

Abstract
Ligands targeting GPCRs can be categorized according to their intrinsic efficacy to trigger a specific, receptor‐mediated response. A ligand endowed with the same level of efficacy as the endogenous agonist can be classified as a full agonist, whereas a compound that displays greater efficacy, that is, higher receptor signalling output than the endogenous agonist, can be called a superagonist. Subsequent to GPCR activation, an intracellular signalling cascade is set in motion, which may generate substantial amplification of the signal. This may obscure superagonism in pharmacological assays and, therefore, the definition of superagonism necessitates a combination of operational approaches, reduction of spare receptors or estimation of receptor activation close to the receptor level to quantify relative agonist efficacies in a particular system.
The first part of this review will compare GPCR superagonism with superagonism in the field of immunology, where this term is well established. In the second part, known GPCR superagonists will be reviewed. Then, the experimental and analytical challenges in the deconvolution of GPCR superagonism will be addressed. Finally, the potential benefit of superagonism is discussed.
The molecular mechanisms behind GPCR superagonism are not completely understood. However, crystallography shows that agonist binding alone is not sufficient for a fully active receptor state and that binding of the G protein is at least equally important. Accordingly, the emerging number of reported superagonists implies that ligand‐induced receptor conformations more active than the ones stabilized by the endogenous agonist are indeed feasible. Superagonists may have therapeutic potential when receptor function is impaired or to induce negative feedback mechanisms.
Linked Articles
This article is part of a themed section on Molecular Pharmacology of G Protein‐Coupled Receptors. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v173.20/issuetoc
Abbreviations
- BRET
bioluminescence resonance energy transfer
- DMR
dynamic mass redistribution
- Emax
maximum‐inducible response, asymptote of the concentration‐effect curve
- Oxo M
Oxotremorine M
- TRH
thyrotropin‐releasing hormone
- [35S]GTPγS
guanosine 5′‐O‐(γ –thio)triphosphate
Tables of Links
| TARGETS |
|---|
| GPCRs a |
| α2A‐adrenoceptors |
| Ghrelin receptors |
| Gonadotropin‐releasing hormone (GnRH) receptors |
| Muscarinic M2 receptors |
| Muscarinic M4 receptors |
| Somatostatin sst4 receptors |
| TRH1 receptors |
| Ligand ‐gated ion channels b |
| 5‐HT3AB receptors |
| GABAA receptors |
| Enzymes c |
| MAP kinase |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a b cAlexander et al., 2013a, 2013b, 2013c).
Introduction
G protein‐coupled receptors (GPCRs) are cell surface receptors, crucial for signal transduction across cell membranes (Millar and Newton, 2010) and represent one of the largest and most diverse protein families in the human genome (Oldham and Hamm, 2008; Alexander et al., 2013a). In humans, GPCRs are encoded by about 800 different genes (Fredriksson et al., 2003; Bjarnadóttir et al., 2006) and mediate most cellular responses to hormones, neurotransmitters and other sensory inputs like vision, olfaction and taste (Rosenbaum et al., 2009). Based on phylogenetic criteria, the large superfamily of human GPCRs can be subdivided into the five main subfamilies Glutamate, Rhodopsin, Adhesion, Frizzled/Taste and Secretin (‘GRAFS’ nomenclature) (Fredriksson et al., 2003), among which the Rhodopsin family (resembling the class A GPCR family in the Kolakowski/NC‐IUPHAR extended nomenclature system (Kolakowski, 1994; Foord et al., 2005)) is by far the largest and most studied subclass. So far, more than one‐third of all drugs target GPCRs (Overington et al., 2006), and an emerging number of still ‘undrugged’ receptors display association with various diseases (e.g. Garland, 2013). This implies that GPCRs may become even more important drug targets in the future and explains why so much effort is still being put into GPCR drug discovery.
The canonical view of GPCR signal transduction is focused on the activation of intracellular heterotrimeric guanine nucleotide binding proteins (G proteins) (Milligan and Kostenis, 2006; Dohlman, 2015). In addition, G protein‐independent signalling pathways are well established (Pierce and Lefkowitz, 2001; Pierce et al., 2002). The ability of a ligand to elicit a receptor‐mediated physiological or pharmacological response is addressed by the term ‘efficacy’ (Stephenson, 1956; Kenakin, 1995a; Kenakin, 2002; Kenakin, 2013; Rajagopal, 2013), and ligands can be categorized accordingly (Kenakin, 1995a; Smith et al., 2011). Historically, the efficacy of a specific ligand is derived from concentration‐effect curves and quantified by the maximum effect (Emax) relative to Emax of a standard compound such as the endogenous agonist (Strange, 2008; Smith et al., 2011; Langmead and Christopoulos, 2013). Test systems include isolated organs, GPCR‐linked second messenger accumulation and regulation of gene expression. In this regard, it is important to note that Emax parameters are assay‐dependent and system‐dependent (Langmead and Christopoulos, 2013). Drugs that induce the maximum response of a system may nevertheless differ in efficacy, because the fraction of receptors required to be agonist‐bound may well differ between different agonists. This fraction depends on the individual efficacy of a given agonist for receptor activation (Nickerson, 1956; Stephenson, 1956; Kenakin, 1985). Notably, in assays, which monitor signalling outcome distal from receptor activation, the signalling response that can be measured has usually a certain assay‐dependent limit, and the signal is often substantially amplified (Kenakin, 2002; Milligan, 2003; Colabufo et al., 2007). As a consequence, the assay will fail to discriminate between agonists that are endowed with high, yet differing, efficacy. Approaches such as the operational model of agonism (Black and Leff, 1983) and partial receptor inactivation (e.g. Furchgott, 1966; Furchgott and Bursztyn, 1967) are therefore necessary to quantify relative estimates of ligand efficacy (e.g. Christopoulos and El‐Fakahany, 1999).
