

Abstract
Objective
Sleep disturbances are prevalent in human tauopathies yet despite the importance of sleep, little is known about its relationship with tau pathology. Here, we investigate this interaction by analyzing sleep and tau pathology throughout tauopathy disease progression in P301S human tau transgenic mice.
Methods
P301S and wild‐type mice were analyzed by electroencephalography (EEG)/electromyography at 3, 6, 9, and 11 months of age for sleep/wake time, EEG power, and homeostatic response. Cortical volume and tau pathology was also assessed by anti‐phospho‐tau AT8 staining.
Results
P301S tau mice had significantly decreased rapid eye movement (REM) sleep at 9 months of age and decreased REM and non‐REM (NREM) sleep as well as increased wakefulness at 11 months. Sleep loss was characterized by fewer wake, REM, and NREM bouts, increased wake bout duration, and decreased sleep bout duration. Decreased REM and NREM sleep was associated with increased brainstem tau pathology in the sublaterodorsal area and parafacial zone, respectively. P301S mice also showed increased EEG power at 6 and 9 months of age and decreased power at 11 months. Decreased EEG power was associated with decreased cortical volume. Despite sleep disturbances, P301S mice maintained homeostatic response to sleep deprivation.
Interpretation
Our results indicate that tau pathology is associated with sleep disturbances that worsen with age and these changes may be related to tau pathology in brainstem sleep regulating regions as well as neurodegeneration. Tau‐induced sleep changes could affect disease progression and be a marker for therapeutic efficacy in this and other tauopathy models.
Introduction
Abnormal phosphorylation and aggregation of the microtubule‐binding protein tau is a hallmark of a class of neurodegenerative diseases known as tauopathies. Tauopathies include diseases such as Alzheimer's disease (AD), which is characterized by aggregation of tau and amyloid‐β (Aβ), some forms of frontotemporal dementia (FTD) such as Pick's disease, and pure tauopathies such as progressive supranuclear palsy (PSP) and corticobasal degeneration. Sleep disturbances are prevalent in tauopathies with up to 76% of FTD and 67% of AD patients exhibiting clinical sleep disturbances of some kind and as many as 20% of PSP patients displaying rapid eye movement (REM) sleep specific disorders.1, 2 Although varied in presentation and pathology, tauopathies are associated with an overall decrease in sleep.3, 4, 5, 6, 7 Sleep disturbance is one of the leading causes of institutionalization in AD, however, the direct effect of tau pathology on sleep remains poorly understood.8
Tau abnormalities are among the earliest observable neurodegenerative pathologies associated with AD and in both AD and PSP tau pathology is prevalent in sleep centers of the brain.9, 10, 11, 12, 13 For example, AD‐type tau pathology is shown to begin accumulating in the brainstem early in life, with 70% of 20–30 year olds studied having abnormal tau pathology in the locus coeruleus (LC).9, 10 Early abnormal tau pathology progresses to other parts of the brainstem and hypothalamus, including many sleep regulating regions, prior to any cortical detection of tau or Aβ aggregation.9, 11 In AD, decreases in sleep efficiency precede the onset of dementia, while reduced sleep and increased sleep fragmentation are associated with an increased risk of developing AD.14, 15, 16 Tau pathology in sleep regions of the brain may play a role in these preclinical AD sleep disturbances. Furthermore, tau pathology develops in sleep‐regulating brain regions throughout the clinical course of AD and PSP, suggesting that tau pathology could also play a role in the severity of sleep disturbances in clinical tauopathy disease progression.6, 12, 13, 17, 18
Sleep disturbances have been identified in mice expressing both tau and amyloid pathology as well as mice expressing tau pathology alone in the forebrain.19, 20, 21, 22 Furthermore, tau knockout mice have decreased sleep suggesting that loss of endogenous tau function may also contribute to sleep disturbances.23 However, the direct effect of tau pathology throughout the brain on sleep has not been studied. To determine if tau pathology alone is sufficient to alter sleep we characterized sleep over time in a mouse model of FTD with Parkinsonism linked to chromosome 17 containing a human tau transgene with the P301S mutation. We further investigate tau pathology in brainstem sleep regions of P301S Tau transgenic (tg) mice over time to determine if pathology in these areas may contribute to changes in sleep.
Methods
Animals
Male P301S mice and wild‐type (WT) littermates were studied at 3–4, 6–7, 9–10, and 11–12 months of age. P301S mice (PS19) contain a human tau transgene with a P301S mutation on the B6C3 background and were obtained from Jackson Labs (Bar Harbor, ME USA).24 Mice were housed in a 12 h light/dark cycle with food ad libitum. All animal procedures and studies were approved by the Animal Studies Committee at Washington University School of Medicine.
EEG sleep/wake monitoring
Electroencephalography (EEG)/electromyography (EMG) sleep/wake monitoring was performed as previously described.22, 25 EEG was recorded by stainless steel bone screws placed over the right frontal bone (Bregma: +1.0 mm, 1.0 mm lateral to midline) and right parietal bone (Bregma: −3.0 mm, 2.5 mm lateral to midline) and EMG recorded from two electrode wires placed on the right and left neck musculature. All signals were grounded to a bone screw electrode placed over the cerebellum midline. Following electrode implantation, mice were housed in 12 h light/dark conditions for 10 days. Mice were transferred to EEG/EMG cages, attached to recording cables, and habituated for 3 days after which EEG/EMG was recorded for 2 days. For sleep deprivation studies, a third day of recording was conducted in which mice were manually kept awake by gentle touching with a paintbrush for 6 h following lights on and further recorded for five more hours to test homeostatic response. During recording, mice were disturbed only during the 1 h prior to lights off, this time was omitted from all analysis. EEG and EMG signals were acquired by a P511K A.C. Preamplifier (Grass‐Telefactor Instruments, Warwick, RI USA), digitized with a DigiData 1440A Data Acquisition System (Molecular Devices, Sunnyvale, CA USA), and recorded digitally using pClamp 10.2 (Molecular Devices).
