Dilution | Concentration (μg/ml) | AR |
|---|---|---|---|---|---|
| Bcl-2 | 124 (mouse) | DAKO Carpentaria, CA | 1:250 | 0.60 | AR, pH 9 |
| CD3 | SP7 (rabbit) | ThermoFisher Fremont, CA | 1:20 | * | No AR, AR, pH 6 or 9 |
| 1:40 | * | ||||
| 1:100 | * | ||||
| CD20 Ab-1 | L26 (mouse) | ThermoFisher | 1:20 | * | No AR, AR, pH 6 or 9 |
| 1:40 | * | ||||
| 1:100 | * | ||||
| LCA (CD45) Ab-2 | 135-4C5 (mouse) | Neomarkers, Fremont, CA | 1:20 | 10 | No AR, AR, pH 6 or 9 |
| 1:40 | 5 | ||||
| 1:100 | 2 | ||||
| Cleaved caspase 3 | D175 (rabbit) | Cell Signaling, Danvers, MA | 1:200 | 0.5 | AR, pH 9 |
| Cytokeratins | AE1/AE3 (mouse) | Millipore Corp., Temecula, CA | 1:5000 | 0.2 | AR, pH 9 |
| E-cadherin | EP700Y (rabbit) | ThermoFisher | 1:20 | * | AR, pH 9 |
| 1:40 | * | ||||
| 1:200 | * | ||||
| 1:400 | * | ||||
| EGFr | 31G7 (mouse) | Zymed, San Francisco, CA | 1:3 | 5 | No AR |
| Estrogen Receptor alpha | 6F11 (mouse) | Santa Cruz Biotechnology, Santa Cruz, CA | 1:20 | * | AR, pH 9 |
| HER2 | Ab-3 (mouse) | Calbiochem, Cambridge, MA | 1:200 | 0.5 | AR, pH 9 |
| Ki67 | SP6 (rabbit) | ThermoFisher, | 1:1000 | * | AR, pH 9 |
| PCNA | PC-10 (mouse) | Santa Cruz Biotechnology | 1:15000 | 0.0067 | AR, pH 9 |
| Progesterone Receptor | PgR1294 (mouse) | DAKO | 1:200 | * | AR, pH 9 |
Concentration not specified by vendor.
Prior to AR, the sections were deparaffinized as described above, washed in deionized water and placed in the AR solution. The two AR solutions were 0.01 M citric acid monohydrate, pH 6, and 10 mM Tris/1 mM EDTA, pH 9. The sections were boiled in one of the AR solutions in a pressure cooker at 15 psi for 10 min. Pressure was allowed to equilibrate and sections were cooled for 10 min, washed in Tris buffer (0.05 M Trizma base (Sigma-Aldrich), 0.15 M NaCl, 1% Triton X-100), pH 7.6, at room temperature before IHC.
Immunohistochemistry (IHC)
After AR (if any), sections were incubated with 3% hydrogen peroxide for 5 min to quench endogenous peroxidase, then rinsed with deionized water followed by Tris buffer, pH 7.6. Sections then were incubated with 3% goat serum for 1 h to block nonspecific staining.
The primary antibodies were diluted in PBE buffer (1% bovine serum albumin, 1 mM EDTA in PBS) and incubated on sections for 1 h at room temperature. The dilution of each antibody based on the stock solution is specified in Table 2. All sections were washed three times in Tris buffer for 5 min each time, then incubated with horseradish peroxidase labeled polymer conjugated secondary antibody goat anti-mouse/goat anti-rabbit (EnVision+ System-HRP; Dako, Carpentaria, CA) for 30 min. As a control for nonspecific binding of the secondary detection system, a slide in which the primary antibody was omitted also was included from each block. After three washes in Tris buffer for 5 min each, a diaminobenzidine (DAB) substrate kit (Biogenex, San Ramon, CA) was used to visualize the antigen/antibody complex. Sections were lightly counterstained with Mayer’s hematoxylin (Sigma-Aldrich) for 30 sec, dehydrated through graded ethanols, cleared in xylene and mounted with Permount (Fisher Scientific, Fair Lawn, NJ).
TdT-mediated dUTP-FITC nick end-labeling (TUNEL) assay
The TUNEL assay was performed using the ApopTag Peroxidase in Situ Apoptosis Detection Kit (Millipore, Temecula, CA) following the manufacturer’s protocol with slight modification. Four-micrometer paraffin sections were mounted on Bond-Rite slides (Richard Allan Scientific, Kalamazoo, MI) at 60° C for 1 h, cleared in three changes of xylene and rehydrated through graded alcohols. Sections were pretreated in 10 mM glycine, pH 3, in a pressure cooker at 15 psi for 1 min. The pressure cooker then was depressurized rapidly and the sections transferred immediately into deionized water for cooling. The sections were quenched with 3% hydrogen peroxide for 5 min, then washed and placed in deionized water overnight at 4° C. The instructions in the kit were followed from this point forward. Specifically, the sections were washed with Tris buffer followed by equilibration buffer for 30 min at room temperature. Slides were drained and the working concentration of terminal deoxynucleotidyl transferase (TdT) diluted in reaction buffer was added. For each tissue block, slides in which no enzyme was added also were prepared. All slides were incubated in a humid chamber for 1 h at 37° C. Slides then were washed in stop/wash buffer for 10 min. After washing in Tris buffer, anti-digoxygenin-peroxidase was added to all slides, which then were incubated at room temperature for 30 min, then washed in Tris buffer. Subsequent steps were identical to the IHC protocol. DAB was used for visualization; slides were counterstained with Mayer’s hematoxylin, dehydrated through graded alcohols, cleared in three changes of xylene and mounted with Permount.
Evaluation
Evaluation of all staining was performed by a board certified pathologist (WEG). We used a semi-quantitative evaluation that used the percentage of stained cells at each level of staining intensity to obtain an immunostaining score. The intensity of immunohistochemical staining for a specific antibody was scored from 0 for no staining to 4 for strongest staining. The percentage of cells at each intensity divided by 100% was multiplied by each level of intensity, the products of which (i.e., for 0, +1, +2, +3 and +4 intensity) were added to give an immunostaining score also ranging from 0 to 4 (Grizzle et al. 1998, Otali et al. 2009).
Results
H & E staining
Hematoxylin stained the nuclei of spleen cells purple. Because most of the cells of the spleen are lymphocytes with scant cytoplasm, staining of their cytoplasm with eosin was not prominent; however, the cytoplasm of non-lymphocytes, including plasma cells and smooth muscle cells, was stained pink (Fig. 1 panel A). In the nine combined cell lines with stroma, the stroma was stained pink with blue strands owing to fragments of nuclear material that remained after preparation of the stroma. Also, some arteries remained in the stroma after mincing and their cytoplasm also was stained pink (Fig. 1, panel B1). The nine combined cell lines did not show the desired consistent eosinophilic cytoplasmic staining. To identify cell lines with more eosinophilic cytoplasm, three squamous carcinoma cell (SCC) lines, A431, CRL5928 and HTB182, were stained using the individual cell line protocols. None of the three cell lines were optimal for H & E staining, but the A431 cell line was the best. H & E staining of A431 cells is shown in Fig. 1, panel B2. The lack of H & E staining of the cytoplasm of the cell lines is likely due to the lack of differentiation in the rapidly growing cells as discussed subsequently. The muscles of the mosquitofish provided a good positive control for H & E staining (Fig. 1, panel C1).