Classification of GPCR ligands in the light of the term ‘superagonism’
Ligands that produce the full biological response in a particular system can be designated full agonists, whereas ligands that produce only a submaximal response even at receptor saturating concentrations are classified as partial agonists (Kenakin, 1985; Rosenbaum et al., 2009; Nygaard et al., 2013). This classification may necessitate ligand re‐classification each time a new ligand with a higher systems response is identified. Additionally, a drug can elicit a full systems response without having full intrinsic efficacy. Therefore, as used in this article, ligands can be also classified according to their efficacy relative to the endogenous agonist: ligands, which have the same intrinsic efficacy for receptor activation as the endogenous agonist in a specific system, can be defined as full agonists. Consequently, compounds, which do not activate the receptor to its full extent even at receptor‐saturating concentrations, are referred to as partial agonists, whereas ligands that display higher efficacy than the endogenous agonist are classed as “superagonists” (Figure 1A) (Smith et al., 2011). Major difficulties in estimating efficacy result from high receptor densities on overexpressing cells and a certain degree of signalling amplification in pharmacological assays. This may result in a high receptor reserve, that is, only a small fraction of receptor available needs to be occupied to evoke a maximum systems response (Nickerson, 1956; Stephenson, 1956; Ariens et al., 1960). Comparison of maximum effects will show that in a system with high receptor density, two agonists with different levels of intrinsic efficacy may both appear as full agonists, whereas in a biological system with low receptor density, one agonist may act as a partial agonist compared with the other (Figure 1B) (e.g. Rajagopal et al., 2011; Langmead and Christopoulos, 2013; Shonberg et al., 2014). As mentioned earlier, this may necessitate application of inactivation methods and functional data analysis with operational approaches to estimate relative ligand efficacies (Christopoulos and El‐Fakahany, 1999).
Figure 1.
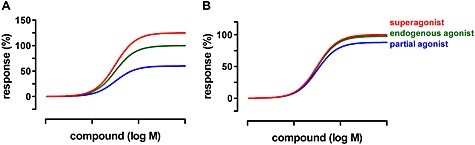
Signal amplification may obscure superagonism. 0% is defined by the basal level of the effect. 100% is the maximum effect of the endogenous agonist, classified as ‘full agonist’. Concentration‐response relationships were simulated by plotting the four‐parameter logistic function (Barlow and Blake, 1989) for a set of receptor ligands differing in capacity for receptor activation using GraphPad Prism 5. (A) In a system with no or weak signal amplification: the maximum effect of the superagonist exceeds the maximum effect of the physiological agonist (taken to define the 100% level). Superagonism is obvious. (B) In a system with powerful signal amplification and in which the endogenous full agonist is taken to define the 100% level of the effect, the superagonism is masked. Note: direct experimental identification of superagonism requires use of a sensitive test system.
Notably, the term efficacy always has a certain ‘quality’, as ligands with the ability to activate the same receptor protein may stabilize different active receptor conformations, which in turn activate different subsets of intracellular adaptor proteins leading to diverse functional responses (Kenakin, 1995b; Deupi and Kobilka, 2010; Kenakin and Christopoulos, 2013). This ‘biased agonism’ or ‘functional selectivity’ has become increasingly interesting for potential therapeutic exploitation in drug discovery (Shonberg et al., 2014).
This article aims to review GPCR agonists, which are able to activate the receptor to an even higher degree than the endogenous agonist does, that is, ‘superagonists’ (Engström et al., 2005; Smith et al., 2011; Schrage et al., 2013), and to highlight the potential exploitation of superagonists for both basic GPCR research and therapeutic use. Of note, application of the term ‘superagonist’ in biomedical data bases and search engines reveals that this designation is most commonly used in immunological research where cell signalling is mainly mediated by kinase‐associated receptors or receptor kinases. In the following section, we will first focus on the term superagonism in immunology regarding similarities and differences compared with GPCR superagonism. We will then report on examples of GPCR superagonism and difficulties in the identification of compounds exceeding the endogenous agonist in efficacy. We would point out that there are also several examples of compounds with supraphysiological efficacy at ligand‐gated ion channels (Carlier et al., 2002; Thompson and Lummis, 2013).
Consequently, we propose that superagonism is evident not only at GPCRs but may also be applicable to all main receptor classes, which mediate physiological actions in response to endogenous agonists.
Up until now, the term ‘superagonist’ has not been addressed in the NC‐IUPHAR nomenclature system (Neubig et al., 2003). In the literature, the term is also used for agonists that are endowed with higher binding affinity compared with the endogenous activator. For example, ‘superagonists’ of the gonadotropin‐releasing hormone receptor, such as the synthetic nonapeptide buserelin, have usually higher biological potency but not necessarily higher efficacy than the endogenous agonist gonadotropin‐releasing hormone (Loumaye et al., 1982; Padula, 2005; Leaños‐Miranda et al., 2006). However, more‐than‐physiological receptor binding does not necessarily accompany more‐than‐physiological efficacy for receptor activation.
Superagonism in immunology
The immune system is crucial for fighting off acute infections and mediating long‐term protection from various pathogens and malignancies. However, if it turns against endogenous or innocuous antigens, it can also cause harm. T cells play a central role in both protective and deleterious immune responses. They can be subdivided in pro‐inflammatory (effector T cells (Teffs)) or suppressive T cells (regulatory T cells (Tregs)). In this regard, compounds, which hyperstimulate either immune‐inhibitory Tregs or pro‐inflammatory Teffs, may be of great therapeutic value for the treatment of autoimmune disorders or anti‐cancer therapy respectively. There are several examples for immunological ‘superagonists’ that, in comparison with the endogenous ligands, display enhanced biological activity for triggering immune cell activation and proliferation (Chen et al., 2000; Beyersdorf et al., 2005; Rubinstein et al., 2006; Abdul‐Alim et al., 2010).
The best established example of immunological superagonists are anti‐inflammatory CD28‐binding antibodies, which were discovered in 1997 (Tacke et al., 1997). In contrast to the natural CD28‐ligands, CD80 or CD86, CD28‐binding antibodies can fully activate anti‐inflammatory Tregs without the need of T cell receptor activation, which is usually required (Lenschow et al., 1996; Tacke et al., 1997; Janeway et al., 2001) (Figure 2A). Binding of the endogenous ligands to CD28 does not elicit a biological response (Figure 2B), suggesting that similar to GPCR superagonism, the conformation of CD28 stabilized by superagonist CD28 antibodies is distinct from that stabilized by CD80 or CD86.
Figure 2.
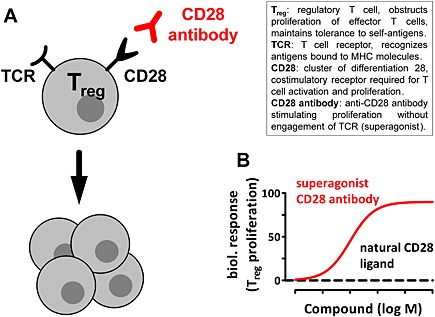
Superagonists of CD28 induce activation and proliferation of regulatory T cells. (A) Antibodies targeting the T cell surface receptor CD28 can induce activation and proliferation of regulatory T cells (Tregs) without the need of T cell receptor activation. (B) Hypothetical concentration‐response curves illustrating CD28‐mediated induction of Treg proliferation by the natural CD28 ligands CD80 or CD86 (black dashed line) or a superagonistic CD28 antibody (red).