EEG/EMG analysis and statistics
EEG/EMG recordings were scored manually for wake, non‐rapid eye movement (NREM) sleep, and REM sleep in 10 second epochs using SleepSign (Kissei Comtec Co., Ltd, Matsumoto Japan) and all statistics performed in Graphpad Prism 5 (GraphPad Software Inc., La Jolla, CA USA) unless otherwise stated. Sleep time was averaged over 2 days (23 h/day) and significance at each age determined by Student's t‐test. Sleep/wake bout number and duration analysis as well as power analysis was completed on day 2 of recording only. Significance in bout number and duration at each time point was determined by Student's t‐test. FFT significance was determined by two‐way analysis of variance (ANOVA) and followed with Bonferroni post hoc when a significant interaction was observed. In power analysis, two P301S outliers were detected using Grubb's test (GraphPad QuickCalcs) between all P301S animals at that time point. Animals excluded from analysis include one 6 month animal for NREM power only and one 11 month animal for REM power only. For sleep deprivation studies, sleep time and power for 5 h following sleep deprivation was normalized to the previous day and analyzed as stated above. All data are expressed as mean ± standard error of the mean (SEM).
Immunohistochemistry
Following the recording session mice were anesthetized with pentobarbatol (200 mg/kg, i.p.) and perfused with cold Dulbecco's phosphate‐buffered saline (PBS) with heparin (3 U/mL). The left brain hemisphere was fixed in 4% paraformaldehyde and cryoprotected in 30% sucrose in PBS. Brain hemispheres were sliced coronally in 30 μm sections from the most rostral region of the cortex caudally through the brain stem using a freezing slice microtome. Sections were stored at −20°C in cryoprotectant (0.2 mol/L PBS, 30% sucrose, 30% ethylene glycol) until use.
Immunohistochemistry was performed in five batches that each contained a control sample for normalization. Every sixth hemisphere section was selected from the rostral cortex through the brainstem and placed in netwells (6 slices/well). Slices were washed three times for 5 min in Tris‐buffered saline (TBS), incubated in 0.3% hydrogen peroxide in TBS for 10 min, and washed three times. Samples were then blocked for 1 h in 3% milk in TBS with 0.25% triton (vol/vol) (TBS‐X) and incubated overnight at 4°C with biotinylated AT8 antibody (1:500, Thermo‐Fisher, Rockford, IL USA) in 1% milk in TBS‐X. AT8 recognizes tau phosphorylated at Ser202 and Thr205.26 Following antibody incubation, slices were washed, bound to avidin using Vectastain ABC elite (Vector Laboratories, Burlingame, CA USA), washed, and developed in DAB (Sigma, St. Louis, MO USA). Slices were mounted on gelatin coated slides and rehydrated in increasing concentrations of ethanol and xylene. All samples were imaged using a Nanozoomer slide scanner (Hamamatsu Photonics, Hamamatsu Japan).
Cortex volume and tau pathology analysis
AT8‐stained slices were used to determine cortex volume and tau pathology. Cortex volume was determined using NDP viewer (Hamamatsu Photonics). Every sixth slice was analyzed from the crossing of the corpus callosum (Bregma: +1.1 mm) to the dorsal end of the hippocampus (Bregma: −3.9 mm) and volume was determined stereologically. Significance was determined by one‐way ANOVA with Tukey's post hoc. To analyze tau pathology, slices were converted to gray scale and analyzed using ImageJ (National Institutes of Health, Bethesda, MD, USA). For each region a threshold was determined to accurately depict positive staining and the percent area covered determined by ImageJ. One slice/animal was used to determine pathology in the sublaterodorsal area (SLD) (approximately Bregma: −5.3 mm) and parafacial zone (PZ) (approximately Bregma: −5.6 mm) and three slices/animal separated by 180 μm used to determine entorhinal cortex and piriform cortex area pathology (approximately Bregma: −1.75, −1.93, −2.11). All % area covered values were normalized to appropriate staining control to eliminate batch variation. Significant differences between age points were determined by one‐way ANOVA with Tukey's post hoc and significant correlation identified by Pearson r.
Results
P301S Tau tg mice have decreased sleep, increased wakefulness, and disrupted sleep architecture that worsens with age
P301S Tau tg mice display a progressive development of tau phosphorylation and aggregation with onset of tau pathology around 5 months of age and brain atrophy beginning around 8 months.24 To determine if tau pathology throughout the brain is sufficient to alter sleep, P301S and WT mice at 3 months (prepathology), 6 months (early pathology), 9 months (late pathology, early atrophy), and 11 months (late pathology, late atrophy) of age were analyzed by EEG/EMG recording and the amount of time spent in wake, REM sleep, and NREM sleep quantified. No significant changes in sleep or wake time were observed between genotypes at 3 or 6 months of age (Fig. 1). At 9 months of age, REM sleep was significantly decreased by 28% in P301S compared to WT mice but no changes were observed in wake or NREM sleep (Fig. 1). Loss of REM sleep worsened at 11 months of age with P301S REM sleep decreased 61% compared to WT mice (Fig. 1B). In addition to changes in REM sleep, 11 month P301S mice showed a significant decrease in NREM sleep and a significant increase in wake (Fig. 1A and C). Despite loss of sleep time, P301S mice maintained circadian sleep rhythms with increased sleep during light periods compared to dark and increased wake during dark periods compared to light, suggesting that these changes in sleep time are not due to lack of circadian regulation (data not shown).
Figure 1.
