

Abstract
Background and Purpose
Palmitoylethanolamide (PEA) is an endogenous congener of anandamide and potentiates its actions at cannabinoid CB 1 and CB 2 receptors, and at transient receptor potential vanilloid type‐1 (TRPV1) channels. The other endocannabinoid, 2‐arachidonoylglycerol (2‐AG), was recently suggested to act as a TRPV1 channel agonist. We investigated if PEA enhanced levels of 2‐AG in vitro or in vivo and 2‐AG activity at TRPV1 channels.
Experimental Approach
Endogenous lipid levels were measured by LC‐MS in (i) human keratinocytes incubated with PEA (10–20 μM, 40 min, 6 and 24 h, 37°C); (ii) the blood of spontaneously A scaris suum hypersensitive beagle dogs given a single oral dose of ultramicronized PEA (30 mg·kg−1, 1, 2, 4 and 8 h from administration); (iii) the blood of healthy volunteers given a single oral dose of micronized PEA (300 mg, 2, 4 and 6 h from administration). Effects of 2‐AG at TRPV1 channels were assessed by measuring intracellular Ca2+ in HEK‐293 cells over‐expressing human TRPV1 channels.
Key Results
PEA elevated 2‐AG levels in keratinocytes (∼3‐fold) and in human and canine plasma (∼2 and ∼20‐fold respectively). 2‐AG dose‐dependently raised intracellular Ca2+ in HEK‐293‐TRPV1 cells in a TRPV1‐dependent manner and desensitized the cells to capsaicin. PEA only slightly enhanced 2‐AG activation of TRPV1 channels, but significantly increased 2‐AG‐induced TRPV1 desensitization to capsaicin (IC 50 from 0.75 ± 0.04 to 0.45 ± 0.02 μM, with PEA 2 μM).
Conclusions and Implications
These observations may explain why several effects of PEA are attenuated by cannabinoid receptor or TRPV1 channel antagonists.
Linked Articles
This article is part of a themed section on Endocannabinoids. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v173.7/issuetoc
Abbreviations
- 2‐AG
2‐arachidonoylglycerol
- AEA
anandamide
- FAAH
fatty acid amide hydrolase
- GPR
orphan GPCR
- OEA
oleoylethanolamide
- PEA
palmitoylethanolamide
- TRPV1
transient receptor potential vanilloid type‐1
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes c |
| CB1 receptors | FAAH, fatty acid amide hydrolase |
| CB2 receptors | Monoacylglycerol lipase (MGL) |
| GPR55 | N‐acyl‐phosphatidylethanolamine‐specific PLD (NAPE‐PLD) |
| GPR119 | Nuclear receptor d |
| Ion Channels b | PPARα (NR1C1) |
| TRPV1 cation channels |
| LIGANDS |
|---|
| 2‐AG, 2‐arachidonoylglycerol |
| AEA, anandamide |
| Capsaicin |
| OEA, oleoylethanolamide |
| PEA, palmitoylethanolamide |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,c dAlexander et al., 2013a, 2013b, 2013c, 2013d).
Introduction
Palmitoylethanolamide (PEA) is an endogenous congener of the endocannabinoid anandamide (AEA) with potent anti‐inflammatory, analgesic and neuroprotective actions in a wide range of biological systems and pathological conditions (Petrosino et al., 2010a). PEA is usually biosynthesized together with AEA through similar anabolic pathways and enzymes, including the one‐step hydrolysis of the corresponding biosynthetic precursors catalysed by N‐acyl‐phosphatidylethanolamine‐specific PLD (Bisogno et al., 1997). PEA is degraded via enzymic hydrolysis of its amide bond through the catalytic action of either of two amidases: (i) fatty acid amide hydrolase (FAAH), which has AEA as its preferred substrate, or, preferentially, (ii) the N‐acylethanolamine acid amidohydrolase, a lysosomal enzyme, which does not hydrolyse AEA (Tsuboi et al., 2005). While, apart from acting as an endogenous agonist of cannabinoid CB1 and CB2 receptors, AEA also activates transient receptor potential vanilloid type‐1 (TRPV1) channels (Zygmunt et al., 1999), PEA has very low affinity for both CB1 and CB2 receptors and only activates TRPV1 channels at very high concentrations (Petrosino et al., 2010a). However, we and others (De Petrocellis et al., 2001; Di Marzo et al., 2001a; Smart et al., 2002; Ho et al., 2008) have previously shown that PEA can enhance the levels and/or actions of AEA, at both CB receptors and TRPV1 channels, a property known as the ‘entourage’ effect, and first described in the 1990s for two congeners of the other endocannabinoid, 2‐arachidonoylglycerol (2‐AG) also inactive at CB receptors (Ben‐Shabat et al., 1998). This property of PEA may explain why some of the pharmacological effects of this compound are antagonized by CB receptor or TRPV1 channel antagonists (Calignano et al., 1998; Costa et al., 2008). PEA can also activate directly PPAR‐α (LoVerme et al., 2005) and orphan GPCRs such as GPR119 and GPR55 (Ryberg et al., 2007; Godlewski et al., 2009), and was recently found to activate TRPV1 channels by acting in part via PPAR‐α (Ambrosino et al., 2013). Very recently, 2‐AG, which was previously thought to be only weakly active at TRPV1 channels (Zygmunt et al., 1999), was shown to act as a physiologically relevant activator of this channel, and to take part in its PLC‐mediated activation (Zygmunt et al., 2013). Interestingly, the authors also provided data suggesting that PEA could potentiate TRPV1 activation by 2‐AG. However, in this study, the effect of 2‐AG at TRPV1 channels was studied by using straightforward electrophysiological and pharmacological approaches (Zygmunt et al., 2013), but not the well‐known property of this channel to control Ca2+ influx and hence intracellular Ca2+ levels (Caterina et al., 1997). More importantly, the ability of 2‐AG to desensitize TRPV1 channels, a property shared by all TRPV1 channel agonists (Brederson et al., 2013), was not shown (Zygmunt et al., 2013). This ability is very important as TRPV1 channel desensitization is considered to be responsible in part for the transient and local analgesic and anti‐inflammatory actions of TRPV1 channel agonists, currently exploited in the clinic for the treatment of neuropathic pain (Brederson et al., 2013). Furthermore, TRPV1 channel desensitization has been suggested to underlie some of the analgesic and anti‐inflammatory effects of PEA, such as the attenuation of contact allergic dermatitis (Petrosino et al., 2010b) and the suppression of neuropathic pain (Costa et al., 2008) in mice.