Examples of immunological pro‐inflammatory superagonists are highly active cytokines complexed to a soluble subunit of their receptor (Fischer et al., 1997; Pflanz et al., 1999; Rubinstein et al., 2006), which trigger increased activation of target cells, such as T cells and natural killer cells, and altered peptide ligands stimulating activation of cytotoxic T cells to a greater extent than the natural peptide (Bakker et al., 1997; Valmori et al., 1998; Chen et al., 2000).
Taken together, distinct classes of immunological superagonists act through diverse mechanisms resulting in a more‐than‐physiological (‘superagonist’) biological activity, that is, enhanced cell growth and proliferation of a subset of immune cells. There are obvious similarities to ‘GPCR superagonism’, which is defined as receptor activation with a superior efficacy in comparison with that of the endogenous agonist (Smith et al., 2011). However, biological activity of immune‐stimulating compounds is assessed by cell growth and proliferation in a multi‐layered interplay between different immunological cell types. Under these complex conditions, compounds with supraphysiological efficacy for activation of a certain receptor are difficult to identify. This is because greater biological activity might be due to an increased affinity for receptor binding or functional selectivity for a certain signalling pathway and, therefore, is not necessarily due to higher efficacy for receptor activation on the receptor protein level.
Class A GPCR superagonists
As discussed earlier, ligands that target GPCRs are usually classified according to their efficacy, that is, their ability to elicit a receptor‐mediated physiological or pharmacological response (Stephenson, 1956; Kenakin, 1995a; Smith et al., 2011). By intuition, the interaction of the endogenous transmitter with its cognate receptor might be assumed as efficacious as possible because it is the consequence of a strong evolutionary force (Langmead and Christopoulos, 2013). This would preclude a more‐than‐physiological ligand efficacy. However, crystallographic efforts with GPCRs in their active state show that agonist binding alone is not sufficient to stabilize a fully active receptor conformation (Rosenbaum et al., 2011) and that binding of a G protein or a G protein‐mimicking nanobody is at least as important to capture the protein in a fully active state (Rasmussen et al., 2011b; Rasmussen et al., 2011a). Moreover, recent studies show that GPCRs exist in ensembles of conformations and that agonists stabilize only a subset of possible conformational states (Deupi and Kobilka, 2007; Kobilka and Deupi, 2007; Kenakin, 2013). Therefore, diverse agonists of a given receptor protein may stabilize different subsets of conformations with distinct efficacies for the activation of specific signalling pathways (Kenakin, 2013). This is also supported by NMR spectroscopy studies showing that even ‘strong’ agonists alone populate a set of conformations similar to the more uniform and fully active conformation generated by a highly efficacious agonist plus a G protein mimicking nanobody (Nygaard et al., 2013). Consequently, at least from a theoretical point of view, supraphysiological efficacy of compounds stabilizing a more uniform conformation than the endogenous agonist should in principle be feasible. Indeed, in the largest and most ‘druggable’ class of GPCRs (the rhodopsin‐like class or class A (Fredriksson et al., 2003; Lagerström and Schiöth, 2008)), a few synthetic compounds have been described that are endowed with greater intrinsic efficacy than the endogenous ligand (Table 1).
Table 1.
Examples of class A (Rhodopsin‐like) GPCR superagonists
| Endogenous ligand | ‘Superagonist’ | GPCR subtype | Experimental evidence for superagonism |
|---|---|---|---|
| SRIF‐14: Ala‐Gly‐Cys‐Lys‐Asn‐Phe‐Phe‐Trp‐Lys‐Thr‐Phe‐Thr‐Ser‐Cys |
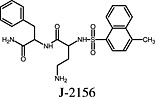 J‐2156 J‐2156 |
Somatostatin sst4 receptor | J‐2156 is a superagonist at the human sst4 receptor as shown by [35S]GTPγS binding assays, which revealed J‐2156 to generate Emax values that were two–three times larger than the Emax values of the two endogenous peptides SRIF‐14 and SRIF‐28 (Engström et al., 2005). |
| SRIF‐28: Ser‐Ala‐Asn‐Ser‐Asn‐Pro‐Ala‐Met‐Ala‐Pro‐Arg‐Glu‐Arg‐Lys‐Ala‐Gly‐Cys‐Lys‐Asn‐Phe‐Phe‐Trp‐Lys‐Thr‐Phe‐Thr‐Ser‐Cys | |||
| Ghrelin: Gly‐Ser‐Ser(n‐octanoyl)‐Phe‐Leu‐Ser‐Pro‐Glu‐His‐Gln‐Arg‐Val‐Gln‐Gln‐Arg‐Lys‐Glu‐Ser‐Lys‐Lys‐Pro‐Pro‐Ala‐Lys‐Leu‐Gln‐Pro‐Arg |
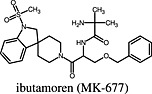 Ibutamoren (MK‐677) Ibutamoren (MK‐677) |
Ghrelin receptor | Ibutamoren (MK‐677) is a superagonist at the ghrelin receptor as it displayed higher Emax values for β‐arrestin activation (discovered in BRET assays) (Holst et al., 2005), for SRE‐mediated transcription assays (Holst et al., 2005), and for the activation of Gαo1 (Bennett et al., 2009) in [35S]GTPγS assays. |
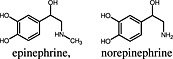 Adrenaline, noradrenaline Adrenaline, noradrenaline |
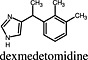 Dexmedetomidine Dexmedetomidine |
α2A‐adrenoceptor | Dexmedetomidine induced higher Emax values for α2A adrenoceptor‐mediated MAP kinase activation than adrenaline (Tan et al., 2002). |
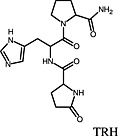 TRH TRH |
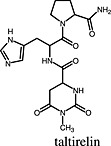 Taltirelin Taltirelin |
Thyrotropin‐releasing hormone TRH1 receptor | Taltirelin is a superagonist at the human TRH1 receptor because it increased cellular IP1 to Emax = 180% of that induced by TRH (Thirunarayanan et al., 2012). |
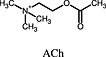 ACh ACh |
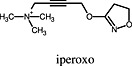 Iperoxo Iperoxo |
Muscarinic M2 cholinoceptor | Iperoxo is a superagonist at muscarinic M2 receptors for Gαi and Gαs –mediated DMR as it displayed higher operational efficacy (τ) (Schrage et al., 2013). Iperoxo induced more pronounced intracellular loop rearrangement than the endogenous agonist ACh (Bock et al., 2012). |
SRIF‐14, SRIF‐28 are the endogenous forms of somatostatin and ligands for the somatostatin receptor
One example is the somatostatin receptor superagonist J‐2156 [(1′S,2S)‐4‐amino‐N‐(1′‐carbamoyl‐2′‐phenylethyl)‐2‐(4″‐methyl‐1″‐naphthalenesulfonylamino)butanamide], which binds to the human somatostatin sst4 receptor with subnanomolar affinity (Engström et al., 2005). J‐2156 induced sst4 receptor‐mediated [35S]GTPγS binding in CHO membranes with a signal two–three times as large as the response induced by the two endogenous peptides somatostatin SRIF‐28 and SRIF‐14. In contrast, maximal inhibition of intracellular cAMP levels, a cellular signalling event that occurs downstream of GPCR/G protein activation, was comparable between J‐2156 and the two natural peptides (Engström et al., 2005). This example illustrates that the assessment of ligand efficacy distal from receptor activation may be difficult because the signal can be substantially amplified inside the cell (e.g. Milligan, 2003; Colabufo et al., 2007). Such signal amplification after activation of a Gs‐coupled receptor is illustrated in Figure 3. One active receptor protein may lead to the activation of several intracellular kinases, which finally shape the cellular response. Therefore, quantification of agonist efficacy by Emax values (i.e. the asymptote of the concentration‐effect curve) is likely to obscure superagonism in an assay system characterized by strong signal amplification.