Fig. 1.
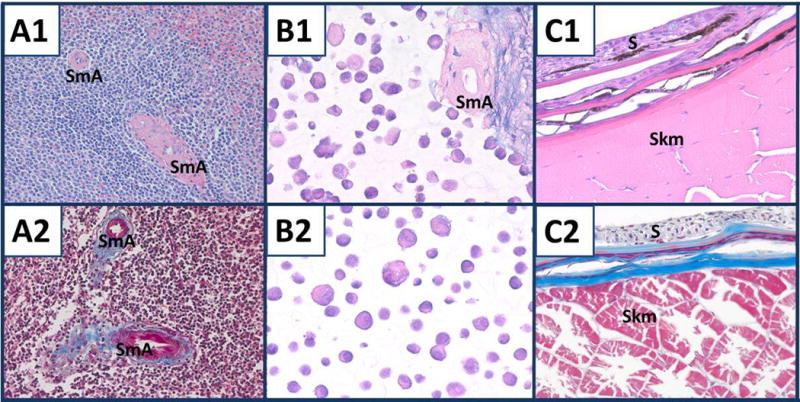
Components of the standard tissue. A1) Human spleen. SmA, two arteries. H & E. × 200. A2) Human spleen with two arteries. Masson trichrome. × 200. B1) Nine combined cell lines with splenic stroma. H & E. × 400. B2) A431 cells. H & E. × 400. C1, C2) Mosquitofish skin (S) and skeletal muscle (Skm). H & E and Masson trichrome, respectively. × 400.
Masson trichrome staining
The nuclei of the lymphocytes in the spleen were stained dark purple by Weigert’s hematoxylin component of the Masson trichrome stain, but their cytoplasm was too scant for prominent staining. By contrast, the cytoplasm of the smooth muscle cells of the arteries exhibited prominent red staining and connective tissue surrounding the arteries were stained blue (Fig. 1, panel A2). In the nine combined cell lines with stroma and the A431 cells, the coloration of nuclear staining varied from red to dark purple (data not shown). There was variation in staining of splenic stroma as well; staining ranged from blue to purple. In the mosquitofish component, collagen fibers were stained blue and muscle red (Fig. 1, panel C2).
Mucin-related staining
A summary of the results of mucin-related staining of the composite tissue is shown in Table 3. The stroma of the intact spleen was stained blue with AB-2.5, but very weakly with AB-1.0. This was accentuated in the fibrous tissue surrounding the splenic arteries. This pattern of staining was expected owing to high glycoprotein content of the stroma of the spleen and the arteries and overall low sulfomucin content of the stroma. The nine combined cell lines were not useful for evaluating mucin stains except for PAS or PASH (Fig. 2); however, AB-2.5 stained the acellular stroma blue and AB-1.0 stained the stroma very light blue (Fig. 2, panels A1, A2). Therefore, only the acellular splenic stroma was useful for evaluating histological stains for detecting mucins, e.g., AB-2.5. Some cells of the nine combined cell lines were stained with PASH, (Fig. 2, panels C1, C2), which means that these cells would be a useful control for PAS or PASH staining. Colloidal iron stained mucin minimally in scattered cells as did the combination stains, colloidal iron-PASH and Alcian blue 2.5-PASH (Fig. 2, panels D1, D2); however, the staining likely was due to staining by PAS/PASH. The colloidal iron stained both the HistoGel™ and 1.2% agar matrices extensively, i.e., entire areas of sections containing either HistoGel™ or 1.2% agar were stained by colloidal iron; the 1.2% agar was stained stronger (Fig. 2, panels D1, D2). Therefore, background staining of the HistoGel™ and 1.2% agar matrices by colloidal iron made evaluation of cells lines that were positive for colloidal iron difficult. We observed very light blue staining of the 1.2% agar matrix compared to HistoGel™ after AB-1.0, AB-2.5 and PASH staining that would not affect the interpretation of these mucin stains. We observed no mucicarmine staining of the 1.2% agar or HistoGel™ matrices.
Table 3.
Mucin staining in components of the standard tissue
| AB-1.0 | AB-2.5 | Mucicarmine | PAS | PASH | Colloidal iron | AB-2.5 + PASH | Colloidal iron + PASH | |
|---|---|---|---|---|---|---|---|---|
| Nine combined cell lines | N | N | N | V | V | N | V | V |
| Stroma | FB | blue | N | NE | W* | V | S | W* |
| Mosquitofish goblet cells | W* | S | S | S | S | S | S | S |
N, negative except for counterstain; FB, faint blue staining; V, some cells staining; W, light blue; W*, moderate to weak staining; S, strong staining. NE, not evaluated.
Fig. 2.
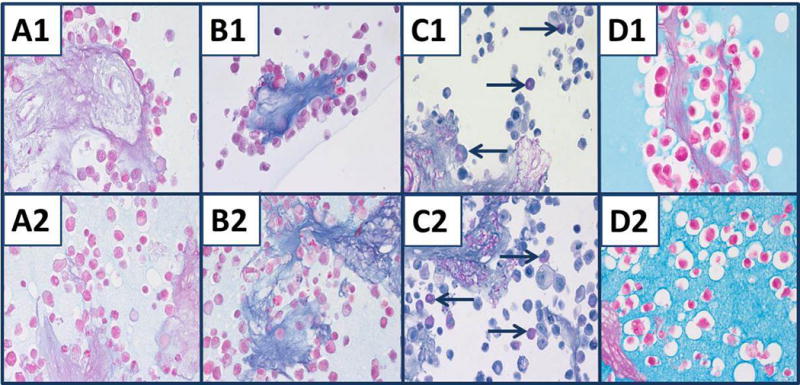
Mucin staining of nine combined cell lines and acellular stroma of spleen. Cell lines and stroma embedded in either HistoGel™ (#1) or 1.2% agar (#2). A1, A2) AB-1.0. B1, B2) AB-2.5 stains stroma of spleen blue, but there is little staining of cells other than nuclear counterstain. C1, C2) Selective cells stained with PASH and PAS of splenic stroma (blue arrows). D1, D2) Colloidal iron does not stain splenic stroma clearly, but does stain the HistoGel™ matrix (D1) and agar matrix (D2). Staining of cells and splenic stroma by colloidal iron is not apparent owing to staining of the matrices, HistoGel™ or 1.2% agar. All panels, original magnification × 400.
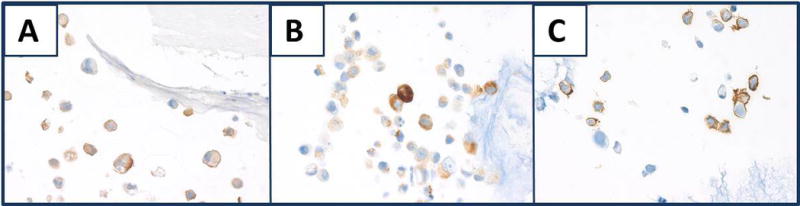
The mosquitofish component of the standard tissue proved to be an excellent control for all histochemical stains for mucin tested. Figure 3 shows goblet cells of the small intestine and the skin was stained strongly with AB-2.5, mucicarmine (data not shown), colloidal iron and the colloidal iron-PASH combination stain, which indicates the presence of mucins, and weak to moderately with AB-1.0, which is consistent with the presence of some sulfomucins. Although the goblet cells of the intestine were stained strongly with colloidal iron (Fig. 3, panel D), there was less staining of the mucin in the goblet cells of the skin. The difference in the mucin of goblet cells of the intestine and goblet cells of the skin was demonstrated by colloidal iron-PASH stain (Fig. 3, panel F). In the intestine, the goblet cells were stained blue-purple (red arrow), which indicated staining with both colloidal iron and PASH, but in some goblet cells of the skin, the colloidal iron staining was overwhelmed by the red color of the PASH stain (blue arrow). In the other types of fish tested, the staining of mucins in the skin was variable; some goblet cells of the skin exhibited staining characteristic of colloidal iron, AB-2.5 or mucicarmine, but others were stained in a pattern consistent with PASH.