On this basis, we undertook the present study with the aim of assessing, first, whether PEA raised 2‐AG levels in vitro or in vivo, particularly under conditions in which the compound was previously shown to produce anti‐inflammatory actions that is in human keratinocytes (Petrosino et al., 2010b) and in a canine model of allergic dermatitis (spontaneous hypersensitivity to Ascaris suum; Cerrato et al., 2012a). We also evaluated this potential property of PEA in human healthy volunteers. Our second aim was to find out if 2‐AG also stimulated TRPV1 channels to allow the elevation of intracellular Ca2+ in HEK‐293 cells over‐expressing the human recombinant TRPV1 channel, and, more importantly, if the endocannabinoid desensitizes these channels to the action of the prototypical TRPV1 channel agonist, capsaicin, to this effect, as previously shown for AEA. Finally, we tested the enhancement, by PEA, of the TRPV1‐mediated elevation in intracellular Ca2+ by 2‐AG, and the desensitization by PEA of this effect.
Methods
Cell cultures and treatments
Human keratinocytes
The immortalized human keratinocyte (HaCaT) cell line was purchased from CLS Cell Lines Service GmbH (Eppelheim, Germany) and cultured in DMEM supplemented with glutamine (2 mM), penicillin (400 U·mL−1), streptomycin (50 mg·mL−1) and 10% FBS in an humidified 5% CO2 atmosphere at 37°C. HaCaT cells were plated into 6‐well culture dishes at a cell density of 9 × 105 cells per well, and after 1 day were treated with PEA (10–20 μM) or vehicle (methanol 0.05%, CTRL) for 40 min, 6 and 24 h and incubated at 37°C in 5% CO2. The cells and supernatants were then analysed for endocannabinoids (AEA, 2‐AG), PEA and oleoylethanolamide (OEA).
HEK‐293 cells
HEK‐293 cells, either wild type or stably transfected with human recombinant TRPV1 cDNA, were grown as monolayers in EMEM medium supplemented with non‐essential amino acids, 10% FBS and 2 mM glutamine, maintained under 5% CO2 at 37°C, plated on 100 mm diameter Petri dishes. Transfected cells were selected using G‐418 (Geneticin; 600 μg·mL−1).
Animals
All animal care and experimental procedures were performed in accordance with European regulations governing the care and treatment of laboratory animals and were approved by the Animal Care and Use Committee of the Universitat Autònoma de Barcelona (CEEAH 1375). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of six animals were used in the experiments described here.
Six male beagle dogs (Harlan Laboratories, Barcelona, Spain) spontaneously hypersensitive against A. suum with mean body weights of 14.0 ± 0.6 kg were used. The animals were fasted overnight before oral administration of an aqueous suspension of ultramicronized PEA (30 mg·kg−1) in carboxymethylcellulose (0.5%). No drugs or additional treatments were given during the study. Dogs were housed in groups of two in the University kennel under a temperature‐controlled environment (21 ± 1°C) with a 12 h/12 h light/dark cycle and fed standard chow for at least 1 week before any manipulation. All animals were monitored during the procedure. Determination of PEA plasma levels was performed immediately before (0) and after 1, 2, 4 and 8 h of PEA administration. Blood samples were obtained through a catheter inserted into a cephalic vein. To determine PEA and endocannabinoid plasma levels, blood samples were collected and placed into spray‐coated lithium‐heparin tubes (BD Vacutainer Becton, Dickinson and Company, Franklin Lakes, NJ, USA). Afterwards, blood samples were centrifuged at 1800× g for 10 min and the collected plasma was stored at −80°C until analysis. Blood samples obtained before treatment were considered as control for each animal.
Human volunteers
The study included 10 healthy subjects, of both sexes (five each), 18 to 54 years of age, with a body mass index between 19 and 30 kg·m−2. Subjects had no history of clinically significant medical illness; no medications were taken for 30 days before the study. Eligible subjects gave their written informed consent. The subjects took 300 mg of micronized PEA (Normast 300 mg, Epitech Group, Saccolongo, PD, Italy) before breakfast at 9:00 am. Blood sample collection was carried out immediately before, and after 1, 2, 4 and 6 h after taking PEA. Blood samples were collected and placed into plastic tubes with EDTA, and then centrifuged at 1800× g for 10 min. The collected plasma was stored at −80°C until analysis.