Figure 3.
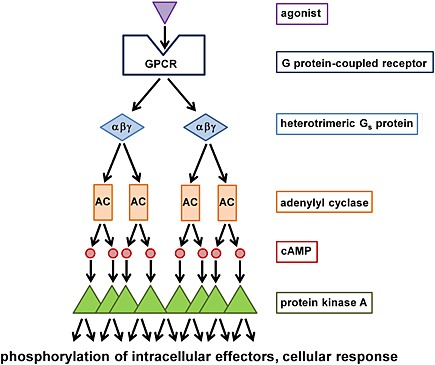
GPCR signalling may undergo substantial cellular amplification. As an example, the diagram shows an intracellular signalling cascade initiated by the activation of a Gs‐linked GPCR. Upon activation, the receptor may activate more than one heterotrimeric G protein, which can activate more than one membrane‐bound adenylyl cyclase isoforms (AC). ACs, in turn, catalyse the generation of several molecules of cAMP. cAMP binds and activates kinases such as protein kinase A, which are responsible for the phosphorylation of various intracellular effector proteins finally shaping the ultimate cellular response.
The ghrelin receptor activates a plethora of signalling pathways, and several compounds that can engage one or the other pathway with supraphysiological efficacy have been described (Holst et al., 2005; Bennett et al., 2009). One of these compounds is ibutamoren (MK‐677), which acts as full agonist regarding Ca++ mobilization and inositol phosphate accumulation but displayed superagonism in ‘serum‐responsive element’ (SRE)‐mediated transcription assays (Holst et al., 2005), β‐arrestin recruitment (Holst et al., 2005) and Gαo1 activation (Bennett et al., 2009). This example shows that superagonism does not necessarily occur in all possible signalling pathways that can be engaged by a certain receptor protein. This is also the case for dexmedetomidine, an α2A‐adrenoceptor agonist, which had higher Emax value for MAP kinase activation than adrenaline but was a partial agonist in [35S]GTPγS binding assays performed with HEK293‐α2A membranes (Tan et al., 2002).
In a study performed by Thirunarayanan et al., the pharmacology of taltirelin, the only analogue of the thyrotropin‐releasing hormone (TRH) approved for use in humans, was investigated (Thirunarayanan et al., 2012). Taltirelin is used in Japan for the treatment of adult spinal muscular atrophy. In comparison with TRH, taltirelin had a lower affinity for the human TRH1 receptor in whole cell binding experiments and also a lower potency, while showing the same Emax for the induction of Ca++ release. A first indicator of supraphysiological efficacy was the higher affinity/potency ratio of taltirelin compared with TRH, which has been used before to estimate efficacy and identify superagonists (Engel et al., 2006). When TRH1 receptor‐induced inositol monophosphate (IP1) accumulation, a cellular event upstream of receptor‐mediated Ca++ release, was quantified, taltirelin stimulated an increase in IP1 production that was 180% of that stimulated by TRH, identifying taltirelin as a superagonist. Similar to the study by Engström et al. (2005), the study by Thirunarayanan et al. demonstrates a substance's superagonism by comparison of Emax values in functional assays detecting events close to the receptor. Of note, in both studies, superagonism was masked when more distal cellular events (in this case Ca++ release) were used to quantify receptor activation (Thirunarayanan et al., 2012).
Challenges in the detection of superagonism: examples from muscarinic receptors
Similar to the receptors discussed earlier, muscarinic ACh receptors belong to the rhodopsin‐like or class A GPCRs (e.g. Kruse et al., 2014). Two structurally similar compounds, iperoxo (Schrage et al., 2013) and oxotremorine M (Oxo M) (Mistry et al., 2005), have been identified as agonists with supraphysiological efficacy at the closely related muscarinic M2 and M4 receptors respectively. Both receptors preferentially activate inhibitory Gi proteins (e.g. Caulfield, 1993; Wess et al., 1997) but for both, M2 (Michal et al., 2001; Bock et al., 2012; Schrage et al., 2013) and M4 receptors (Dittman et al., 1994; Mistry et al., 2005), the activation of stimulatory Gs proteins has also been reported. Of note, Oxo M and iperoxo (Table 1) are bulkier than the endogenous agonist ACh and may therefore form more interactions with the receptor, which in turn leads to higher efficacy (Schrage et al., 2013).
In the study performed by Mistry et al. (2005), the classical muscarinic agonist Oxo M and methacholine (used as a surrogate for the endogenous agonist ACh in this study) shared the same maximum for M2 and M4 receptor‐mediated Gi and Gs activation. The relative efficacy values (Erel), according to the method of Ehlert (1985), quantified the coupling of receptor occupancy to adenylyl cyclase functional responses and revealed that, for both Gi and Gs pathways, Oxo M was a full agonist at M2 but a superagonist at M4 receptors, relative to methacholine. However, although highly similar, methacholine is structurally distinct from the endogenous agonist ACh, the latter not being included in this study (Mistry et al., 2005). Consequently, the greater efficacy of Oxo M compared with that of ACh remains to be demonstrated.