Fig. 3.
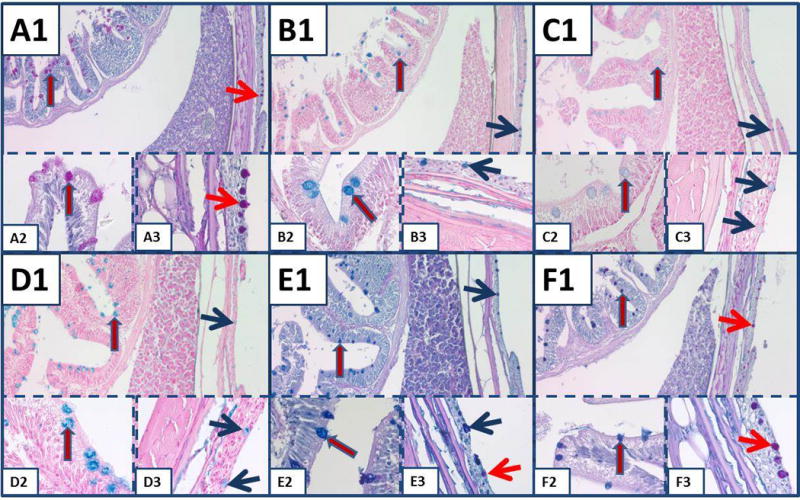
Mucin staining in the small intestine and skin of mosquitofish. A) PASH stains goblet cells of small intestine (red arrow blue outline) and goblet cells of the skin (red arrow). Similar arrows are used for the same identifications in subsequent panels, but colors demonstrating the goblet cells of the skin are red or blue to differentiate staining. B) AB-2.5. C) AB-1.0. D) Colloidal iron. E) AB-2.5-PASH. In panel E3, note the different types of mucin in the goblet cells of the skin. F) Colloidal iron-PASH. In (F), different types of mucin are stained in goblet cells of skin (red) compared to goblet cells of intestine (blue). All panels, original magnification × 100; insets × 630.
IHC staining
Results of immunostaining separate components of the standard tissue, i.e., the nine combined cell lines mixed with splenic stroma, some tissues of the mosquitofish, fungus and the intact spleen as well as the individual cell lines without splenic stroma with specific antibodies are shown in Table 4. The membrane staining of HER2, EGFr, cytokeratins AE1/AE3 and E-cadherin varied among the cell lines as shown in Fig. 4; in some cell lines and in fungus, we found no expression of these molecules. Examples of HER2 staining shown in Fig. 5, panels A1, A2, A3, include SKOV3, MiaPaca 2 and DU145 cell lines, respectively. MDA-MB-231, DU145, SU-DHL-4 and SU-DHL-6 stained weakly for HER2, while HTB182 (lung) and Ly3 (B-cell lymphoma) showed no immunostaining. EGFr immunostaining is shown in Fig. 5, panels B1 (A431), B2 (LNCaP) and B3 (MCF7). We observed no immunostaining of EGFr in CRL5928 (lung), MiaPaca 2 (pancreas) or in any of the three B-cell lymphoma cell lines. Immunostaining for cytokeratins AE1/AE3 is shown in Fig. 5, panels C1, C2, C3, which are MCF7, HTB182 and MiaPaca 2 cells, respectively. We observed no staining of cytokeratins AE1/AE3 in MiaPaca 2 or in the three lymphoma cell lines.
Table 4.
Molecular characteristics of cell lines, mosquitofish and selected tissues and their interaction with specific antibodies
| Cell line/specimen | EGFr 5 μg/ml | Ki67 (SP6) 1:1000 | E-Cad 1:20 | PR 1:200 | ERα 1:20 | HER2 0.5 μg/ml | Cytokeratin AE1/AE3 0.2 μg/ml | PCNA 0.0067 μg/ml |
|---|---|---|---|---|---|---|---|---|
| A431 | +3 | +2 | +2 | 0 | 0 | +1 | +3 | +3 |
| Fungus | ||||||||
| Human spleen | 0 | +1 | V* | 0 | 0 | V +3 | 0 | V +3 |
| Mosquitofish | 0 | N | L | 0 | N | Skm/L | S | Sp/L/C |
| Nine combined cell lines | V +2 |
V +2 |
V +3 |
V +3 |
V +3 |
V +4 |
V +4 |
V +3 |
| MCF-7 | +1 | +3 | +3 | +3 | +3 | +1 | +3 | +3 |
| MDA-MB-231 | +3 | +3 | 0 | 0 | 0 | 0 | +2 | +3 |
| CRL5928 | 0 | +3 | +3 | 0 | 0 | +3 | +3 | +3 |
| HTB182 | 0 | +3 | +2 | 0 | 0 | 0 | +3 | +3 |
| Ly3 | 0 | +3 | 0 | 0 | 0 | 0 | 0 | +3 |
| SU-DHL-4 | 0 | +3 | 0 | +1 | 0 | 0 | 0 | +3 |
| SU-DHL-6 | 0 | +3 | 0 | 0 | 0 | 0 | 0 | +3 |
| SKOV3 | +3 | +3 | 0 | 0 | 0 | +3 | +2 | +3 |
| MiaPaca 2 | +2 | +2 | 0 | 0 | +2 | 0 | +3 | |
| DU145 | +3 | +3 | +1 | 0 | 0 | 0 | +3 | +3 |
| LNCaP | +2 | +3 | 0 | 0 | 0 | +1 | +2 | +3 |
Patterns of staining of specific molecular marker based on immunostaining scores: (0) no staining, (+1) 0.5–1.0, (+2) 1.1–2.0, (+3) 2.1–3.0, (+4) 3.1–4.0. C, colon; L, liver; Skm, skeletal muscle; S, skin; Sp, spleen; E-Cad, E-cadherin; V, variable because of nine combined cell lines with some cells showing (0) immunostaining score in a range to (+4), i.e., strongest staining cells.
Uncommon staining; N, not evaluated.
Fig. 4.