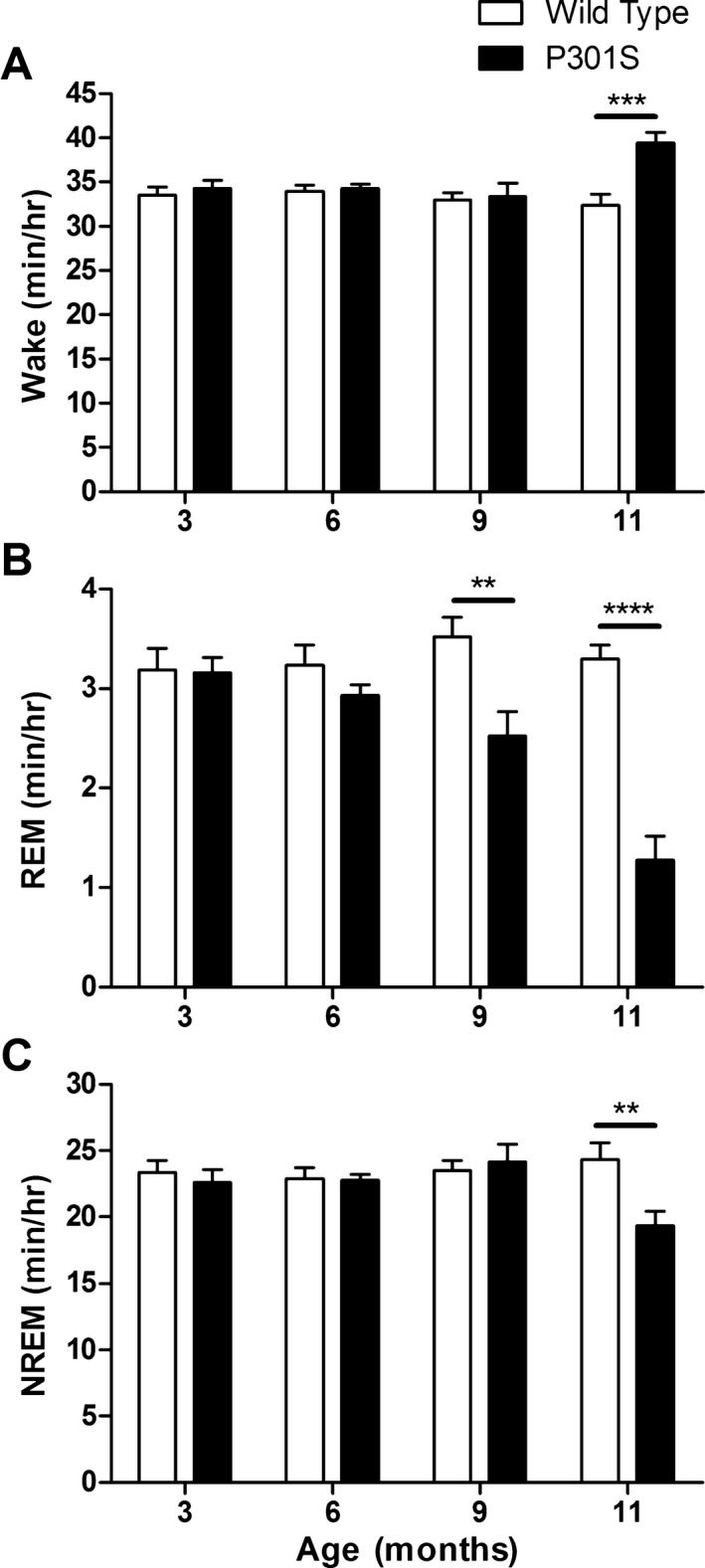
P301S Tau tg mice have decreased sleep that worsens with age. Analysis of the average minutes/hour spent in wake (A), REM sleep (B), and NREM sleep (C) for P301S and WT mice at 3 (n = 6), 6 (n = 9), 9 (n = 11 P301S, 10 WT), and 11 months of age (n = 14). P301S mice had a significant loss of REM sleep at 9 months of age that worsened at 11 months (B). Eleven‐month‐old P301S mice also spent significantly less time in NREM sleep and increased time in wake (A and C). **P < 0.01, ***P < 0.001, ****P < 0.0001. Student's t‐test at each age. All values represent mean ± SEM. REM, rapid eye movement; NREM, non‐REM; WT, wild‐type.
The observed changes in sleep time were associated with altered wake, REM, and NREM bout number as well as bout duration. P301S mice had significantly fewer wake bouts and significantly longer wake bout duration at both 9 and 11 months of age (Fig. 2A and D). These results demonstrate that P301S mice do not transition out of wake into sleep states as often as WT mice, suggesting a breakdown in sleep‐generating/wake‐inhibiting pathways in P301S mice. Furthermore, decreased REM sleep in P301S mice (Fig. 1B) was associated with fewer REM bouts at 9 and 11 months of age and a shorter REM bout duration at 11 months (Fig. 2B and E). NREM bouts were also affected with significantly fewer entries into NREM sleep at 9 and 11 months of age, which was countered by increased NREM bout duration at 9 months but not at 11 months (Fig. 2C and F). Taken together, these changes demonstrate that sleep architecture is significantly disrupted in P301S mice beginning by 9 months of age and sleep disturbances worsen with disease progression.
Figure 2.
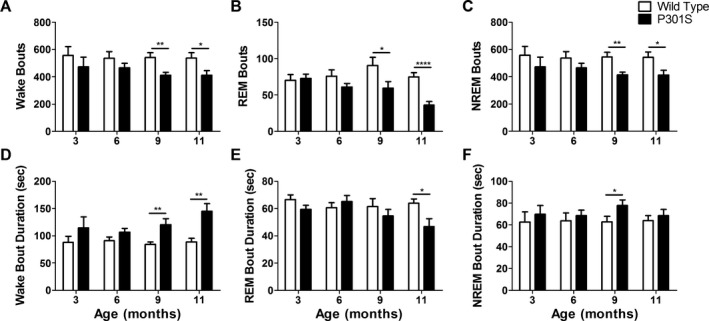
Wake, REM, and NREM bouts are altered in P301S mice. The number of wake (A), REM (B), and NREM (C) bouts was significantly decreased in P301S mice compared to WT mice at 9 (n = 11 P301S, 10 WT) and 11 months of age (n = 14) but not at 3 (n = 6) and 6 months (n = 9). (D) Wake bout duration was significantly increased at 9 and 11 months of age. (E) P301S REM bout duration was unchanged at 3, 6, and 9 months of age but significantly decreased at 11 months. (F) P301S NREM bout duration was significantly increased at 9 months of age but unchanged at 3, 6, and 11 months. *P < 0.05, **P < 0.01, ****P < 0.0001. Student's t‐test at each age. All values represent mean ± SEM. REM, rapid eye movement; NREM, non‐REM; WT, wild‐type.