Measurement of 2‐AG, AEA, PEA and OEA levels
Cells and supernatants were homogenized in a solution of chloroform/methanol/Tris‐HCl 50 mM pH 7.4 (2/1/1 by vol.) containing 10 pmol of [2H]8‐AEA, and 5 pmol of [2H]5‐2‐AG, [2H]4‐PEA and [2H]2‐OEA as internal standards (Devane et al., 1992; Bisogno et al., 1997). The lipid‐containing organic phase was dried down, weighed and pre‐purified by open‐bed chromatography on silica gel. Fractions obtained by eluting the column with a solution of chloroform/methanol (90:10 by vol.) were analysed by LC‐atmospheric pressure chemical ionization‐MS (LC‐APCI‐MS) by using a Shimadzu (Shimadzu, Kyoto, Japan) HPLC apparatus (LC‐10ADVP) coupled to a Shimadzu (LCMS‐2020) quadrupole MS via a Shimadzu APCI interface. LC‐APCI‐MS analyses of AEA, 2‐AG, PEA and OEA were carried out in the selected ion monitoring mode (Di Marzo et al., 2001b), using m/z values of 356 and 348 (molecular ions +1 for deuterated and undeuterated AEA), 384.35 and 379.35 (molecular ions +1 for deuterated and undeuterated 2‐AG), 304 and 300 (molecular ions +1 for deuterated and undeuterated PEA), 328 and 326 (molecular ions +1 for deuterated and undeuterated OEA). AEA, 2‐AG, PEA and OEA levels were calculated on the basis of their area ratio with the internal deuterated standard signal areas, and their amounts (pmol) were normalized per mg of lipid extract or per mL of volume.
Experiments on intracellular Ca2+ elevation
The effect of 2‐AG (Tocris Bioscience, Minneapolis, MN, USA) on intracellular Ca2+ was determined by using Fluo‐4 (Molecular Probes, Eugene, OR, USA), a selective intracellular fluorescent probe for Ca2+ ions. HEK‐293 cells stably transfected with human recombinant TRPV1 cDNA were loaded for 1 h at room temperature with the methyl ester Fluo4‐AM 4 μM; containing 0.02% Pluoronic F‐127 (Molecular Probes) in EMEM without FBS, then were washed twice in Tyrode's buffer (145 mM NaCl, 2.5 mM KCl, 1.5 mM CaCl2, 1.2 mM MgCl2, 10 mM D‐glucose and 10 mM HEPES, pH 7.4), resuspended in Tyrode's buffer and transferred to the quartz cuvette of the spectrofluorimeter (Perkin‐Elmer LS50B; Perkin‐Elmer Life, Waltham, MA, USA) under continuous stirring. Experiments were carried out by measuring cell fluorescence at 25°C (excitation λ = 488 nm; emission λ = 516 nm) before and after the addition of compounds. The effects of 2‐AG on TRPV1 channels was determined by normalizing its effect to the maximum Ca2+ influx effect on [Ca2+]i observed after the application of 4 μM ionomycin (Sigma Aldrich, St Louis, MO, USA). Potency was expressed as the concentration of 2‐AG exerting the half‐maximal agonistic effect (i.e. half‐maximal increases in [Ca2+]i) (EC50). Desensitizing behaviour was evaluated against capsaicin (0.1 μM) by adding 2‐AG, PEA or adelmidrol at different concentrations in the quartz cuvette containing cells 5 min before stimulation of cells with capsaicin 0.1 μM. Data are expressed as the concentration exerting a half‐maximal inhibition of agonist‐induced [Ca2+]i elevation (IC50). The effect on [Ca2+]i exerted by capsaicin 0.1 μM alone was taken as 100%. The selective TRPV1 channel antagonist 5′‐iodo‐resiniferatoxin (IRTX, 0.1 μM) was added 5 min before 2‐AG. PEA (1–5 μM) or adelmidrol (1–50 μM) were also added to the cells 5 min before 2‐AG to study their effects on 2‐AG‐induced activation of the TRPV1‐mediated elevation of intracellular Ca2+. However, in some experiments on 2‐AG‐induced desensitization of the TRPV1‐mediated elevation of intracellular Ca2+ by capsaicin (0.1 μM), PEA (2 μM) was added together with, or also 1 or 2 min before, 2‐AG (0.2 μM).
Data analysis
For the determination of endocannabinoids and PEA and OEA, group means were compared using the Student's t‐test for the experiments in vitro and one‐way anova followed by Dunnett's multiple comparison test for data obtained in canine or human plasma. All determinations were performed at least in triplicate. Dose–response curves were fitted by a sigmoidal regression with variable slope. Curve fitting and parameter estimation were performed with GraphPad Prism (GraphPad Software Inc., San Diego, CA). Group means were compared using one‐way anova followed by Bonferroni's test.
Materials
Deuterated standards – [2H]8‐AEA, [2H]5‐2‐AG, [2H]4‐PEA and [2H]2‐OEA – and IRTX were purchased from Cayman Chemicals. Unlabelled AEA and OEA were purchased from TOCRIS Bioscience. Capsaicin and unlabelled 2‐AG and capsaicin were purchased from ENZO Life Sciences. Adelmidrol and unlabelled PEA and adelmidrol were provided by the Epitech Group.