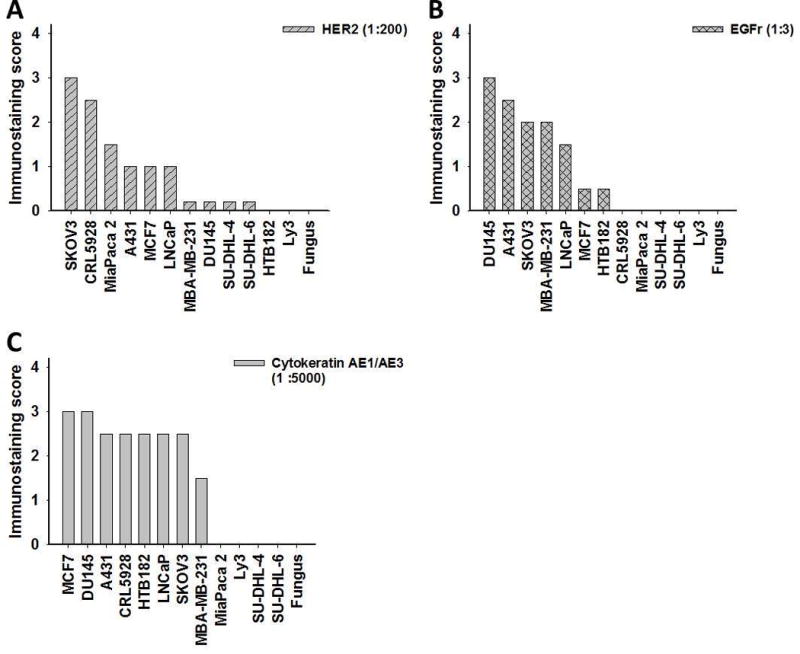
Immunostaining scores of antibodies. A) HER2. B) EGFr. C) Cytokeratins AE1/AE3 in 12 cell lines and fungus.
Fig. 5.
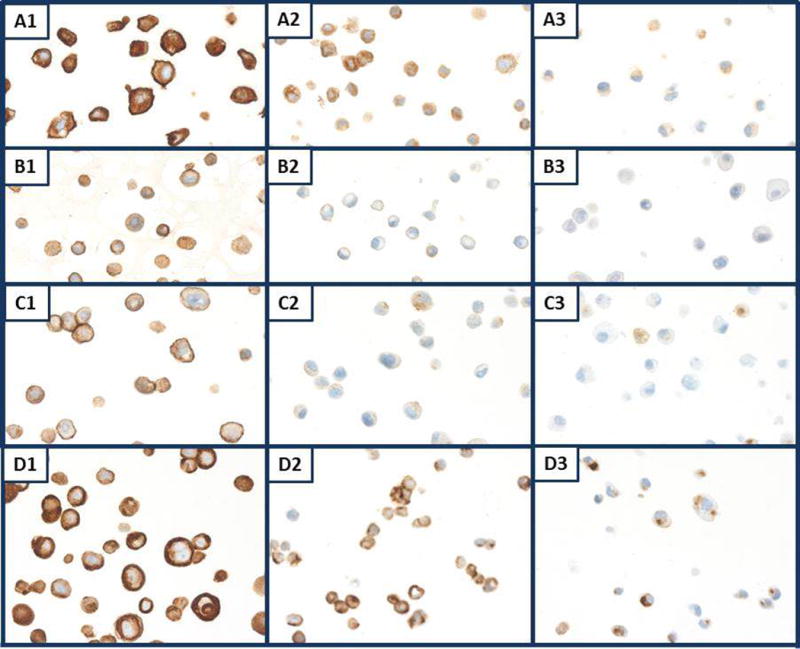
Individual cell lines immunostained for membrane markers. A1, A2, A3) SKOV3, MiaPaca 2 and DU145 cells, respectively, immunostained for HER2. B1, B2, B3) A431, LNCaP, MCF7 cells, respectively, immunostained for EGFr. C1, C2, C3) CRL5928, A431, DU145 cells, respectively, immunostained for E-cadherin. D1, D2, D3) MCF7, HTB182, MiaPaca 2 cells, respectively, immunostained for cytokeratin AE1/AE3. All panels, original magnification × 400.
The individual cell lines may be useful for antibody titration. Each positive cell line exhibited decreased staining intensity as the concentration of E-cadherin was reduced (Fig. 6). At a dilution of the primary antibodies of 1:200, some cell lines reached a plateau in staining intensity after which there frequently was only a small increase in immunostaining. Membrane staining for EGFr, HER2 and E-cadherin of scattered cells of the nine combined cell lines and stroma is shown in Fig. 7, panels A, B, C, respectively.
Fig. 6.
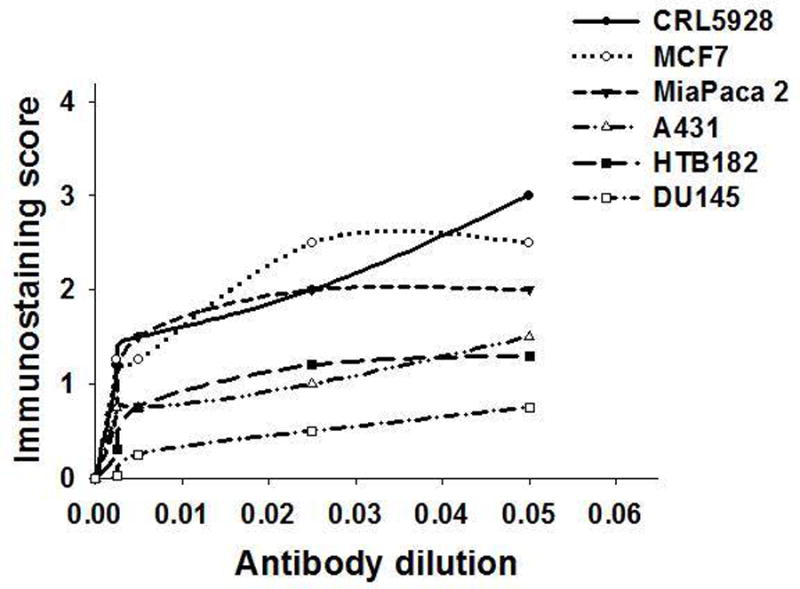
Antibody titration curves of six cell lines stained for E-cadherin at different dilutions after AR with 10 mM Tris/1 mM EDTA, pH 9, boiled in a pressure cooker at 15 psi for 10 min.
Fig. 7.
Membrane staining of nine combined cell lines with splenic stroma. A, B, C) Cell lines were stained for EGFr, HER2 and E-cadherin, respectively, while some cells lines in the combined nine cell lines exhibited membrane staining while others did not owing to cellular dilution. All panels, original magnification × 400.
Immunostaining for PCNA and Ki67 (SP6) in the nine combined cell lines showed that most cells stained with either marker (Fig. 8, panels A, B, respectively) as would be expected in a proliferating cell line. Cells that were not stained likely suffered growth inhibition in areas of confluence. For immunostaining of estrogen receptor alpha (ERα) and progesterone receptor (PR), scattered cells were stained and their numbers were proportional to the one cell line of the combined nine that expresses ERα and PR, i.e., MCF7 (Table 4). It is of interest that there was relatively weak staining for PR in the SU-DHL-4 cells (Table 4).
Fig. 8.