Decreased REM and NREM sleep in P301S Tau tg mice is associated with tau pathology in brainstem sleep regulating regions
In tauopathies, sleep regulating regions of the brain accumulate tau pathology and in AD specifically, tau pathology develops in many regions prior to cortical tau or amyloid aggregation.9, 11, 12, 13 To test if tau pathology in sleep centers of the brain may contribute to sleep disturbances in P301S Tau tg mice we analyzed tau pathology, as identified by AT8 staining, in 3‐, 6‐, 9‐, and 11‐month‐old P301S mice. Many brain regions contribute to the maintenance of sleep/wake architecture.27 Due to the observed decrease in REM and NREM sleep as well as decreased sleep bouts in P301S mice, we analyzed tau pathology in two brain regions known to play a substantial role in generating REM and NREM sleep. The SLD is a REM‐generating region located in the pons/brainstem (Fig. 3A) and loss of glutamatergic neurons in this region results in decreased REM sleep.28, 29 P301S mice displayed a significant increase in tau pathology in the SLD at 9 and 11 months of age, the ages at which REM sleep was decreased in these mice (Fig. 3B, one‐way ANOVA, P < 0.0001). Furthermore, SLD AT8 staining was negatively correlated with total REM sleep time (Fig. 3C, Pearson r, P < 0.0001). The GABAergic PZ, located in the brainstem (Fig. 3D), is a slow wave sleep‐ or NREM sleep‐promoting region.30 P301S mice showed a significant increase in PZ tau pathology at 11 months of age, the same age at which NREM sleep time was significantly decreased (Fig. 3E, one‐way ANOVA, P < 0.0001). PZ AT8 pathology was also negatively correlated with total NREM sleep time (Fig. 3F, Pearson r, P < 0.0001). These results suggest that brainstem tau pathology may play a role in the sleep loss observed in P301S mice. Due to the global neuronal expression of the P301S Tau transgene and progressive pathology development in P301S mice, correlation between sleep time and tau pathology was not limited to these two brainstem regions. However, AT8 pathology in the entorhinal cortex, a forebrain region heavily affected with tau pathology, as well as the piriform cortex did not correlate with REM or NREM sleep times in P301S mice (data not shown, Pearson r, P > 0.05). The entorhinal cortex is well studied in AD and is an early location of tau pathology progression in P301S mice as well as human forebrain AD tau pathology.9, 24 The lack of association between sleep time and entorhinal or piriform cortex tau pathology further strengthens the possible role of brainstem pathology in P301S sleep disturbances.
Figure 3.
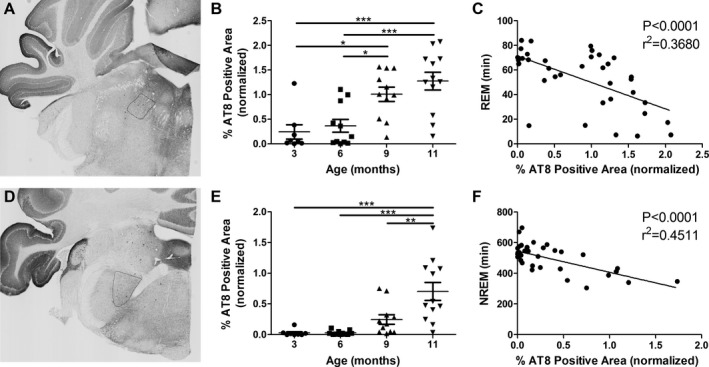
Decreased REM and NREM sleep in P301S Tau tg mice is associated with tau pathology development in sleep regulating regions of the brain. (A) Representative image of the SLD, a REM sleep‐generating region of the brain. (B) AT8 staining in the SLD was significantly increased at 9 (n = 11) and 11 (n = 12) months of age compared to 3 (n = 8) and 6 (n = 11) months. (C) Time in REM sleep was negatively correlated with AT8 SLD staining (n = 38). (D) Representative image of the PZ, a slow wave/NREM sleep‐promoting region. (E) AT8 staining in the PZ was significantly increased at 11 month of age and (F) NREM sleep time negatively correlated with AT8 PZ staining. *P < 0.05, **P < 0.01, ***P < 0.001. One‐way ANOVA with Tukey's post hoc, all values represent mean ± SEM (B and E). Pearson r (C and F). All AT8 staining values were normalized to a batch control. REM, rapid eye movement; NREM, non‐REM; SLD, sublaterodorsal area; PZ, parafacial zone; ANOVA, analysis of variance.
P301S Tau tg mice exhibit changes in EEG delta and theta power
EEG power from 1 to 20 Hz was determined for wake, REM, and NREM vigilance states in P301S and WT mice. No differences in EEG power were observed at 3 months of age between P301S and WT mice (Fig. 4A–C). At 6 months of age, P301S NREM EEG power was significantly increased in the delta frequencies (1–4 Hz) compared to WT but wake and REM sleep were not significantly altered (Fig. 4D–F, two‐way ANOVA, NREM interaction: P = 0.019, genotype: P < 0.0001). P301S NREM EEG delta power remained significantly increased at 9 months of age and REM sleep EEG power was also significantly increased in the theta frequencies (4–8 Hz) (Fig. 4H and I, two‐way ANOVA, REM interaction: P = 0.0179, NREM interaction: P = 0.0005, REM and NREM genotype: P < 0.0001). Wake EEG power was not affected at 9 months of age (Fig. 4G). However, at 11 months of age, P301S mice showed a marked and significant decrease in delta EEG power during wake and NREM sleep, and theta EEG power during REM sleep compared to WT mice (Fig. 4J–L, two‐way ANOVA, Wake interaction: P = 0.0031, REM interaction: P = 0.001, NREM interaction: P = 0.0163, all genotype: P < 0.0001). EEG power in WT mice did not change with age (Fig. 5A–C) and is therefore not responsible for the observed changes in P301S EEG power. However, in P301S Tau tg mice, 11‐month‐old animals had significantly decreased EEG power compared to all other age points in the delta and theta frequencies of wake and NREM sleep and in theta frequencies only for REM sleep (Fig. 5D–F, two‐way ANOVA, all interaction and age: P < 0.0001). These measures were not significantly different when comparing 3, 6, and 9 month P301S mice.
Figure 4.