Recently, the muscarinic agonist iperoxo, a derivative of Oxo M, which binds to muscarinic receptors with outstanding affinity (Dallanoce et al., 1999; Antony et al., 2009; Schrage et al., 2013; Schrage et al., 2014) and has been extensively exploited as a building block for muscarinic ortho‐allosteric hybrid compounds (Disingrini et al., 2006; Antony et al., 2009; Bock et al., 2012; Bock et al., 2014; Matera et al., 2014; Chen et al., 2015), was identified as a superagonist at M2 receptors. In the study by Schrage et al., (2013), iperoxo was a highly potent agonist for the activation of M2 receptors stably expressed in CHO cells in two assays. Whole cell label‐free dynamic mass redistribution (DMR) revealed highly potent activation of Gi and Gs mediated pathways. Likewise, [35S]GTPγS binding experiments performed with CHO‐M2 membranes revealed that the potency for Gi activation by iperoxo was 100‐fold higher than that of ACh. However, the maximum effect of iperoxo did not differ from the effects of ACh and Oxo M. As both assays may be subject to substantial signal amplification, the operational model of agonism was employed to analyse functional data from DMR and [35S]GTPγS experiments. The operational efficacy parameter τ incorporates efficacy, receptor density and coupling efficiency within a system (Black and Leff, 1983; Rajagopal et al., 2011; Kenakin and Christopoulos, 2013). The value of τ was significantly greater for iperoxo than for ACh and Oxo M, both for the Gi and the Gs pathways. Therefore, iperoxo was classified as a superagonist. However, like other analytical methods, the operational model of agonism may lack a certain robustness with which the model can be fitted to the data (Langmead and Christopoulos, 2013), and this is especially true for highly efficacious agonists (Rajagopal, 2013). To improve robustness of operational efficacies, the apparent agonist equilibrium binding constants KA, derived from whole cell radioligand binding experiments, were included in the analysis. However, binding affinity may not reflect the functional affinity within the trimeric agonist–receptor–transducer complex (Kenakin et al., 2012; Kenakin and Christopoulos, 2013; Langmead and Christopoulos, 2013) and, therefore, may insert a certain ‘system bias’ into the estimation of operational efficacies. Nevertheless, this approach suggested that iperoxo's operational efficacy was greater than that of ACh and Oxo M. (Schrage et al., 2013). In two other studies, in which functional data of iperoxo and ACh were fitted to the operational model of agonism without fixing KA values, iperoxo and ACh showed equal operational efficacy in [35S]GTPγS and cAMP accumulation assays (Bock et al., 2012), but iperoxo had greater operational efficacy compared with ACh in DMR experiments (Bock et al., 2014). To test further for superagonism, the M2 receptor density was reduced in order to eliminate spare receptors, by alkylation with the irreversible antagonist phenoxybenzamine (Furchgott, 1966). Under these conditions, the maximum DMR response of ACh was more compromised by receptor alkylation than the Emax of iperoxo (Schrage et al., 2013). However, alkylation experiments may be difficult as they do not reflect equilibrium conditions and are sensitive to the time of measurement and ground state, for example, receptor number, of the system. Nevertheless, inactivation methods (Furchgott, 1966; Furchgott and Bursztyn, 1967) are most useful to estimate agonist affinity and relative efficacy and may be applied for both partial and highly efficacious agonists (Christopoulos and El‐Fakahany, 1999).
Finally, when M2 receptor activation was measured directly at the receptor level by the quantification of intracellular loop rearrangement in Förster resonance energy transfer assays, iperoxo displayed a greater agonist‐induced change of receptor conformation than ACh (Bock et al., 2012). Taken together, findings from various experimental approaches suggest that iperoxo has supraphysiological efficacy at the muscarinic M2 receptor subtype. Moreover, iperoxo provides a paradigm to demonstrate the usefulness of superagonism in experimental pharmacology: the compound served to crystallize the muscarinic M2 receptor in its active state (Kruse et al., 2013). In addition, the tritiated form of iperoxo is the first radioagonist to probe all five muscarinic receptor subtypes (Schrage et al., 2014).
In summary, operational approaches to quantify agonist coupling efficiency are useful to probe for GPCR superagonism. However, operational efficacies should be considered with caution regarding data robustness. To overcome the lack of robustness, one might be tempted to reduce flexibility from the operational model by including the experimentally measured, apparent agonist equilibrium binding constant KA. However, this may insert system bias to the data. If researchers aim at the identification of superagonism, it is useful to apply additional routes to check for supraphysiological efficacy such as receptor alkylation experiments, estimation of receptor activation close to the receptor level or the direct probing of agonist‐induced conformational transitions.
Superagonism at other receptor classes
There are several examples (Carlier et al., 2002; Ihara et al., 2004; Brown et al., 2006; Thompson and Lummis, 2013) for supraphysiological agonist efficacy at ligand‐gated ion channels. The study of ion channel activation by ion fluxes may allow a rather direct approach to measure agonism at the receptor level without complicating signal amplification. GABA is the major inhibitory transmitter in the vertebrate central nervous system and is an agonist at three different GABA receptor subtypes. In 2002, Carlier et al. identified a superagonist at the GABAA receptor, a ligand‐gated chloride ion channel. This compound, a GABA amide dimer (compound 5b) was 33‐fold less potent than GABA but induced a chloride uptake that was 49% higher than that achieved by GABA (Carlier et al., 2002). Another example is the 5‐HT3 receptor at which m‐chlorophenylbiguanide is a superagonist. In 5‐HT3AB receptor heteromers it produced a response 2.6‐fold higher than that of 5‐HT (Thompson and Lummis, 2013).
There are also reports that assign superagonism to other targets such as receptor tyrosine kinases (Puddicombe et al., 1996; Thomas et al., 2008) or receptors with transcriptional activity (Hourai et al., 2008). However, the functional readouts for these receptors are often distal of receptor activation, that is, cell growth/proliferation and gene expression. Such ‘down‐stream readouts’ may be subject to amplification, cross‐regulation of pathways or signal convergence. Therefore, it is not surprising that many of these studies are identifying ligands with higher affinity, but not necessarily higher efficacy, than the endogenous ligand.