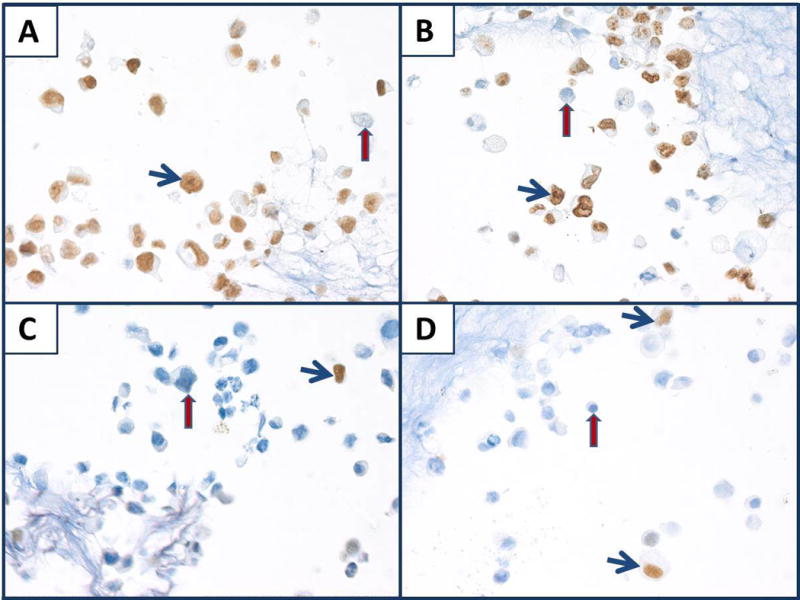
Nuclear staining of nine combined cell lines mixed with splenic stroma. A, B, C, D) Immunostaining for PCNA, Ki67 (SP6), ERα, PR, respectively. Note staining (blue arrows) and non-staining (red arrows). All panels, original magnification × 400.
The three diffuse B-cell lymphoma cell lines were immunostained using CD20 and CD45. There was minimal staining of CD20 in the Ly3 cell line even after AR using either 0.01 M citric acid, pH 6, or 10 mM Tris/1 mM EDTA, pH 9 (Fig. 9, panel A). Conversely, SU-DHL-4 and SU-DHL-6 B-cell lymphomas were stained strongly for CD20 with or without AR. We observed CD45 immunostaining in the Ly3 B-cell lymphoma cell line at a relatively high concentration (10 μg/ml) after AR using 10 mM Tris/1 mM EDTA, pH 9. The SU-DHL-6 B-cell lymphoma cell line exhibited staining for CD45 at all concentrations with or without AR; by contrast, SU-DHL-4 exhibited no staining for CD45 with or without AR (Fig. 9, panel B).
Fig. 9.
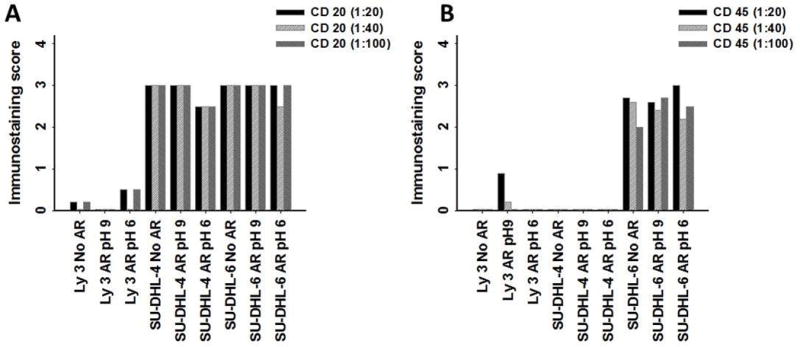
Antibody titration of lymphoma cell lines with and without AR. A, B) Immunostaining scores for CD20 and CD45, respectively.
CD3, a T cell marker, also was tested on all three B-cell lymphoma cell lines with or without AR. As expected, we observed no CD3 staining in any of the three lymphoma cell lines when used at concentration that did not produce background staining with other cell lines (Table 5).
Table 5.
Molecular characteristics of lymphoma and lymphoma cell lines, and their interaction with specific monoclonal antibodies
| Cell line | CD3 1:20, no AR |
Bcl-2 1:250, AR, pH 9 |
CD20 1:20, AR, pH 9 |
LCA (CD45) 1:20, AR, pH 9 |
|---|---|---|---|---|
| Lymphoma | 0 | +1 | +1 | +1 |
| Ly3 | 0 | +3 | 0 | +1 |
| SU-DHL-4 | 0 | +3 | +3 | 0 |
| SU-DHL-6 | 0 | 0 | +3 | +3 |
Pattern of staining of the specific molecular biomarkers based on immunostaining scores: (0) no staining, (+1) 0.5–1.0, (+2) 1.1–2.0, (+3), 2.1–3.0, (+4) 3.1–4.0.
TdT-mediated dUTP-FITC nick end-labeling (TUNEL) assay (apoptosis marker)
Only a few cells of the nine combined cell lines showed immunostaining for cleaved caspase 3 (Fig. 10, panel A1). By contrast, the TUNEL assay exhibited staining in more of the cell lines as well as strong staining of the stroma due to DNA damage from preparation of the stroma (Fig. 10, panel A2). The lymphoma cell lines, SU-DHL-6 and SU-DHL-4, showed greater numbers of cells with morphologic features of apoptosis; these cells also had a higher proportion that stained for cleaved caspase 3 and TUNEL, (Fig. 10, panels B1, B2), respectively. In the mosquitofish intestine, several cells showed positive staining for both cleaved caspase 3 and TUNEL (Fig. 10, panels C1, C2), respectively.
Fig. 10.
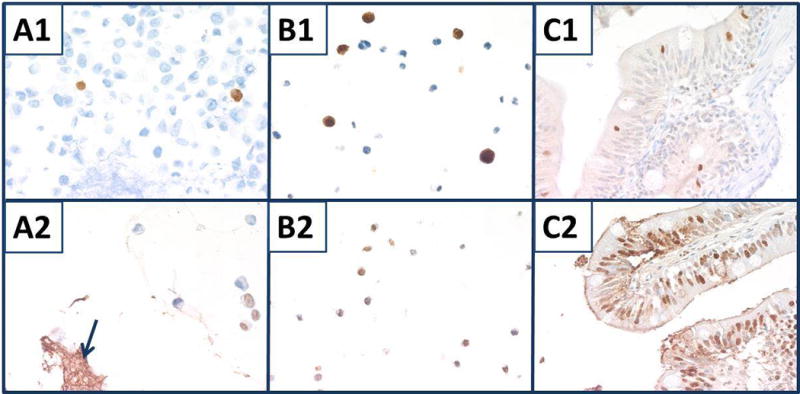
Staining for apoptosis. Top panels were stained for cleaved caspase 3. A1) Nine combined cell lines with splenic stroma. B1) SU-DHL-6. C1) Mosquitofish intestine. Bottom panels were stained with TUNEL assay. A2) Nine combined cell lines with splenic stroma (blue arrow). B2) SU-DHL-4. C2) Mosquitofish bowel. All panels, original magnification × 400.
Figure 11, panels A–C are examples of cross reactivity of mosquitofish tissue that stained for the antibodies, PCNA in intestine (A) and HER2 in liver (B) and skeletal muscle (C). HER2 staining of the liver suggests that molecules other than HER2 may be stained.
Fig. 11.
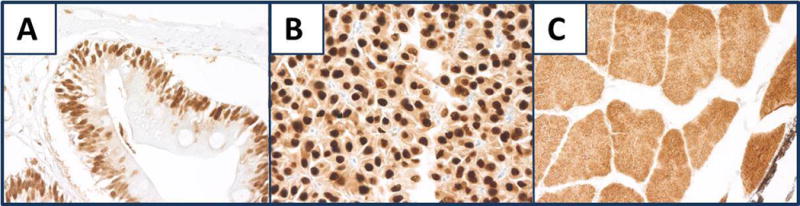
Mosquitofish tissues. A) Intestine PCNA staining. B, C) Staining for HER2 in liver and skeletal muscle, respectively,. All panels, original magnification × 400.