Results
PEA elevates 2‐AG levels in human keratinocytes
We measured the effect of PEA (10 or 20 μM, 40 min, 6h and 24 h incubation at 37°C) on 2‐AG levels in human HaCaT keratinocytes, where PEA was previously shown to produce TRPV1‐mediated anti‐inflammatory actions (Petrosino et al., 2010b). At all sampling points, PEA (10 μM) elevated by ∼2.5–3‐fold the amounts of 2‐AG, compared with vehicle‐treated keratinocytes (Figure 1A). PEA also elevated AEA levels by ∼5‐fold, although only after 6 h and at the higher concentration (20 μM; Figure 1B). No consistent effect was observed on OEA levels (data not shown).
Figure 1.
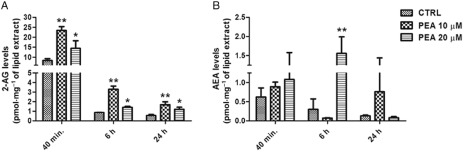
Concentrations of 2‐AG (A) and AEA (B) in HaCaT cells incubated with PEA (10 or 20 μM), or vehicle (CTRL) for 40 min, 6 and 24 h. Data are means ± SE of n = 6 separate determinations. For 2‐AG levels: **P < 0.01 for CTRL versus PEA 10 μM; *P < 0.05 for CTRL versus PEA 20 μM. For AEA levels: **P < 0.01 for CTRL versus PEA 20 μM. Student's t‐test was used.
PEA elevates plasma 2‐AG levels in dogs
A single oral dose of ultramicronized PEA (30 mg·kg−1) was administered orally to spontaneously A. suum hypersensitive beagle dogs, in which PEA had been shown to produce skin anti‐inflammatory actions (Cerrato et al., 2012a), and blood collected 1, 2, 4 and 8 h after administration for endocannabinoid, PEA and OEA analysis. As expected, plasma PEA levels were increased following administration, with similar levels being reached after 1 and 2 h from administration, more than five‐fold higher than the baseline levels (Figure 2A). This corresponded to the maximum anti‐inflammatory action of PEA (in terms of reduction of wheal induced by either A. suum extract or anti‐canine IgE; Cerrato et al., 2012a). Importantly, at the same time points but with a slight delay, the plasma levels of 2‐AG were also significantly elevated by up to ∼20‐fold (Figure 2B), whereas only non‐statistically significant trends for increases were observed for AEA and OEA levels (Figure 2C and D).
Figure 2.
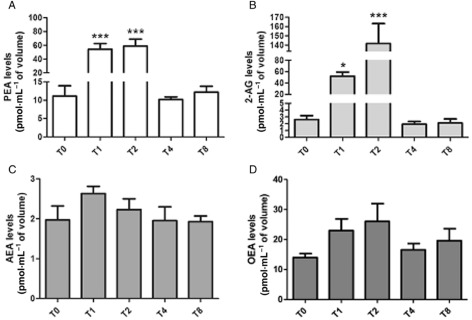
Concentrations of PEA (A), 2‐AG (B), AEA (C) and OEA (D) in plasma of dogs before (T0), and 1 (T1), 2 (T2), 4 (T4) and 8 (T8) h after an oral administration of ultramicronized PEA 30 mg·kg−1. Data are means ± SE of n = 4 separate determinations. For PEA levels: ***P < 0.001 for T0 versus T1 and T2. For 2‐AG levels: *P < 0.05 for T0 versus T1; ***P < 0.001 for T0 versus T2. One‐way anova followed by Dunnett's multiple comparison test.
PEA elevates plasma 2‐AG levels in healthy human volunteers
We administered a single oral dose of micronized PEA (300 mg) to human volunteers and measured plasma endocannabinoid levels 2, 4 and 6 h later. We observed that, following a peak of plasma PEA levels at 2 h; Figure 3A), the plasma levels of 2‐AG were significantly elevated by up to two‐fold after 4 and 6 h (Figure 3B). No effect on AEA or OEA levels was observed (Figure 3C and D).
Figure 3.
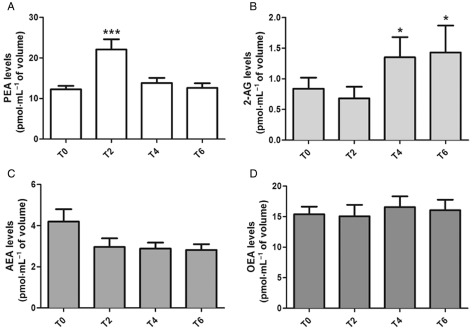
Concentrations of PEA (A), 2‐AG (B), AEA (C) and OEA (D) in plasma of human volunteers before (T0), and 2 (T2), 4 (T4) and 6 (T6) h after an oral administration of micronized PEA 300 mg. Data are means ± SE of n = 10 separate determinations. For PEA levels: ***P < 0.001 for T0 versus T2. For 2‐AG levels: *P < 0.05 for T0 versus T4 and T6. One‐way anova followed by Dunnett's multiple comparison test.