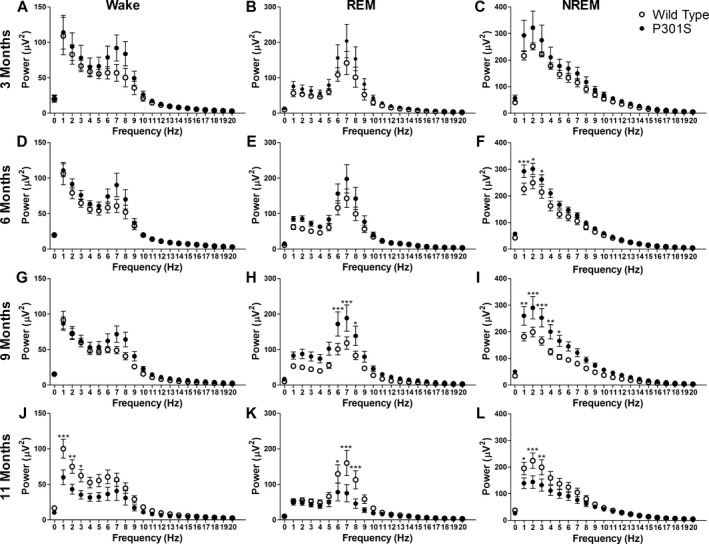
EEG power is altered in P301S Tau tg mice. Analysis of EEG power during wake, REM sleep, and NREM sleep at 3 (n = 6 P301S and WT), 6 (n = 8 P301S [D and E], n = 7 P301S [F], n = 8 WT), 9 (n = 11 P301S, n = 10 WT), and 11 months of age (n = 12 P301S [J and L], n = 11 P301S [K], n = 12 WT) in P301S and WT mice. EEG power was not significantly altered at 3 months of age in wake (A), REM (B) or NREM sleep (C). EEG power was unchanged at 6 months of age during wake (D) and REM sleep (E), but power in P301S mice was significantly increased during NREM sleep in the delta frequencies (1–4 Hz) (C). Nine‐month‐old P301S mice exhibited significantly increased EEG power in the theta frequencies (4–8 Hz) during REM sleep (H) and delta frequencies in NREM sleep (I) but power was not significantly affected during wake (G). At 11 months of age, P301S delta EEG power was significantly decreased in wake (J) and NREM sleep (L) and theta EEG power significantly decreased during REM sleep (K). *P < 0.05, **P < 0.01, ***P < 0.001. Two‐way ANOVA with Bonferroni post hoc. All values represent mean ± SEM. EEG, electroencephalography; REM, rapid eye movement; NREM, non‐REM; WT, wild‐type; ANOVA, analysis of variance.
Figure 5.
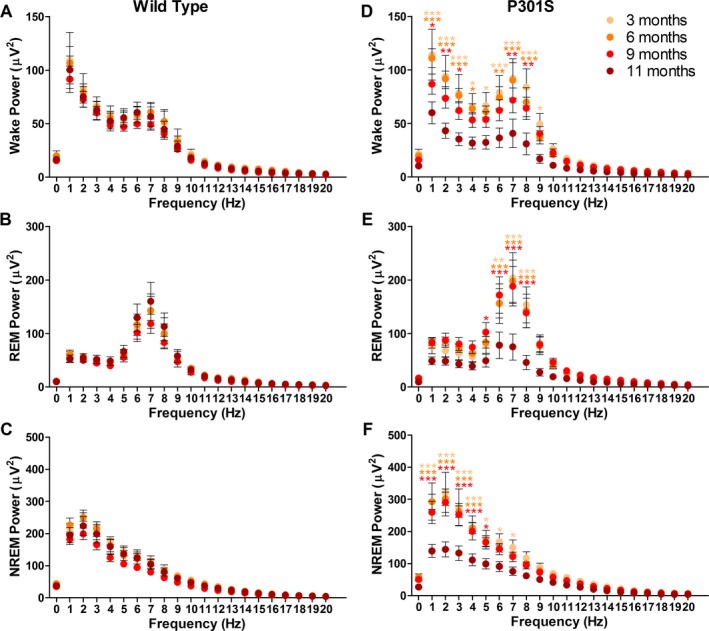
EEG power does not change with age in WT mice but is significantly decreased in 11‐month‐old P301S mice. WT mice at 3, 6, 9, and 11 months of age showed no changes in wake (A), REM (B), or NREM (C) EEG power with age. However, P301S mice had significantly decreased wake (D), REM (E), and NREM (F) EEG power at 11 months of age compared to 3, 6, and 9 month P301S mice. *P < 0.05, **P < 0.01, ***P < 0.001. Two‐way ANOVA. All significance compared to 11 month P301S mice by Bonferroni post hoc (D‐F). All values represent mean ± SEM. EEG, electroencephalography; REM, rapid eye movement; NREM, non‐REM; ANOVA, analysis of variance; WT, wild‐type.
Cortex volume is associated with EEG power in P301S Tau tg mice
P301S Tau tg mice have neurodegeneration and develop brain atrophy beginning around 8 months of age that results in loss of cortex volume by 12 months.24 To test if the observed decrease in P301S EEG power at 11 months of age is associated with atrophy we analyzed hemispheric cortical volume at 3, 6, 9, and 11 months of age. In this study, 11‐month‐old P301S mice had significantly decreased cortical hemispheric volume compared to 3‐ and 6‐month‐old P301S mice, the same age at which decreased EEG power was observed in these mice (Fig. 6A, one‐way ANOVA, P < 0.0012). Furthermore, cortical hemispheric volume was significantly correlated with NREM EEG delta power in P301S mice of all ages combined, demonstrating a possible interaction between cortical volume and EEG power (Fig. 6B, Pearson r, P = 0.0002,). NREM delta power was analyzed due to the profound and early changes observed at this frequency range in P301S mice (Fig. 4). When analyzed at independent age points, P301S cortical hemispheric volume was significantly correlated with NREM delta power at 11 month of age, but this correlation was not significant at 9 months (Fig. 6C and D, Pearson r, 11 months: P = 0.0005, 9 months: P = 0.1345). The observed correlation at 11 months of age when EEG power is significantly decreased but not at 9 months when EEG power is elevated suggests that neurodegeneration‐induced brain atrophy may play a role in the loss of EEG power observed at 11 months of age but may not explain the increased EEG power observed at earlier time points.
Figure 6.
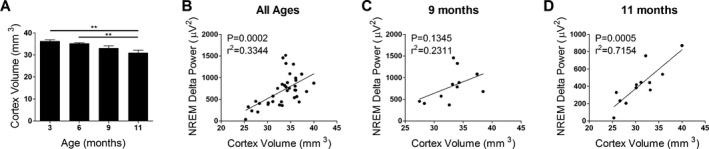
Cortical hemispheric volume decreases with age and correlates with NREM delta power in P301S Tau tg mice. (A) Analysis of cortical hemispheric volume in 3 (n = 8), 6 (n = 11), 9 (n = 11), and 11 (n = 12) month‐old P301S mice showed a significant decrease at 11 months. Cortical hemispheric volume was significantly correlated with NREM delta power when all P301S mice ages were combined (B, n = 36) and 11 months of age only (D, n = 12), but not at 9 months of age (C, n = 11), suggesting that cortical volume loss may play a role in the decreased EEG power observed at 11 months of age. **P < 0.01. One‐way ANOVA with Tukey's post hoc, all values represent mean ± SEM (A). Pearson r (B–D). NREM, non‐rapid eye movement; EEG, electroencephalography; ANOVA, analysis of variance.