Discussion
Histopathological staining of tissues typically varies among institutions that perform the staining in part because controls for histology and IHC vary. Tissue blocks used for controls are exhausted rapidly owing to repeated sectioning. A common problem for standardizing histological staining, therefore, is the lack of a consistent control tissue that can be used over time. We addressed this problem by developing a standard tissue that contains five components: 1) nine combined cell lines in a mixture of stroma of human spleen, 2) a squamous cancer cell line, A431 (without splenic stroma), 3) a small portion of intact normal human spleen, 4) a fungus and 5) sections of mosquitofish. We determined that each component of the standard tissue provided information that aided determination of the quality and characteristics of histology and IHC methods and was useful as a control for assay validation, quality assurance (QA) and evaluation of new approaches to fixation and tissue processing.
Currently, antibodies may be marketed for “experimental use only” and frequently they do not work under the conditions specified by the vendor. Even the same clone from the same manufacturer, but marketed by different vendors, may behave differently in the same assay owing to vendor modifications such as dilutions and additives. Also, vendors frequently recommend a concentration or dilution that produces staining of maximal intensity when a much lower concentration would work as well or better because of less background staining or cross reactivity (Fig. 11). Use of a standard tissue could save resources by permitting selection of an appropriate concentration and facilitating discussions with vendors concerning the sensitivities and specificities of the antibodies they market.
Although generally we obtained intact sections from our standard tissue block, occasionally we obtained torn sections owing to the bones in the fish. To reduce tissue tearing, sections of mosquitofish and other types of small fish, e.g., guppies,) after fixation in 10% NBF can be exposed to a decalcifying agent such as 10% formic acid for at least 6 h prior to processing and embedment. We chose the mosquitofish owing to ease of collection, wide availability and convenient size. The mosquitofish are similar to a large guppy; it is a surface feeder and proliferates rapidly in captivity. Some histology facilities may choose not to collect these specimens in the wild; in these cases, we recommend obtaining “feeder” fish, i.e., fish fed to larger carnivorous fish, from a pet store. We experimented with three such fish (unpublished observations): the goldfish, Carassius auratus (too large), the rosy red minnow, Pimephales promelas, a minnow variant developed as a feeder fish, and the common guppy, Poecillidae reticulata. When processing fish, the tissues must be fixed quickly, because the gastrointestinal system undergoes autolysis rapidly, generally within minutes after death. To appreciate the anatomy of small fish, we recommend a book on the anatomy and histology of the zebrafish (Danio rerio), which commonly are used for experimentation (Holden et al. 2012).
Histochemical and IHC staining of sections of our standard tissue block prepared up to 1 year earlier showed no detectable changes during this period. This indicates that our protocol for preparing standard tissue, which includes fixation in 10% NBF for 2 h followed by tissue processing is acceptable for histochemical or immunohistochemical staining for at least 1 year.
Our construction of a standard tissue allows investigators to add or remove cell lines for specific purposes, especially for research. For example, for dermatology, cell lines characteristic of skin cancers, e.g., melanomas, could be substituted for breast cancer cell lines. Therefore, our nine combined cell lines can be adapted to organs of particular interest.
H & E is the most frequently used stain in histology laboratories and its use is required for pathological diagnosis. Hematoxylin stains primarily nuclear components with varying intensity from blue to dark purple, while eosin stains cytoplasm and most connective tissue fibers varying shades and intensities of orange, pink or red (Gamble 2008). Because H & E is important for diagnosing pathology, its standardization is critical, especially for telepathology, because pathologists must have confidence in the tinctorial characteristics of the stains.
We found that a small piece of intact normal spleen was an important component of the standard tissue as a control for H & E staining. Hematoxylin staining can be assessed by the staining of the normal lymphocytes of the spleen and eosin staining can be assessed by the staining of the smooth muscle of splenic arteries. A “normal” spleen from a surgical case can provide thousands of small areas of spleen and therefore is virtually inexhaustible. The histology of the spleen used in a standard tissue, however, should be evaluated before addition to a standard tissue block so that areas of blood pooling and other traumatic damage can be avoided. Neither the squamous cancer cell line, A431, nor the nine combined cell lines were satisfactory for assessing H & E staining. We suggest that this was because some differentiation is lost either secondary to rapid proliferation during cell culture (Freshney 2005) or to the poorly differentiated cells that initially grow as cell cultures are developed and established (Fogh and Sykes 1972; Freshney 2005).
Two adjacent cross sections of the mosquitofish from just caudal to the head provide skin, skeletal muscle, kidney, liver, bowel, spinal cord and sometimes spleen, pancreas, ovary or testis samples for the standard tissue. H & E staining of the mosquitofish is similar to that of human tissues, but is somewhat variable among liver, bowel, skeletal muscle, skin, exocrine pancreas and spleen. The skeletal muscle, which has large eosinophilic sarcoplasm, especially is useful for standardizing staining with eosin, while the other tissues provided nuclei for standardization of hematoxylin staining.
The Masson trichrome stain is used to identify collagen and, because of its blue staining, to differentiate it from the red staining of smooth muscle and other cells; nuclei stain purple-black, erythrocytes, fibrin, cytoplasm of cells and muscle are stained red and collagen is stained blue. The intact human spleen, which contains arteries surrounded by stroma, is a good control for the trichrome stain; the cells and connective tissues stain as expected. Trichrome staining in the mosquitofish is similar to that of human tissue, i.e., collagen fibers are stained blue, muscle red and the heads of sperm in male fish are stained dark purple.
The spleen is a useful control for some mucin stains. Because of the accumulation of iron in splenic macrophages as senescent red blood cells are removed by the spleen, the intact spleen serves as an indicator that the last portion of the colloidal iron stain (i.e., ferrocyanide-HCl reaction) is working. Also, the stroma of the spleen contains sufficient glycoproteins to be stained by AB-2.5 as we showed with the stroma mixed with the nine combined cell lines. Finally, most spleen contains plasma cells that are stained by PAS/PASH.
There was very little mucin staining of the cell lines in the standard tissue even though many were adenocarcinoma; this likely was due to the decreased production of some molecules associated with differentiation, such as mucin in rapidly proliferating cells. Our interpretation is supported by strong staining of pancreatic cancers by colloidal iron, mucicarmine, AB-1.0 and AB-2.5 (data not shown). The mosquitofish is an excellent positive control for all mucin stains, especially the PAS/PASH, AB-2.5, colloidal iron and mucicarmine stains, and also can serve as an adequate positive control for AB-1.0. The guppy, goldfish and rosy red minnow can serve as equivalent positive controls (unpublished observations). As demonstrated in Fig. 3, the goblet cells of the intestine and skin of the mosquitofish are stained strongly with AB-2.5, colloidal iron, PASH and mucicarmine (mucicarmine results not shown). Sulfomucins were present in goblet cells of the intestine and skin of the mosquitofish as shown by the weak to moderate, but consistent staining by AB-1.0. The combined stains, colloidal iron plus PASH and AB-2.5 plus PASH, also stained all goblet cells; however, there were some differences in staining by colloidal iron plus PASH with the goblet cells of the mosquitofish intestine, which were stained blue-purple and the goblet cells of the skin, which were stained the typical dark red of PAS. This likely represents different combinations of mucins in the goblet cells related to different functions. We found different staining patterns for the goblet cells of the skin in the goldfish, guppy and rosy red minnow (unpublished observations).