PEA enhances 2‐AG activation and desensitization of TRPV1‐mediated intracellular Ca2+ elevation
We measured intracellular Ca2+ in HEK‐293 cells over‐expressing human TRPV1 channels. In agreement with previous data using patch‐clamp electrophysiology and mesenteric artery vasodilation, 2‐AG dose‐dependently elevated intracellular Ca2+ in these cells (Table 1), whereas it failed to evoke similar Ca2+ responses in untransfected HEK‐293 cells (Figure 4A). The selective TRPV1 channel antagonist IRTX (0.1 μM) blocked the Ca2+ response induced by 2‐AG (Figure 4A). Importantly, 2‐AG also desensitized TRPV1 channels to the effect of capsaicin (0.1 μM) on intracellular Ca2+ (IC50 values in Table 1; Figure 4B). The 2‐AG was more than three times less potent than AEA at both activating and desensitizing TRPV1 channels (Table 1). PEA (1–5 μM) alone had no effect on activation or desensitization of TRPV1 channels (data not shown). On the contrary, PEA slightly enhanced 2‐AG activation of TRPV1‐mediated intracellular Ca2+ increase, this effect being statistically significant only at 2 μM (Figure 5A, Table 1); but significantly increased by nearly two‐fold the 2‐AG‐induced TRPV1 channel desensitization to the effect of capsaicin 0.1 μM (Figure 5B and Table 1). The efficacy of capsaicin, expressed as percent of the effect of ionomycin 4 μM, was 70 ± 1%. We also assessed the effects of the time of pre‐exposure to PEA on its interaction with 2‐AG. The results, expressed as % of the response observed with capsaicin (0.1 μM) + vehicle (Table 2), showed no change in the PEA effect on 2‐AG‐induced TRPV1 channel desensitization when PEA was pre‐incubated 1 and 2 min before 2‐AG, compared with the effect observed with PEA pre‐incubated 5 min before 2‐AG. However, when PEA (2 μM) and 2‐AG (0.2 μM) were incubated simultaneously (Table 2), although a significant effect was still observed on TRPV1 channel desensitization, compared with 2‐AG alone, this effect was weaker than that observed when PEA was pre‐incubated 1, 2 or 5 min before 2‐AG, as described above (Table 2).
Table 1.
Effects of PEA on elevation of intracellular Ca2+ through TRPV1 channels induced by 2‐AG
| EC50 | IC50 (vs. capsaicin 0.1 μM) | % efficacy (vs. ionomycin 4 μM) | |
|---|---|---|---|
| 2‐AG | 0.85 ± 0.06 | 0.75 ± 0.03 | 59.1 ± 0.3 |
| PEA, 5 μM + 2‐AG | 0.74 ± 0.04 | 0.61 ± 0.05 | 56.6 ± 0.2 |
| PEA 2 μM + 2‐AG | 0.52 ± 0.02 | 0.45 ± 0.02* | 53.2 ± 0.2 |
| PEA 1 μM + 2‐AG | 0.92 ± 0.20 | 0.65 ± 0.04 | 56.9 ± 0.8 |
| Adelmidrol 50 μM + 2‐AG | 0.79 ± 0.02 | 0.77 ± 0.04 | 60.5 ± 0.2 |
| AEA | 0.27 ± 0.01 | 0.21 ± 0.06 | 53.8 ± 0.2 |
The EC50 values (μM) shown are for 2‐AG alone and after PEA or adelmidrol pre‐incubation for 5 min before 2‐AG, as well as for anandamide. The IC50 values (μM) shown are for TRPV1 channel desensitization by 2‐AG in the presence of PEA or adelmidrol, pre‐incubated 5 min before capsaicin, and for TRPV1 channel desensitization by AEA. The efficacy of the compounds or their combinations are reported as % of the maximum effect observed with ionomycin (4 μM). All the experiments were performed in HEK‐293 cells over‐expressing human TRPV1 channels. Data are means ± SE of n = 4 separate determinations. *P < 0.05 versus 2‐AG only; one‐way anova followed by Bonferroni's test.
Figure 4.
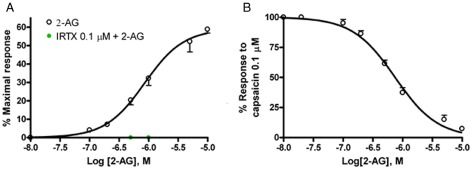
(A) Effect of 2‐AG on elevation of intracellular Ca2+ in HEK‐293 cells over‐expressing human TRPV1 channels. The effect of a 5 min pre‐incubation with the TRPV1 antagonist IRTX on the response to is also shown. (B) Desensitization by 5 min pre‐incubation with 2‐AG of capsaicin (0.1 μM)‐induced Ca2+ elevation in HEK‐293 cells over‐expressing the human TRPV1 channels. The effects of capsaicin (0.1 μM) alone were set as 100%. Data are means ± SE of n = 4 separate determinations. Dose–response curves were fitted by a sigmoidal regression with variable slope. Curve fitting and parameter estimation were performed with GraphPad Prism.
Figure 5.
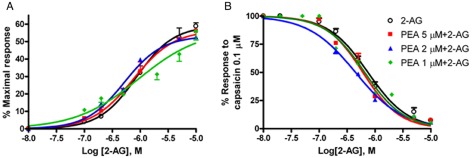
(A) Effect of PEA (1–5 μM) on intracellular Ca2+ elevation by 2‐AG in HEK‐293 cells over‐expressing human TRPV1 channels. PEA was pre‐incubated with cells 5 min prior to 2‐AG treatment. (B) Effect of PEA (1–5 μM) on 2‐AG‐induced TRPV1 channel desensitization of the effect of capsaicin (0.1 μM) in HEK‐293 cells over‐expressing human TRPV1 channels. PEA was pre‐incubated with cells 5 min prior to 2‐AG treatment. The effect on intracellular Ca2+ of capsaicin (0.1 μM) alone were set as 100%. Data are means ± SE of n = 4 separate determinations. Dose–response curves were fitted by a sigmoidal regression with variable slope. Curve fitting and parameter estimation were performed with GraphPad Prism.