Conclusion
This review aims to give an overview of superagonism in different receptor classes, that is, GPCRs, catalytic receptors associated with kinases, ion channels and other target receptors, with a focus on GPCR superagonism. Superagonists may have great value as tools in experimental pharmacology or may serve to overcome loss‐of‐function receptor mutants, stimulate inhibitory receptors or induce negative feedback mechanisms.
Although not yet officially recognized by the NC‐IUPHAR (Neubig et al., 2003), there are several examples for agonists with a more‐than‐physiological efficacy especially at GPCRs (Tan et al., 2002; Engström et al., 2005; Holst et al., 2005; Mistry et al., 2005; Engel et al., 2006; Bennett et al., 2009; Schrage et al., 2013). These examples show that the interaction of a receptor with its endogenous ligand was not necessarily designed by nature to be as efficacious as possible. More likely, the evolutionary forces drove receptors and endogenous ligands to a well‐working machinery that is efficient but leaves substantial flexibility in biological networks to fine‐tune responses. Therefore, we would predict that compounds with supraphysiological efficacy might be generated for several members of the GPCR family. Moreover, superagonists have also been identified for ligand‐gated ion channels (Carlier et al., 2002; Ihara et al., 2004; Brown et al., 2006; Thompson and Lummis, 2013). This, and the frequent identification of superagonists for catalytic, kinase‐associated receptors in immunology (Chen et al., 2000; Beyersdorf et al., 2005; Rubinstein et al., 2006; Abdul‐Alim et al., 2010), implies that ligands with supraphysiological efficacy can be achieved for several, if not all, receptor classes.
Conflict of interest
None.
Acknowledgement
A. D. M. was supported by the research training group 1873, Deutsche Forschungsgemeinschaft (DFG).
Schrage, R. , De Min, A. , Hochheiser, K. , Kostenis, E. , and Mohr, K. (2016) Superagonism at G protein‐coupled receptors and beyond. British Journal of Pharmacology, 173: 3018–3027. doi: 10.1111/bph.13278.
References
- Abdul‐Alim CS, Li Y, Yee C (2010). Conditional superagonist CTL ligands for the promotion of tumor‐specific CTL responses. J Immunol 184: 6514–6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013a). The Concise Guide to PHARMACOLOGY 2013/14: G Protein‐Coupled Receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: Ligand‐Gated Ion Channels. Br J Pharmacol 170: 1582–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013c). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony J et al. (2009). Dualsteric GPCR targeting: a novel route to binding and signaling pathway selectivity. FASEB J 23: 442–450. [DOI] [PubMed] [Google Scholar]
- Ariens EJ, van Rossum J, Koopman PC (1960). Receptor reserve and threshold phenomena. I. Theory and experiments with autonomic drugs tested on isolated organs. Arch Int Pharmacodyn Ther (Archives internationales de pharmacodynamie et de thérapie) 127: 459–478. [PubMed] [Google Scholar]
- Bakker AB et al. (1997). Analogues of CTL epitopes with improved MHC class‐I binding capacity elicit anti‐melanoma CTL recognizing the wild‐type epitope. Int J Cancer 70: 302–309. [DOI] [PubMed] [Google Scholar]
- Barlow R, Blake JF (1989). Hill coefficients and the logistic equation. Trends Pharmacol Sci 10: 440–441. [DOI] [PubMed] [Google Scholar]
- Bennett KA, Langmead CJ, Wise A, Milligan G (2009). Growth hormone secretagogues and growth hormone releasing peptides act as orthosteric super‐agonists but not allosteric regulators for activation of the G protein Galpha(o1) by the ghrelin receptor. Mol Pharmacol 76: 802–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyersdorf N, Hanke T, Kerkau T, Hünig T (2005). Superagonistic anti‐CD28 antibodies: potent activators of regulatory T cells for the therapy of autoimmune diseases. Ann Rheum Dis 64: iv91–iv95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjarnadóttir TK, Gloriam DE, Hellstrand SH, Kristiansson H, Fredriksson R, Schiöth HB (2006). Comprehensive repertoire and phylogenetic analysis of the G protein‐coupled receptors in human and mouse. Genomics 88: 263–273. [DOI] [PubMed] [Google Scholar]
- Black JW, Leff P (1983). Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci 220: 141–162. [DOI] [PubMed] [Google Scholar]
- Bock A et al. (2012). The allosteric vestibule of a seven transmembrane helical receptor controls G‐protein coupling. Nat Commun 3: 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock A et al. (2014). Dynamic ligand binding dictates partial agonism at a G protein‐coupled receptor. Nat Chem Biol 10: 18–20. [DOI] [PubMed] [Google Scholar]
- Brown LA, Ihara M, Buckingham SD, Matsuda K, Sattelle DB (2006). Neonicotinoid insecticides display partial and super agonist actions on native insect nicotinic acetylcholine receptors. J Neurochem 99: 608–615. [DOI] [PubMed] [Google Scholar]
- Carlier PR, Chow ES, Barlow RL, Bloomquist JR (2002). Discovery of non‐zwitterionic GABA(A) receptor full agonists and a superagonist. Bioorg Med Chem Lett 12: 1985–1988. [DOI] [PubMed] [Google Scholar]
- Caulfield MP (1993). Muscarinic receptors – characterization, coupling and function. Pharmacol Ther 58: 319–379. [DOI] [PubMed] [Google Scholar]
- Chen JL et al. (2000). Identification of NY‐ESO‐1 peptide analogues capable of improved stimulation of tumor‐reactive CTL. J Immunol 165: 948–955. [DOI] [PubMed] [Google Scholar]
- Chen X et al. (2015). Rational design of partial agonists for the muscarinic m1 acetylcholine receptor. J Med Chem 58: 560–576. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, El‐Fakahany EE (1999). Qualitative and quantitative assessment of relative agonist efficacy. Biochem Pharmacol 58: 735–748. [DOI] [PubMed] [Google Scholar]
- Colabufo NA, Perrone GM, Contino M, Berardi F, Perrone R (2007). Receptor–drug interaction: europium employment for studying the biochemical pathway of G‐protein‐coupled receptor activation. Met Based Drugs 2007: 12635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallanoce C et al. (1999). Synthesis and functional characterization of novel derivatives related to oxotremorine and oxotremorine‐M. Bioorg Med Chem 7: 1539–1547. [DOI] [PubMed] [Google Scholar]
- Deupi X, Kobilka BK (2007). Activation of G protein‐coupled receptors. Adv Protein Chem 74: 137–166. [DOI] [PubMed] [Google Scholar]