Our standard tissue should be useful for initial IHC assays and especially for research using antibodies that have not been approved for clinical use by the FDA. Potential applications include comparing variability and function in immunostaining among lots and types of antibodies (e.g., is the antibody lot equivalent to a prior lot), establishing optimal dilutions of antibodies, selecting the optimal method for AR for primary antibodies, and evaluating the effects of fixation and tissue processing. We investigated selected markers of proliferation (Ki67 (SP6), PCNA), targets of therapy (ERα, PR, HER2) and a marker of cellular characteristics, E-cadherin, a marker of epithelial to mesenchymal transition.
It should be possible to use cell lines embedded in a tissue microarray to construct a standard curve to validate assay comparisons within or between laboratories. Camp et al. (2008) reported the intensity of immunostaining for EGFr as follows MDA-MB-231 > A431 = SKOV3 > MCF7. Although our results of immunostaining for EGFr in our individual cell lines were DU145 > A431 > SKOV3 > MDA-MB-231 > MCF7, a plot of our values lies within the standard curve of the Camp et al. (2008) study. Our nine combined cell lines could be a general control for tissue arrays and individual cell lines can be used to create a standard curve for immunostaining of paraffin embedded tissues.
An earlier study of reproducibility of assays of samples from patients with breast cancer used formalin fixed cell lines that were embedded in paraffin (Rhodes et al. 2002). These investigators reported strong HER2/neu expression in membranes of SKOV3 cells, but no expression in MCF7 or MDA-MB-231 cells. We observed similar levels of immunostaining for HER2 in SKOV3 cells; however, weak, but clearly detectable immunostaining for HER2 was observed in MCF7 cells and barely detectable staining was observed in MDA-MB-231 cells. The difference between our results and those of (Rhodes et al. 2002) likely is due to differences in AR and/or different sensitivities of secondary detection methods. We used 10 mM Tris/1 mM EDTA/l, pH 9, for 10 min compared to 10 mM/l citrate, pH 6, or 10 mM/l, pH 7, for 2 min and 2.5 min, respectively used in the study by Rhodes et al. (2002). In general, we found that AR at pH 9 is more effective and resulted in stronger immunostaining for most antibodies than at pH 6 or 7.
Different cell lines have varying thresholds for detection of selected molecules or the same antibody concentration and AR method (Fig. 6, 9, panels A, B). Use of our standard tissue would help determine the optimal concentration of an antibody for IHC analysis of formalin fixed tissue. To illustrate titration curves, we immunostained individual cell lines with varying concentrations of E-cadherin. The curves indicated that as the antibody concentration increased, the immunostaining score increased to a level that resulted in strong staining, beyond which there was little change, except for squamous cancer cell line CRL5928 (lung).
Andersson et al. (2006) investigated immunostaining of cultured cells embedded in agarose gel, then processed to paraffin. These investigators reported stainingin several organelles and cytoplasmic staining of cytokeratins AE1/AE3 in HaCaT epithelial cells as expected. Epithelial cell lines including adenocarcinomas and squamous cell carcinomas contain variable levels of cytokeratins. Our study quantified earlier observations based on immunostaining scores using multiple cell lines.
Immunohistochemical staining for cytokeratins AE1/AE3 in the mosquitofish skin exhibited a pattern consistent with human skin. Our observation suggests that some tissues of the two vertebrates, fish and mammals, also may be related molecularly and in agreement with the concept that some organ systems of the fish maybe analogous to other vertebrates.
Markers of proliferation, such as Ki67 and PCNA, typically are expressed in all proliferating cell lines in our standard tissue. Cultured cells processed to paraffin and immunostained with Ki67 (clone MIB-1) have been used previously to ensure uniformity during a transition from AR using a microwave to AR using a vegetable steamer (Ruby and McNally 1996). In our study, the immunostaining scores for both Ki67 (SP6) and PCNA after AR ranged from 2.0 to 3.0 in our standard tissue, which indicates that the standard tissue can be used as a reference for markers of proliferation for inter-or intra-laboratory controls.
Pellets of cultured MCF7 cells have been included with breast tissues in tissue cassettes as an internal standard for immunohistochemical staining for ERα (Riera et al. 1999). Of 12 cell lines studied individually without combination with other cells or mixed with splenic stroma, only the MCF7 cells were stained with antibodies to ERα (Table 4); therefore, the moderate to strong immunostaining of ERα in the nine combined human cell lines mixed with splenic stroma as a component of our standard tissue was due to the MCF7 cells; therefore, the pellet of the nine combined cell lines and stroma also can be used as an internal control.
Lymphoma cell lines express hematopoietic markers, such as leukocyte common antigen (CD45), but usually not epithelial markers, such as E-cadherin, so these cell lines can be used in standard tissues as negative controls for many, but not all, epithelial related molecules. We emphasize that the staining of hematopoietic markers in the spleen, tonsil and a B-cell lymphoma was greater than that in the individual lymphoma cell lines or in the nine combined cell lines that were immunostained simultaneously at the same concentration. The intact spleen serves as both a negative control for many antigens and a positive control for hematopoietic related antigens, such as leukocyte common antigen (LCA or CD45), CD3 and CD20.
We observed strong TUNEL staining of the splenic stroma that was mixed with the nine cell lines; this may be explained by the mincing and washing required for preparation. It is likely that DNA was sheared from the cells within the stroma that were destroyed during preparation. The mosquitofish intestine also was a useful positive control for cleaved caspase 3 and the TUNEL assay, although the latter was the more sensitive method (Fig. 10, panels C1, C2). The SU-DHL-4 and SU-DHL-6 lymphoma cell lines also serve as excellent controls for markers of apoptosis, because these cell lines exhibit extensive apoptosis during their growth; however, Ly3 does not. Specifically for immunostaining with the TUNEL and cleaved caspase 3 assays, large numbers of apoptotic SU-DHL-4 and SU-DHL-6 cells, respectively, were observed in the individual lymphoma cell lines without splenic stroma. In the nine combined cells mixed with splenic stroma of the standard tissue; however, the proportion of apoptotic cells was less likely due to dilution by the addition of other cells.
The use of the mosquitofish to test the cross reactivity and specificity of antibodies is beyond the scope of our study; however, it is likely that the TUNEL assay would recognize damaged DNA in all species. When staining is observed in the mosquitofish using antibodies to cleaved caspase 3, E-cadherin, cytokeratins AE1/AE3 and HER2, the molecules that are detected by the antibodies is uncertain, especially because some intracellular patterns of immunostaining, such as that of HER2, are different from those observed in human tissues. Also, it is unknown whether the antigens in the mosquitofish function in the same way as the analogous molecules in humans. This is especially likely to be true for polyclonal antibodies, which typically react with a greater number of epitopes.
We have developed a standard tissue that obviates the exhaustion and variability of typical control tissues. Our standard tissue can be used in research and can serve as a consistent long term control for most histochemical stains and many IHC stains.