Table 2.
Time course of the modification by PEA of the desensitization of TRPV1 channels to capsaicin by exposure to 2‐AG
| % of the response of capsaicin (0.1 μM) | |
|---|---|
| +2‐AG, 0.2 μM | 77.2 ± 3.4 |
| +PEA 2 μM + 2‐AG, 0.2 μM (simultaneously) | 66.0 ± 1.5* |
| +PEA, 2 μM (1 min before) + 2‐AG, 0.2 μM | 56.6 ± 1.7* |
| +PEA, 2 μM (2 min before) + 2‐AG, 0.2 μM | 55.8 ± 3.5* |
| +PEA, 2 μM (5 min before) + 2‐AG, 0.2 μM | 56.0 ± 4.2* |
Intracellular Ca2+ was elevated through TRPV1 channels in response to capsaicin and this response was reduced by pre‐exposure to 2‐AG alone. Adding PEA to the 2‐AG – at the same time or as a pretreatment (1, 2 or 5 min), increased the desensitization by 2‐AG. All the experiments were performed in HEK‐293 cells over‐expressing human TRPV1 channels. Data are means ± SE of n = 4 separate determinations. *P < 0.05 versus 2‐AG only; one‐way anova followed by Bonferroni's test.
Adelmidrol, a synthetic analogue of PEA with anti‐inflammatory actions in dogs (Cerrato et al., 2012b), had no effect on 2‐AG‐induced activation or desensitization of the TRPV1‐mediated elevation of intracellular Ca2+ in the range of concentrations of 1 to 50 μM (Table 1). Adelmidrol alone produced no stimulatory or inhibitory effect on TRPV1 channels (data not shown).
Discussion
The findings reported here indicate that, in addition to the previously observed similar effect on AEA, PEA exerts significant ‘entourage’ effects also on 2‐AG, by enhancing both its levels in vitro and in vivo, and its actions at TRPV1 channels in vitro. These observations may help explaining why, for example, several anti‐inflammatory or analgesic effects of PEA can be attenuated by cannabinoid receptor antagonists or, particularly, TRPV1 channel antagonists (Calignano et al., 1998; Lambert and Di Marzo, 1999; Costa et al., 2008; Petrosino et al., 2010b; Citraro et al., 2013), and add yet another mechanism of action to the multifaceted pharmacological properties of this lipid mediator.
Although shown and discussed here together, the results of the present in vitro and in vivo experiments cannot be compared. The in vitro experiments show that, in a single cell line, and as a consequence of interactions that can occur only in this context, PEA can increase 2‐AG levels. The in vivo experiments suggest that such an effect may occur also in the many cell types that contribute to determine plasma 2‐AG levels (of which keratinocytes might be a minimal component), but may also be the result of different sequential interactions and effects in different cell types, whose effects on the measured levels cannot be easily assessed.
We observed that, at least in the human keratinocyte culture used here, as well as in vivo in beagle dogs, that is, two experimental systems in which PEA was previously shown to produce anti‐inflammatory actions by inhibiting the release of the inflammatory cytokine MCP‐2 and reducing the wheal induced by allergic reactions, respectively (Petrosino et al., 2010b; Cerrato et al., 2012a), the effect on 2‐AG levels was greater, overall, than on AEA levels. While 2‐AG levels were elevated by several fold, thus potentially leading to activation of CB1 and CB2 receptors, as well as activation and desensitization of TRPV1 channels (see below), AEA levels were not significantly elevated in vivo and were elevated in keratinocytes only at the highest concentration of PEA used and only at one of the three incubation times evaluated. Also in human volunteers, oral administration of PEA elevated 2‐AG levels, although this effect (∼2‐fold elevation) was much smaller than in dogs (∼20‐fold elevation). This smaller enhancement, however, was somehow proportionate to the smaller peak of PEA levels found following oral administration in human plasma (a ∼2‐fold elevation vs. baseline, 2 h after administration), compared with dog plasma (∼5‐fold elevation, 1–2 h after administration). This difference, in turn, apart from species‐specific phenomena, might be due to the simple fact that a lower total amount of PEA was acutely administered to human volunteers (about ∼5 mg·kg−1 vs. 30 mg·kg−1 in dogs), and this was a micronized formulation as opposed to the ultramicronized PEA (which is more likely to have higher oral bioavailability) that was given to dogs. Finally, the human volunteers employed for this study were healthy, whereas the dogs were hypersensitive against A. suum, and the bioavailability of PEA might be different during pathological conditions. At any rate, the present data indicate that the ‘entourage’ effect of PEA on 2‐AG levels might be higher than on AEA levels. While the latter effect was previously ascribed to a inhibitory action on the expression and/or activity of the major AEA‐metabolizing enzyme (FAAH; Bisogno et al., 1997; Di Marzo et al., 2001a; Ho et al., 2008), it is unlikely that this was the only mechanism responsible for raised 2‐AG levels after PEA, as (i) the effect of PEA in keratinocytes was already maximum after a 40 min incubation, which is not sufficient to induce transcriptional modulation; (ii) 2‐AG is degraded by several hydrolases, and to a largest extent by monoacylglycerol lipase (Blankman et al., 2007), which is not inhibited by PEA – however, FAAH may play a role in 2‐AG hydrolysis under certain circumstances (Goparaju et al., 1998); and (iii) in most of the experiments presented here the stimulatory action of PEA on 2‐AG levels was not accompanied by a corresponding elevation in the levels of the two preferred substrates for FAAH, AEA and OEA. It is also unlikely that the effect of PEA on 2‐AG levels was due to activation of PPARα or GPR55 and/or GPR119 (LoVerme et al., 2005; Ryberg et al., 2007; Godlewski et al., 2009) as 2‐AG release from diacylglycerols is known to be stimulated by receptors coupled to Gq/11 and none of these three receptors belong to this category. Given the promiscuity of PEA at different molecular targets, the possibility that this compound activates an as yet unidentified Gq/11‐coupled receptor, thereby stimulating 2‐AG biosynthesis, cannot be excluded.