Eleven‐month‐old P301S Tau tg mice maintain homeostatic response to sleep deprivation
Sleep is homeostatically regulated in that prolonged wakefulness leads to a compensatory increase in sleep as well as increased NREM sleep slow wave activity.27 Since P301S mice have decreased sleep time and decreased EEG power at 11 months of age, we further characterized homeostatic sleep response in these mice as another way to assess their sleep/wake cycle. Sleep and wake were analyzed in P301S and WT mice following 6 h of manual sleep deprivation. When sleep and wake time was normalized to the previous baseline day of recording, 11‐month‐old P301S mice had less of a decrease in wake than WT mice, but changes in REM and NREM sleep were not significantly different (Fig. 7A–C). Eleven‐month P301S mice maintained an elevated level of sleep similar to controls following sleep deprivation. Furthermore, NREM EEG power normalized to the previous day was increased in a similar manner in the delta frequencies of both 11‐month‐old P301S and WT mice (Fig. 7D). These results demonstrate that even at advanced stages of disease when sleep time and EEG power is significantly decreased, P301S mice maintain homeostatic sleep responses.
Figure 7.
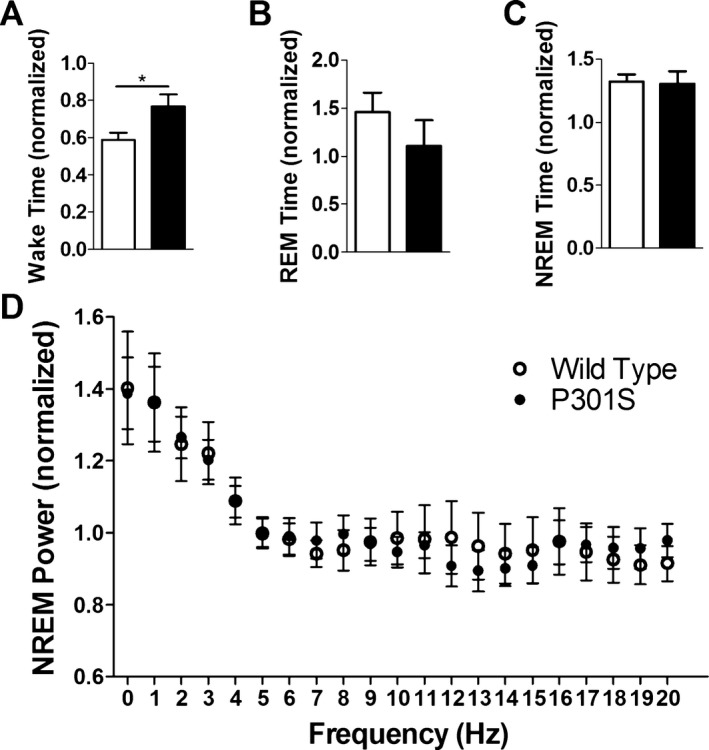
P301S Tau tg mice maintain homeostatic response to sleep deprivation. Eleven‐month‐old P301S and WT mice (n = 6) were sleep deprived for 6 h and EEG analyzed for 5 h following sleep deprivation. All data were normalized to the time matched previous day of recording. Time spent in wake (A), REM sleep (B), and NREM sleep (C) following sleep deprivation. (D) NREM EEG power was similarly increased in the delta frequencies of both P301S and WT mice. *P < 0.05. Student's t‐test (A–C). Two‐way ANOVA (D). All values represent mean ± SEM. EEG, electroencephalography; REM, rapid eye movement; NREM, non‐REM; ANOVA, analysis of variance; WT, wild‐type.
Discussion
Sleep is an important biological function that is commonly altered in neurodegenerative disease.18, 31 Despite the importance of sleep and the prevalence of sleep disturbances observed across tauopathies, little is known about the effects of disease associated tau aggregation throughout the brain on sleep. Here, we demonstrate that a mouse model expressing human P301S Tau throughout the brain and tau pathology in both forebrain and brainstem regions, exhibits changes in sleep with disease progression. We have shown that P301S mice have a progressive decrease in sleep and increase in wakefulness, and that loss of sleep is associated with tau pathology in brainstem sleep regulating regions. Particularly interesting is the early decrease in REM sleep. Furthermore, P301S mice have changes in EEG power over time that are associated with decreased cortical volume. Despite changes in sleep and EEG power, P301S mice maintain homeostatic response to sleep deprivation.
Changes in sleep time and association with brainstem sleep regions
FTD, PSP, and AD patients all have decreased sleep and we have demonstrated that this phenotype is recapitulated in the P301S tauopathy mouse model.3, 4, 5, 6, 7 The progression of sleep disturbances over time has not been reported in tauopathy mouse models and we show here that sleep deficits worsen with age and disease progression. Sleep analysis of a different FTD model expressing tau specifically in the forebrain (PLB2Tau) has also reported loss of NREM sleep and increases in wake, but unlike P301S mice shown here, changes in REM sleep were not observed in PLB2Tau mice.20 Sleep changes in PLB2Tau mice support our findings that tau pathology is sufficient to induce sleep changes and further suggests that the REM sleep disruption we observe in P301S mice may be due to brainstem pathology present in P301S but not PLB2Tau models.20 This is supported by our observation that AT8‐positive tau pathology in the SLD is negatively correlated with REM sleep time. Tau knock‐out mice also only show changes in wake and NREM sleep, suggesting a role for tau pathology, and not tau loss of function, in REM sleep disruption.23 In addition to REM sleep, areas in the brainstem also regulate NREM sleep and wake. Despite the presence of NREM decreases in PLB2Tau forebrain expressing mice, our results highlight that the effect of tau pathology on brainstem regions such as the PZ, which negatively correlated with NREM sleep time in P301S mice, should not be overlooked.20
The brainstem and other sleep‐regulating regions of the brain are affected by tau pathology in taupathies.9, 10, 11, 12, 32 In AD, abnormal tau pathology can be seen in brainstem sleep/wake regions such the LC, dorsal raphe, and parabrachial nucleus as well as hypothalamic sleep regions prior to the onset of cortical tau or amyloid.9, 10, 11 Other sleep regions, such as the pedunculopontine tegmental nucleus (PPT), develop tau pathology in clinical stages of disease.17 In AD, neuronal loss and tau neurofibrillary tangles occur in the PPT, but not amyloid pathology.13, 17 In PSP, REM sleep is highly affected and loss of REM sleep is suggested to result from tau pathology‐induced degeneration of the PPT, demonstrating the importance of brainstem regions in sleep and their vulnerability to tau pathology.6, 13 Early tau abnormalities in the brainstem are not recapitulated in mouse models and despite pathological analysis of some brainstem regions in human clinical AD, many specific sleep regions, including the SLD and PZ analyzed here, have yet to be studied. However, the association of pathology onset in sleep‐generating brainstem regions with the onset of sleep loss as well as significant correlation between these factors is strengthened by the proximity of these brain regions to others known to be affected by AD tau pathology and is suggestive of a role for brainstem tau pathology in P301S sleep disturbances.