- Deupi X, Kobilka BK (2010). Energy landscapes as a tool to integrate GPCR structure, dynamics, and function. Physiology (Bethesda) 25: 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disingrini T et al. (2006). Design, synthesis, and action of oxotremorine‐related hybrid‐type allosteric modulators of muscarinic acetylcholine receptors. J Med Chem 49: 366–372. [DOI] [PubMed] [Google Scholar]
- Dittman AH et al. (1994). A novel mechanism for coupling of M4 muscarinic acetylcholine receptors to calmodulin‐sensitive adenylyl cyclases: crossover from G protein‐coupled inhibition to stimulation. Biochemistry 33: 943–951. [DOI] [PubMed] [Google Scholar]
- Dohlman HG (2015). Thematic minireview series: cell biology of G protein signaling. J Biol Chem 290: 6679–6680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlert FJ (1985). The relationship between muscarinic receptor occupancy and adenylate cyclase inhibition in the rabbit myocardium. Mol Pharmacol 28: 410–421. [PubMed] [Google Scholar]
- Engel S et al. (2006). Low affinity analogs of thyrotropin‐releasing hormone are super‐agonists. J Biol Chem 281: 13103–13109. [DOI] [PubMed] [Google Scholar]
- Engström M, Tomperi J, El‐Darwish K, Ahman M, Savola J, Wurster S (2005). Superagonism at the human somatostatin receptor subtype 4. J Pharmacol Exp Ther 312: 332–338. [DOI] [PubMed] [Google Scholar]
- Fischer M et al. (1997). I. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat Biotechnol 15: 142–145. [DOI] [PubMed] [Google Scholar]
- Foord SM et al. (2005). International Union of Pharmacology. XLVI. G protein‐coupled receptor list. Pharmacol Rev 57: 279–288. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Lagerström MC, Lundin L, Schiöth HB (2003). The G‐protein‐coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 63: 1256–1272. [DOI] [PubMed] [Google Scholar]
- Furchgott RF (1966). The use of B‐haloalkylamines in the differentiation of receptors and in the determination of dissociation constants of receptor‐agonist complexes In: Harper NJ, Simmonds AB. (eds). Advances in Drug Research, Vol. 3 Academic Press: New York, pp. 21–55. [Google Scholar]
- Furchgott RF, Bursztyn P (1967). Comparison of dissociation constants and of relative efficacies of selected agonists acting on parasympathetic receptors. Ann NY Acad Sci 144: 882–899. [Google Scholar]
- Garland SL (2013). Are GPCRs still a source of new targets? J Biomol Screen 18: 947–966. [DOI] [PubMed] [Google Scholar]
- Holst B, Brandt E, Bach A, Heding A, Schwartz TW (2005). Nonpeptide and peptide growth hormone secretagogues act both as ghrelin receptor agonist and as positive or negative allosteric modulators of ghrelin signaling. Mol Endocrinol 19: 2400–2411. [DOI] [PubMed] [Google Scholar]
- Hourai S et al. (2008). Structure‐based design of a superagonist ligand for the vitamin D nuclear receptor. Chem Biol 15: 383–392. [DOI] [PubMed] [Google Scholar]
- Ihara M, Matsuda K, Shimomura M, Sattelle DB, Komai K (2004). Super agonist actions of clothianidin and related compounds on the SAD beta 2 nicotinic acetylcholine receptor expressed in Xenopus laevis oocytes. Biosci Biotechnol Biochem 68: 761–763. [DOI] [PubMed] [Google Scholar]
- Janeway CA, Traver P, Walport M, Shlomchik MJ (2001). Immunobiology. The Immune System in Health and Disease. Garland Pub: New York. [Google Scholar]
- Kenakin T (1985). The quantification of relative efficacy of agonists. J Pharmacol Methods (Journal of pharmacological methods) 13: 281–308. [DOI] [PubMed] [Google Scholar]
- Kenakin T (1995a). Agonist‐receptor efficacy. I: mechanisms of efficacy and receptor promiscuity. Trends Pharmacol Sci 16: 188–192. [DOI] [PubMed] [Google Scholar]
- Kenakin T (1995b). Agonist‐receptor efficacy. II. Agonist trafficking of receptor signals. Trends Pharmacol Sci 16: 232–238. [DOI] [PubMed] [Google Scholar]
- Kenakin T (2002). Efficacy at G‐protein‐coupled receptors. Nat Rev Drug Discov 1: 103–110. [DOI] [PubMed] [Google Scholar]
- Kenakin T (2013). New concepts in pharmacological efficacy at 7TM receptors: IUPHAR review 2. B J Pharmacol 168: 554–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Christopoulos A (2013). Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov 12: 205–216. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Watson C, Muniz‐Medina V, Christopoulos A, Novick S (2012). A simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci 3: 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka BK, Deupi X (2007). Conformational complexity of G‐protein‐coupled receptors. Trends Pharmacol Sci 28: 397–406. [DOI] [PubMed] [Google Scholar]
- Kolakowski LF (1994). GCRDb: a G‐protein‐coupled receptor database. Recept Channels 2: 1–7. [PubMed] [Google Scholar]
- Kruse AC et al. (2013). Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504: 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Kobilka BK, Gautam D, Sexton PM, Christopoulos A, Wess J (2014). Muscarinic acetylcholine receptors: novel opportunities for drug development. Nat Rev Drug Discov 13: 549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerström MC, Schiöth HB (2008). Structural diversity of G protein‐coupled receptors and significance for drug discovery. Nat Rev Drug Discov 7: 339–357. [DOI] [PubMed] [Google Scholar]
- Langmead CJ, Christopoulos A (2013). Supra‐physiological efficacy at GPCRs: superstition or super agonists? B J Pharmacol 169: 353–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaños‐Miranda A, Ulloa‐Aguirre A, Cervini LA, Janovick JA, Rivier J, Conn PM (2006). Identification of new gonadotrophin‐releasing hormone partial agonists. J Endocrinol 189: 509–517. [DOI] [PubMed] [Google Scholar]
- Lenschow DJ, Walunas TL, Bluestone JA (1996). CD28/B7 system of T cell costimulation. Annu Rev Immunol 14: 233–258. [DOI] [PubMed] [Google Scholar]
- Loumaye E, Naor Z, Catt KJ (1982). Binding affinity and biological activity of gonadotropin releasing hormone agonists in isolated pituitary cells. Endocrinology 111: 730–736. [DOI] [PubMed] [Google Scholar]
- Matera C et al. (2014). Bis(ammonio)alkane‐type agonists of muscarinic acetylcholine receptors: synthesis, in vitro functional characterization, and in vivo evaluation of their analgesic activity. Eur J Med Chem 75: 222–232. [DOI] [PubMed] [Google Scholar]