Whatever its mechanism of action, the marked effect of PEA on 2‐AG levels is likely to enhance several of the CB1 and CB2 receptor and TRPV1 channel‐mediated actions of this compound. In fact, although 2‐AG can activate TRPV1 channels (Zygmunt et al., 1999) as a physiologically relevant activator of this channel and can participate in the PLC‐mediated activation of TRPV1 channels (Zygmunt et al., 2013), the ability of 2‐AG to desensitize TRPV1 channels, a property shared by all TRPV1 channel agonists (Brederson et al., 2013), was not shown in earlier experiments (Zygmunt et al., 2013). A possible explanation for the failure of the previous study to show such inhibitory action of 2‐AG might rely in the high concentration (2 μM) of capsaicin used by the authors (Zygmunt et al., 2013). This is a concentration that causes a maximum elevation of intracellular calcium and hence inhibitory effects are less likely to be detected. In contrast, we used, here, a submaximal concentration of capsaicin, causing only about 75% of capsaicin maximal effect, and we could show that PEA not only elevated the levels of 2‐AG, but also significantly enhanced its capability of desensitizing human recombinant TRPV1 channels in vitro, by nearly halving the concentration of 2‐AG necessary to produce, after a 1, or 2 or 5 min pre‐incubation, a 50% inhibition of capsaicin‐induced TRPV1 channel activation. In view of the present observation that PEA also doubles the amounts of circulating 2‐AG in humans, this finding opens the possibility that administration of this compound may cause a fourfold potentiation of the tonic desensitization of TRPV1 channels, which, considering the role played by this channel in pain and inflammation, may lead to potent TRPV1‐mediated analgesic and anti‐inflammatory actions. Understanding whether or not PEA potentiates the effects of 2‐AG at TRPV1 channels by acting as an allosteric enhancer of its binding to this channel, as previously suggested for PEA potentiation of AEA actions at TRPV1 channels (De Petrocellis et al., 2001), will require specific studies. The present observation that PEA potentiation of 2‐AG desensitization of TRPV1 channels is reduced, but still present, if the two compounds are given to cells together, argues in favour of an allosteric mechanism of action. We have also tested here the effect of adelmidrol, a synthetic analogue of PEA with anti‐inflammatory actions in dogs (Cerrato et al., 2012b). Our results show that adelmidrol, in the range of concentrations from 1 to 50 μM, pre‐incubated 5 min before 2‐AG, had no effect on 2‐AG‐induced activation or desensitization of the TRPV1‐mediated elevation of intracellular Ca2+.
In conclusion, the present study extends to 2‐AG the previously described ‘entourage’ effects of PEA (De Petrocellis et al., 2001; Di Marzo et al., 2001a; Smart et al., 2002; Ho et al., 2008), and provides an additional explanation as to why many effects of this pleiotropic mediator are attenuated by antagonists of TRPV1 channels and of CB1 and CB2 receptors. The understanding of the mechanism through which PEA produces these effects will require further investigation.
Author contributions
S. P., L. D. and V. D. carried designed the experiments and drafted the manuscript. A. S. M. and L. D. P. carried out the experiments on intracellular calcium elevation. S. P. carried out the measurement of 2‐AG, AEA, PEA and OEA levels. M. F. carried out the experiments in healthy human volunteers. A. P. and S. C. carried out the experiments in dogs. All authors have read and approved the final manuscript.
Conflict of interest
A. S. M. receives unrestricted research support from Epitech Italia S.r.l. S. P. and M. F. are employees of Epitech Group S.r.l.
Acknowledgements
This work was supported by Progetto Operativo Nazionale (PON01_02512).