Here, we analyzed the SLD and PZ based on their ability to modulate REM and NREM sleep and our results demonstrate, for the first time, the likely importance of brainstem tau pathology in tau‐induced sleep deficits. Eliminating glutamatergic signaling in the SLD significantly decreases REM sleep by ~40%, similar to what was observed here in P301S mice.28 However, SLD Vglut knockdown also increases sleep bout frequency, which we did not observe in the P301S Tau tg model.28 These similarities and differences demonstrate that tau pathology may affect many regions of the brain, a combination of which is likely responsible for sleep/wake changes observed in P301S mice. In P301S mice we also observed fewer sleep bouts as well as fewer and longer wake bouts. The decreased transition to sleep from wake suggests breakdown of sleep‐promoting/wake‐inhibiting pathways. The PZ promotes slow wave sleep by inhibiting the wake‐promoting parabrachial nucleus and its basal forebrain cortical circuit.30 Thus, loss of PZ function is a possible mechanism for tau pathology‐induced decrease in sleep bouts and this is supported by the negative correlation between NREM sleep time and PZ tau pathology. It is likely that other sleep regions of the brain, including brainstem and thalamic regions, are also affected by tau pathology and contribute to the observed sleep disturbances. More work is needed to determine if tau pathology in the SLD, PZ, or other sleep regions alone is sufficient to induce changes in sleep and if tau pathology‐induced sleep changes can affect disease progression.
EEG power and cortical volume
Here, we demonstrated that P301S mice have changes in EEG power, with elevated EEG power compared to WT at 6 and 9 months of age and significantly decreased power at 11 months. These changes are not due to changes in WT EEG power but to a significant decrease in P301S EEG power at 11 months of age that corresponds with a significant decrease in cortical volume. Changes in EEG power over time have not previously been reported in tauopathy mice. Increased EEG power during disease progression could represent a compensatory response to pathological tau aggregation. Although more work is needed to understand the mechanism of increased power in P301S mice, the observed correlation between cortex volume and EEG power demonstrates that neurodegenerative cortical atrophy may be responsible for the severe decrease in EEG power observed in 11‐month‐old P301S mice. Furthermore, we have shown that even at severe stages of disease when sleep time and EEG power are decreased, P301S mice maintain a homeostatic response to sleep deprivation. We surprisingly observed similar increases in REM and NREM sleep time as well as increased NREM slow wave delta power in aged P301S and WT mice. The mechanisms and exact sleep factors resulting in homeostatic regulation of sleep are not fully elucidated. Adenosine build up and signaling in the basal forebrain and other regions is known to play a role, but it is likely not the only factor.27 Despite this, alterations in sleep and EEG power are well documented.27 The maintenance of homeostatic response of NREM EEG power in P301S aged mice may indicate that despite regional brain atrophy and decreased EEG power, the existing neurons and neural networks maintain the ability to respond and increase activity to homeostatic sleep‐drive cues.
Conclusions and Implications
We have demonstrated for the first time that tau pathology throughout the brain, including both the forebrain and brainstem, can progressively cause sleep disruption and changes in EEG power over time. We have further shown that the brainstem may play an important role in the development of sleep disturbances. Previously, Aβ pathology has been shown to not only induce sleep disruption but pathology is also accelerated by sleep deprivation and decreased by sleep improvement.22, 25, 33 Therefore, tau‐induced decreases in REM and NREM sleep may directly affect amyloid AD pathology, interact with Aβ‐induced sleep problems, and could play a role in worsening disease. In the brain interstitial fluid, levels of tau, like Aβ, are increased with elevated neuronal activity, suggesting that increased wakefulness may elevate tau secretion and disease spreading in tauopathies.25, 34, 35 Whether changes in sleep affect tau pathology directly, as has been seen with Aβ, remains to be studied.25, 33 However, even if changes in sleep do not directly affect tau pathology, decreasing sleep could play a role in accelerating other AD disease mechanisms such as amyloid pathology and secondarily affect tau. Furthermore, quantitative assessment of sleep and EEG power in the P301S Tau tg model could be a useful method to functionally assess the effects of anti‐tau and other neuroprotective therapies, in addition to evaluation of cognitive and motor outcomes. P301S mice and the sleep disturbances observed could therefore serve as a valuable way to assess for disease progression and as a functional tool in assessing disease outcome.
Author Contributions
J. K. H. and D. M. H. conceived, designed, and analyzed the studies as well as wrote and edited the manuscript. All authors contributed to designing and performing experiments.
Conflict of Interest
D. M. H. reports grants from NIH, Tau Consortium, The JPB Foundation, Cure Alzheimer's Fund, Eli Lilly, Denali, AbbVie, and C2N Diagnostics during the conduct of the study. D. M. H. is on the scientific advisory board of C2N Diagnostics, Denali, and Neurophage and consults for Genentech, AbbVie, and Eli Lilly. D. M. H. is a co‐inventor on a patent licensed by Washington University to C2N Diagnostics for anti‐tau antibodies. C2N Diagnostics has licensed these antibodies to AbbVie. D. M. H. is a cofounder of C2N Diagnostics and has equity in the company.
Acknowledgments
The authors thank Clifford Saper (Harvard University) for guidance and expertise regarding sleep regulation and neuroanatomy. This work was funded by grants from the National Institute of Health‐NINDS F32NS089381 (J. K. H.) and P01NS074969 (D. M. H.) as well as the Tau Consortium (D. M. H.), Cure Alzheimer's Fund (D. M. H.), and the JPB Foundation (D. M. H.).