Acknowledgments
This manuscript is based upon a presentation by WEG at the Biological Stain Commission 2014 Annual Meeting. It is supported in part by the following grants: the Cooperative Human Tissue Network (1UM1CA183728), the UAB Pancreatic (2P50CA101955) and Breast (5P50CA089019) SPORES, the DOD Grant (W81XWH-10-1-0543), the UAB Comprehensive Cancer Center Core Support Grant (P30CA13148),the U54 MSM/TU/UAB Comprehensive Cancer Center Partnership (2U54CA118948) and NCI Institutional National Research Service Award (T32) (5T32CA183926-02).
Footnotes
Declaration of interest: The authors report no conflict of interest. The authors alone are responsible for the content and writing of this paper.
References
- Andersson AC, Stromberg S, Backvall H, Kampf C, Uhlen M, Wester K, Ponten F. Analysis of protein expression in cell microarrays: a tool for antibody-based proteomics. J Histochem Cytochem. 2006;54:1413–1423. doi: 10.1369/jhc.6A7001.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke NG, McCaffrey D, Mackle E. Contamination of histology biopsy specimen-a potential source of error for surgeons: a case report. Cases J. 2009;2:7619. doi: 10.4076/1757-1626-2-7619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callis G. Bone. In: Bancroft J, Gamble M, editors. Theory and Practice of Histological Techniques. 6th. Churchill Livingstone; Philadelphia, PA: 2008. pp. 333–363. [Google Scholar]
- Camp RL, Neumeister V, Rimm DL. A decade of tissue microarrays: progress in the discovery and validation of cancer biomarkers. J Clin Oncol. 2008;26:5630–5637. doi: 10.1200/JCO.2008.17.3567. [DOI] [PubMed] [Google Scholar]
- Drexler HG, Dirks W, MacLeod RAF, Quentmeier H, Steube KG, Uphoff CC, editors. DSMZ Catalogue of Human and Animal Cell Lines. 2014 www.dsmz.de.
- Fogh J, Sykes JA. A comparison of methods for morphological studies of cultured cells. In Vitro. 1972;7:206–227. doi: 10.1007/BF02615977. [DOI] [PubMed] [Google Scholar]
- Freshney RI. Differentiation Culture of Animal Cells. John Wiley & Sons, Inc; Hoboken, NJ: 2005. pp. 269–278. [Google Scholar]
- Gamble M. The hematoxylins and eosin. In: Bancroft J, Gamble M, editors. Theory and Practice of Histological Techniques. 6th. Churchill Livingston; Philadelphia, PA: 2008. pp. 121–134. [Google Scholar]
- Gill GW, Frost JK, Miller KA. A new formula for a half-oxidized hematoxylin solution that neither overstains nor requires differentiation. Acta Cytol. 1974;18:300–311. [PubMed] [Google Scholar]
- Goldstein NS, Hewitt SM, Taylor CR, Yaziji H, Hicks DG, Members of ad-Hoc Committee on Immunohistochemistry Recommendations for improved standardization of immunohistochemistry. Appl Immunohistochem Mol Morphol. 2007;15:124–133. doi: 10.1097/PAI.0b013e31804c7283. [DOI] [PubMed] [Google Scholar]
- Grizzle EW, Myers RB, Manne U, Srivastava S. Immunohistochemical Evaluation of Biomarkers in Prostatic and Colorectal Neoplasia, vol 14: Tumor Marker Protocols. Humana Press Inc; Totowa, NJ: 1998. pp. 161–179. [Google Scholar]
- Holden JA, Layfield LL, Matthews JL. The Zebrafish: Atlas of Macroscopic and Microscopic Anatomy. Cambridge University Press; New York: 2012. [Google Scholar]
- Lev R, Spicer SS. Specific staining of sulphate groups with Alcian blue at low ph. J Histochem Cytochem. 1964;12:309. doi: 10.1177/12.4.309. [DOI] [PubMed] [Google Scholar]
- Margolis B, Rhee SG, Felder S, Mervic M, Lyall R, Levitzki A, Ullrich A, Zilberstein A, Schlessinger J. EGF induces tyrosine phosphorylation of phospholipase C-II: a potential mechanism for EGF receptor signaling. Cell. 1989;57:1101–1107. doi: 10.1016/0092-8674(89)90047-0. [DOI] [PubMed] [Google Scholar]
- Masson P. Some histological methods. Trichrome stainings and their preliminary technique. Bull Int Assoc Med. 1929;12:75–90. [Google Scholar]
- McManus JF. Histological demonstration of mucin after periodic acid. Nature. 1946;158:202. doi: 10.1038/158202a0. [DOI] [PubMed] [Google Scholar]
- McManus JF. Histological and histochemical uses of periodic acid. Stain Technol. 1948;23:99–108. doi: 10.3109/10520294809106232. [DOI] [PubMed] [Google Scholar]
- Mowry RW. Alcian blue techniques for histochemical study of acid carbohydrates. J Histochem Cytochem. 1956;4:407–408. [Google Scholar]
- Mowry RW. Improved procedure for the staining of acidic polysaccharides by Muller’s colloidal (hydrous) ferric oxide and its combination with the feulgen and the periodic acid-schiff reactions. Lab Invest. 1958;7:566–576. [PubMed] [Google Scholar]
- Mowry RW. The special value of methods that color both acidic and vicinal hydroxyl groups in the hitochemical study of mucins, with revised directions for the colloidal iron stain, and the use of Alcian blue 8GX and their combination with the periodic acid-schiff reaction. Ann Ny Acad Sci. 1963;106:402–423. [Google Scholar]
- Muller G. Simplification of the reaction after Hale (1946) Acta Histochem. 1955;2:68–70. [PubMed] [Google Scholar]
- Otali D, Stockard CR, Oelschlager DK, Wan W, Manne U, Watts SA, Grizzle WE. Combined effects of formalin fixation and tissue processing on immunorecognition. Biotech & Histochem. 2009;84:223–247. doi: 10.3109/10520290903039094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes A, Jasani B, Couturier J, McKinley MJ, Morgan JM, Dodson AR, Navabi H, Miller KD, Balaton AJ. A formalin-fixed, paraffin-processed cell line standard for quality control of immunohistochemical assay of HER-2/neu expression in breast cancer. Am J Clin Pathol. 2002;117:81–89. doi: 10.1309/4NCM-QJ9W-QM0J-6QJE. [DOI] [PubMed] [Google Scholar]
- Riera J, Simpson JF, Tamayo R, Battifora H. Use of cultured cells as a control for quantitative immunocytochemical analysis of estrogen receptor in breast cancer. The Quicgel method. Am J Clin Pathol. 1999;111:329–335. doi: 10.1093/ajcp/111.3.329. [DOI] [PubMed] [Google Scholar]
- Ruby SG, McNally AC. Quality control of proliferation marker (MIB-1) in image analysis systems utilizing cell culture-based control materials. Am J Clin Pathol. 1996;106:634–639. doi: 10.1093/ajcp/106.5.634. [DOI] [PubMed] [Google Scholar]
- Southgate HW. Note on preparing mucicarmine. J Pathol Bacteriol. 1927;30:729. [Google Scholar]
- Taylor CR. An exaltation of experts: concerted efforts in the standardization of immunohistochemistry. Hum Pathol. 1994;25:2–11. doi: 10.1016/0046-8177(94)90164-3. [DOI] [PubMed] [Google Scholar]