Petrosino, S. , Schiano Moriello, A. , Cerrato, S. , Fusco, M. , Puigdemont, A. , De Petrocellis, L. , and Di Marzo, V. (2016) The anti‐inflammatory mediator palmitoylethanolamide enhances the levels of 2‐arachidonoyl‐glycerol and potentiates its actions at TRPV1 cation channels. Br J Pharmacol, 173: 1154–1162. doi: 10.1111/bph.13084.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013a). The Concise Guide to PHARMACOLOGY 2013/14: G Protein‐Coupled Receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol 170: 1607–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013c). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013d). The Concise Guide to PHARMACOLOGY 2013/14: Nuclear Hormone Receptors. Br J Pharmacol 170, 1652–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosino P, Soldovieri MV, Russo C, Taglialatela M (2013). Activation and desensitization of TRPV1 channels in sensory neurons by the PPARα agonist palmitoylethanolamide. Br J Pharmacol 168: 1430–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Shabat S, Fride E, Sheskin T, Tamiri T, Rhee MH, Vogel Z et al (1998). An entourage effect: inactive endogenous fatty acid glycerol esters enhance 2‐arachidonoyl‐glycerol cannabinoid activity. Eur J Pharmacol 353: 23–31. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Maurelli S, Melck D, De Petrocellis L, Di Marzo V (1997). Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J Biol Chem 272: 3315–3323. [DOI] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF (2007). A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2‐arachidonoylglycerol. Chem Biol 14: 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brederson JD, Kym PR, Szallasi A (2013). Targeting TRP channels for pain relief. Eur J Pharmacol 716: 61–76. [DOI] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, Piomelli D (1998). Control of pain initiation by endogenous cannabinoids. Nature 394: 277–281. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997). The capsaicin receptor: a heat‐activated ion channel in the pain pathway. Nature 389: 816–824. [DOI] [PubMed] [Google Scholar]
- Cerrato S, Brazis P, Della Valle MF, Miolo A, Petrosino S, Di Marzo V et al (2012a). Effects of palmitoylethanolamide on the cutaneous allergic inflammatory response in Ascaris hypersensitive Beagle dogs. Vet J 191: 377–382. [DOI] [PubMed] [Google Scholar]
- Cerrato S, Brazis P, Della Valle MF, Miolo A, Puigdemont A (2012b). Inhibitory effect of topical adelmidrol on antigen‐induced skin wheal and mast cell behavior in a canine model of allergic dermatitis. BMC Vet Res 8: 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citraro R, Russo E, Scicchitano F, van Rijn CM, Cosco D, Avagliano C et al (2013). Antiepileptic action of N‐palmitoylethanolamine through CB1 and PPAR‐α receptor activation in a genetic model of absence epilepsy. Neuropharmacology 69: 115–126. [DOI] [PubMed] [Google Scholar]
- Costa B, Comelli F, Bettoni I, Colleoni M, Giagnoni G (2008). The endogenous fatty acid amide, palmitoylethanolamide, has anti‐allodynic and anti‐hyperalgesic effects in a murine model of neuropathic pain: involvement of CB(1), TRPV1 and PPARgamma receptors and neurotrophic factors. Pain 139: 541–550. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Davis JB, Di Marzo V (2001). Palmitoylethanolamide enhances anandamide stimulation of human vanilloid VR1 receptors. FEBS Lett 506: 253–256. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G et al (1992). Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258: 1946–1949. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Melck D, Orlando P, Bisogno T, Zagoory O, Bifulco M et al (2001a). Palmitoylethanolamide inhibits the expression of fatty acid amide hydrolase and enhances the anti‐proliferative effect of anandamide in human breast cancer cells. Biochem J 358: 249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Goparaju SK, Wang L, Liu J, Bátkai S, Járai Z et al (2001b). Leptin‐regulated endocannabinoids are involved in maintaining food intake. Nature 410: 822–825. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godlewski G, Offertáler L, Wagner JA, Kunos G (2009). Receptors for acylethanolamides‐GPR55 and GPR119. Prostaglandins Other Lipid Mediat 89: 105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goparaju SK, Ueda N, Yamaguchi H, Yamamoto S (1998). Anandamide amidohydrolase reacting with 2‐arachidonoylglycerol, another cannabinoid receptor ligand. FEBS Lett 422: 69–73. [DOI] [PubMed] [Google Scholar]
- Ho WS, Barrett DA, Randall MD (2008). ‘Entourage' effects of N‐palmitoylethanolamide and N‐oleoylethanolamide on vasorelaxation to anandamide occur through TRPV1 receptors. Br J Pharmacol 155: 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert DM, Di Marzo V (1999). The palmitoylethanolamide and oleamide enigmas: are these two fatty acid amides cannabimimetic? Curr Med Chem 6: 757–773. [PubMed] [Google Scholar]
- LoVerme J, La Rana G, Russo R, Calignano A, Piomelli D (2005). The search for the palmitoylethanolamide receptor. Life Sci 77: 1685–1698. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al; NC‐IUPHAR (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosino S, Iuvone T, Di Marzo V (2010a). N‐palmitoyl‐ethanolamine: biochemistry and new therapeutic opportunities. Biochimie 92: 724–727. [DOI] [PubMed] [Google Scholar]
- Petrosino S, Cristino L, Karsak M, Gaffal E, Ueda N, Tüting T et al (2010b). Protective role of palmitoylethanolamide in contact allergic dermatitis. Allergy 65: 698–711. [DOI] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjögren S, Hjorth S, Hermansson NO, Leonova J et al (2007). The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol 152: 1092–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart D, Jonsson KO, Vandevoorde S, Lambert DM, Fowler CJ (2002). ‘Entourage' effects of N‐acyl ethanolamines at human vanilloid receptors. Comparison of effects upon anandamide‐induced vanilloid receptor activation and upon anandamide metabolism. Br J Pharmacol 136: 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi K, Sun YX, Okamoto Y, Araki N, Tonai T, Ueda N (2005). Molecular characterization of N‐acylethanolamine‐hydrolyzing acid amidase, a novel member of the choloylglycine hydrolase family with structural and functional similarity to acid ceramidase. J Biol Chem 280: 11082–11092. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sørgård M, Di Marzo V et al (1999). Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400: 452–457. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Ermund A, Movahed P, Andersson DA, Simonsen C, Jönsson BA et al (2013). Monoacylglycerols activate TRPV1 – a link between phospholipase C and TRPV1. PLoS ONE 8: e81618. [DOI] [PMC free article] [PubMed] [Google Scholar]