- Michal P, Lysíková M, Tucek S (2001). Dual effects of muscarinic M(2) acetylcholine receptors on the synthesis of cyclic AMP in CHO cells: dependence on time, receptor density and receptor agonists. B J Pharmacol 132: 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar RP, Newton CL (2010). The year in G protein‐coupled receptor research. Mol Endocrinol 24: 261–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G (2003). Principles: extending the utility of [35S]GTP gamma S binding assays. Trends Pharmacol Sci 24: 87–90. [DOI] [PubMed] [Google Scholar]
- Milligan G, Kostenis E (2006). Heterotrimeric G‐proteins: a short history. B J Pharmacol 147: S46–S55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry R, Dowling MR, Challiss RA (2005). An investigation of whether agonist‐selective receptor conformations occur with respect to M2 and M4 muscarinic acetylcholine receptor signalling via Gi/o and Gs proteins. B J Pharmacol 144: 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubig RR, Spedding M, Kenakin T, Christopoulos A (2003). International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on terms and symbols in quantitative pharmacology. Pharmacol Rev 55: 597–606. [DOI] [PubMed] [Google Scholar]
- Nickerson M (1956). Receptor occupancy and tissue response. Nature 178: 697–698. [DOI] [PubMed] [Google Scholar]
- Nygaard R et al. (2013). The dynamic process of β(2)‐adrenergic receptor activation. Cell 152: 532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham WM, Hamm HE (2008). Heterotrimeric G protein activation by G‐protein‐coupled receptors. Nat Rev Mol Cell Biol 9: 60–71. [DOI] [PubMed] [Google Scholar]
- Overington JP, Al‐Lazikani B, Hopkins AL (2006). How many drug targets are there? Nat Rev Drug Discov 5: 993–996. [DOI] [PubMed] [Google Scholar]
- Padula AM (2005). GnRH analogues – agonists and antagonists. Anim Reprod Sci 88: 115–126. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 42: D1098–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflanz S et al. (1999). A fusion protein of interleukin‐11 and soluble interleukin‐11 receptor acts as a superagonist on cells expressing gp130. FEBS Lett 450: 117–122. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Lefkowitz RJ (2001). Classical and new roles of beta‐arrestins in the regulation of G‐protein‐coupled receptors. Nat Rev Neurosci 2: 727–733. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ (2002). Seven‐transmembrane receptors. Nat Rev Mol Cell Biol 3: 639–650. [DOI] [PubMed] [Google Scholar]
- Puddicombe SM, Wood L, Chamberlin SG, Davies DE (1996). The interaction of an epidermal growth factor/transforming growth factor alpha tail chimera with the human epidermal growth factor receptor reveals unexpected complexities. J Biol Chem 271: 30392–30397. [DOI] [PubMed] [Google Scholar]
- Rajagopal S et al. (2011). Quantifying ligand bias at seven‐transmembrane receptors. Mol Pharmacol 80: 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S (2013). Quantifying biased agonism: understanding the links between affinity and efficacy. Nat Rev Drug Discov 12: 483. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG et al. (2011a). Crystal structure of the β2 adrenergic receptor‐Gs protein complex. Nature 477: 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG et al. (2011b). Structure of a nanobody‐stabilized active state of the β(2) adrenoceptor. Nature 469: 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM et al. (2011). Structure and function of an irreversible agonist‐β(2) adrenoceptor complex. Nature 469: 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Rasmussen SG, Kobilka BK (2009). The structure and function of G‐protein‐coupled receptors. Nature 459: 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein MP et al. (2006). Converting IL‐15 to a superagonist by binding to soluble IL‐15R{alpha}. Proc Natl Acad Sci U S A 103: 9166–9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrage R et al. (2013). Agonists with supraphysiological efficacy at the muscarinic M2 ACh receptor. B J Pharmacol 169: 357–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrage R et al. (2014). New insight into active muscarinic receptors with the novel radioagonist [3H]iperoxo. Biochem Pharmacol 90: 307–319. [DOI] [PubMed] [Google Scholar]
- Shonberg J, Lopez L, Scammells PJ, Christopoulos A, Capuano B, Lane JR (2014). Biased agonism at G protein‐coupled receptors: the promise and the challenges – a medicinal chemistry perspective. Med Res Rev 34: 1286–1330. [DOI] [PubMed] [Google Scholar]
- Smith NJ, Bennett KA, Milligan G (2011). When simple agonism is not enough: emerging modalities of GPCR ligands. Mol Cell Endocrinol 331: 241–247. [DOI] [PubMed] [Google Scholar]
- Stephenson RP (1956). A modification of receptor theory. Br J Pharmacol Chemother 11: 379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange PG (2008). Agonist binding, agonist affinity and agonist efficacy at G protein‐coupled receptors. B J Pharmacol 153: 1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacke M, Hanke G, Hanke T, Hünig T (1997). CD28‐mediated induction of proliferation in resting T cells in vitro and in vivo without engagement of the T cell receptor: evidence for functionally distinct forms of CD28. Eur J Immunol 27: 239–247. [DOI] [PubMed] [Google Scholar]
- Tan CM, Wilson MH, MacMillan LB, Kobilka BK, Limbird LE (2002). Heterozygous alpha 2A‐adrenergic receptor mice unveil unique therapeutic benefits of partial agonists. Proc Natl Acad Sci U S A 99: 12471–12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirunarayanan N, Raaka BM, Gershengorn MC (2012). Taltirelin is a superagonist at the human thyrotropin‐releasing hormone receptor. Front Endocrinol (Lausanne) 3: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas TP et al. (2008). Dendrimer‐epidermal growth factor conjugate displays superagonist activity. Biomacromolecules 9: 603–609. [DOI] [PubMed] [Google Scholar]
- Thompson AJ, Lummis SC (2013). A single channel mutation alters agonist efficacy at 5‐HT3A and 5‐HT3AB receptors. B J Pharmacol 170: 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valmori D et al. (1998). Enhanced generation of specific tumor‐reactive CTL in vitro by selected Melan‐A/MART‐1 immunodominant peptide analogues. J Immunol 160: 1750–1758. [PubMed] [Google Scholar]
- Wess J, Liu J, Blin N, Yun J, Lerche C, Kostenis E (1997). Structural basis of receptor/G protein coupling selectivity studied with muscarinic receptors as model systems. Life Sci 60: 1007–1014. [DOI] [PubMed] [Google Scholar]