References
- 1. Guarnieri B, Adorni F, Musicco M, et al. Prevalence of sleep disturbances in mild cognitive impairment and dementing disorders: a multicenter Italian clinical cross‐sectional study on 431 patients. Dement Geriatr Cogn Disord 2012;33:50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arnulf I, Merino‐Andreu M, Bloch F, et al. REM sleep behavior disorder and REM sleep without atonia in patients with progressive supranuclear palsy. Sleep 2005;28:349–354. [PubMed] [Google Scholar]
- 3. Anderson KN, Hatfield C, Kipps C, et al. Disrupted sleep and circadian patterns in frontotemporal dementia. Eur J Neurol 2009;16:317–323. [DOI] [PubMed] [Google Scholar]
- 4. Bonakis A, Economou NT, Paparrigopoulos T, et al. Sleep in frontotemporal dementia is equally or possibly more disrupted, and at an earlier stage, when compared to sleep in Alzheimer's disease. J Alzheimers Dis 2014;38:85–91. [DOI] [PubMed] [Google Scholar]
- 5. Bonanni E, Maestri M, Tognoni G, et al. Daytime sleepiness in mild and moderate Alzheimer's disease and its relationship with cognitive impairment. J Sleep Res 2005;14:311–317. [DOI] [PubMed] [Google Scholar]
- 6. Montplaisir J, Petit D, Décary A, et al. Sleep and quantitative EEG in patients with progressive supranuclear palsy. Neurology 1997;49:999–1003. [DOI] [PubMed] [Google Scholar]
- 7. Vitiello MV, Prinz PN, Williams DE, et al. Sleep disturbances in patients with mild‐stage Alzheimer's disease. J Gerontol 1990;45:M131–M138. [DOI] [PubMed] [Google Scholar]
- 8. Bianchetti A, Scuratti A, Zanetti O, et al. Predictors of mortality and institutionalization in Alzheimer disease patients 1 year after discharge from an Alzheimer dementia unit. Dementia 1995;6:108–112. [DOI] [PubMed] [Google Scholar]
- 9. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 2011;70:960–969. [DOI] [PubMed] [Google Scholar]
- 10. Braak H, Del Tredici K. The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol 2011;121:171–181. [DOI] [PubMed] [Google Scholar]
- 11. Stratmann K, Heinsen H, Korf HW, et al. Precortical phase of Alzheimer's disease (AD)‐related tau cytoskeletal pathology. Brain Pathol 2016;26:371–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dickson DW. Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol 1999;246(Suppl 2):II6–II15. [DOI] [PubMed] [Google Scholar]
- 13. Jellinger K. The pedunculopontine nucleus in Parkinson's disease, progressive supranuclear palsy and Alzheimer's disease. J Neurol Neurosurg Psychiatry 1988;51:540–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ju YE, McLeland JS, Toedebusch CD, et al. Sleep quality and preclinical Alzheimer disease. JAMA Neurol 2013;70:587–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hahn EA, Wang HX, Andel R, Fratiglioni L. A change in sleep pattern may predict Alzheimer disease. Am J Geriatr Psychiatry 2013;22:1262–1271. [DOI] [PubMed] [Google Scholar]
- 16. Lim AS, Kowgier M, Yu L, et al. Sleep fragmentation and the risk of incident Alzheimer's disease and cognitive decline in older persons. Sleep 2013;36:1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Parvizi J, Van Hoesen GW, Damasio A. The selective vulnerability of brainstem nuclei to Alzheimer's disease. Ann Neurol 2001;49:53–66. [DOI] [PubMed] [Google Scholar]
- 18. Stern AL, Naidoo N. Wake‐active neurons across aging and neurodegeneration: a potential role for sleep disturbances in promoting disease. Springerplus 2015;4:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jyoti A, Plano A, Riedel G, Platt B. Progressive age‐related changes in sleep and EEG profiles in the PLB1 Triple mouse model of Alzheimer's disease. Neurobiol Aging 2015;36:2768–2784. [DOI] [PubMed] [Google Scholar]
- 20. Koss DJ, Robinson L, Drever BD, et al. Mutant Tau knock‐in mice display frontotemporal dementia relevant behaviour and histopathology. Neurobiol Dis 2016;91:105–123. [DOI] [PubMed] [Google Scholar]
- 21. Platt B, Drever B, Koss D, et al. Abnormal cognition, sleep, EEG and brain metabolism in a novel knock‐in Alzheimer mouse, PLB1. PLoS One 2011;6:e27068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roh JH, Huang Y, Bero AW, et al. Disruption of the sleep‐wake cycle and diurnal fluctuation of β‐amyloid in mice with Alzheimer's disease pathology. Sci Transl Med 2012;4:150ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cantero JL, Hita‐Yañez E, Moreno‐Lopez B, et al. Tau protein role in sleep‐wake cycle. J Alzheimers Dis 2010;21:411–421. [DOI] [PubMed] [Google Scholar]
- 24. Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007;53:337–351. [DOI] [PubMed] [Google Scholar]
- 25. Kang JE, Lim MM, Bateman RJ, et al. Amyloid‐beta dynamics are regulated by orexin and the sleep‐wake cycle. Science 2009;326:1005–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett 1995;189:167–169. [DOI] [PubMed] [Google Scholar]
- 27. Saper CB, Fuller PM, Pedersen NP, et al. Sleep state switching. Neuron 2010;68:1023–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krenzer M, Anaclet C, Vetrivelan R, et al. Brainstem and spinal cord circuitry regulating REM sleep and muscle atonia. PLoS One 2011;6:e24998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lu J, Sherman D, Devor M, Saper CB. A putative flip‐flop switch for control of REM sleep. Nature 2006;441:589–594. [DOI] [PubMed] [Google Scholar]
- 30. Anaclet C, Ferrari L, Arrigoni E, et al. The GABAergic parafacial zone is a medullary slow wave sleep‐promoting center. Nat Neurosci 2014;17:1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Petit D, Gagnon JF, Fantini ML, et al. Sleep and quantitative EEG in neurodegenerative disorders. J Psychosom Res 2004;56:487–496. [DOI] [PubMed] [Google Scholar]
- 32. Pan XD, Chen XC. Clinic, neuropathology and molecular genetics of frontotemporal dementia: a mini‐review. Transl Neurodegener 2013;2:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roh JH, Jiang H, Finn MB, et al. Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer's disease. J Exp Med 2014;211:2487–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yamada K, Holth JK, Liao F, et al. Neuronal activity regulates extracellular tau in vivo. J Exp Med 2014;211:387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cirrito JR, Yamada KA, Finn MB, et al. Synaptic activity regulates interstitial fluid amyloid‐beta levels in vivo. Neuron 2005;48:913–922. [DOI] [PubMed] [Google Scholar]