

Abstract
The restraints in common usage today have been obtained based on small molecule X‐ray crystal structures available 25 years ago and recent reports have shown that the values of bond lengths and valence angles can be, in fact, significantly different from those stored in libraries, for example for the peptide bond or the histidine ring geometry. We showed that almost 50% of outliers found in protein validation reports released in the Protein Data Bank on 23 March 2016 come from geometry of guanidine groups in arginines. Therefore, structures of small molecules and atomic resolution protein crystal structures have been used to derive new target values for the geometry of this group. The most significant difference was found for NE‐CZ‐NH1 and NE‐CZ‐NH2 angles, showing that the guanidinium group is not symmetric. The NE‐CZ‐NH1 angle is larger, 121.5(10)˚, than NE‐CZ‐NH2, 119.2(10)˚, due to the repulsive interaction between NH1 and CD1 atom.
Keywords: X‐ray crystal structures, stereochemical restrains, structure validation, guanidinium geometry, arginine residues
Introduction
During recent submissions of several crystal structures to the PDB1 we noticed that most complaints in the validation reports concern the bond angles within the guanidine moieties of arginine residues. We have inspected PDB validation reports for all 211 crystal structures released in the PDB on March 23, 2016, and confirmed that the bond angles around the CZ atom of guanidines are the parameters most often disagreeing with the standard target values used during the validation process.
The guanidinium group is always treated as protonated, having the +1 charge delocalized over the four atoms, NE, CZ, NH1 and NH2, This description is based on the fact that its pKa value is 13.8,2 much higher than the physiological conditions and those used in crystallographic and NMR analyses. This moiety at the terminus of the arginine side chain is connected with the rest of the residue by the CD atom, and the whole group of five atoms is planar. The nitrogen atom NH1 is positioned cis with respect to the CD and the NH2 atom is in the trans orientation (Fig. 1). Practically all target values of bond lengths and angles, used in refinement and validation of protein structures in macromolecular crystallography and NMR are extracted from the compendia created by Engh and Huber in 19913 and 19994 (here abbreviated as EH91 and EH99). In both of them, the guanidinium group is represented as a symmetric moiety, with two identical angles, NE‐CZ‐NH1 and NE‐CZ‐NH2, equal to 120.0º and 120.3º in EH91 and EH99, respectively (Table 1). It is somewhat surprising that the NH1‐CZ‐NH2 angle in EH91 is defined as 119.7º, leading to non‐planarity around the CZ atom. In EH99 this angle is 119.4º, which conforms to the planarity of the whole guanidinium group. The target geometry values used in the CCP4 library5 and by the most popular refinement and display programs, REFMAC,6 phenix.refine,7 and COOT8 are adopted from the EH91 set, whereas the EH99 set is used in the PDB validation process.
Figure 1.
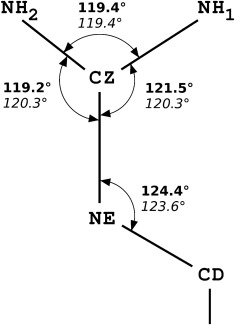
A scheme of the guanidine group in arginines. Numerical values shown correspond to valence angles of the guanidine group obtained from the new CSD survey (top, bold) in comparison to the EH99 values (bottom, italic).
Table 1.
Bond Lengths and Angles with Standard Uncertainties in the Guanidine Groups in Arginines
| N | CD‐NE | NE‐CZ | CZ‐NH1 | CZ‐NH2 | D‐E‐Z | E‐Z‐H1 | E‐Z‐H2 | H1‐Z‐H2 | |
|---|---|---|---|---|---|---|---|---|---|
| ARG resolution <1.0 Å, B<10 Å2 | |||||||||
| Average | 916 | 1.458 | 1.327 | 1.325 | 1.328 | 124.9 | 121.3 | 119.2 | 119.6 |
| RMSD | 916 | 0.012 | 0.011 | 0.013 | 0.012 | 1.4 | 1.0 | 1.0 | 10 |
| Min | 1.390 | 1.267 | 1.266 | 1.294 | 119.4 | 118.2 | 114.2 | 113.6 | |
| Max | 1.520 | 1.384 | 1.386 | 1.394 | 130.2 | 124.6 | 123.0 | 126.1 | |
| ARG resolution 2.50 Å, B<20 Å2 | |||||||||
| Aveage | 8183 | 1.458 | 1.332 | 1.328 | 1.327 | 124.5 | 120.8 | 119.6 | 119.5 |
| RMSD | 8183 | 0.012 | 0.011 | 0.010 | 0.011 | 2.3 | 1.5 | 1.4 | 1.3 |
| Min | 1.343 | 1.276 | 1.264 | 1.253 | 102.8 | 108.6 | 103.0 | 97.8 | |
| Max | 1.594 | 1.487 | 1.413 | 1.469 | 161.3 | 139.0 | 138.0 | 133.5 | |
| Frag/Struct | |||||||||
| CCP4 | 1.460 | 1.329 | 1.326 | 1.326 | 124.2 | 120.0 | 120.0 | 119.7 | |
| EH91 | 1.460 | 1.329 | 1.326 | 1.326 | 123.6 | 120.0 | 120.0 | 119.7 | |
| RMSD | 0.018 | 0.014 | 0.018 | 0.018 | 1.5 | 1.9 | 1.9 | 1.8 | |
| EH99 | 71/98 | 1.460 | 1.326 | 1.326 | 1.326 | 123.6 | 120.3 | 120.3 | 119.4 |
| RMSD | 0.017 | 0.013 | 0.013 | 0.013 | 1.4 | 0.5 | 0.5 | 1.1 | |
| CSD | |||||||||
| Average | 435/148 | 1.456 | 1.326 | 1.323 | 1.329 | 124.4 | 121.5 | 119.2 | 119.4 |
| RMSD | 0.014 | 0.011 | 0.014 | 0.013 | 1.4 | 1.0 | 0.9 | 1.3 |
EH91 and EH99 refer to the stereochemical target libraries provided in two compilations by Engh and Huber in 1991 (EH91) and 2001 (EH99). The PDB statistics correspond to Arg residues from crystal structures refined against resolution higher than 1.0Å and with B factor lower than 10Å2. The second PDB statistics is based on protein structures with data resolution declared as 2.5 Å and Arg residues with B factor smaller than 20 Å2. The CSD entries were selected using the R‐value criterion (R ≤ 7.5%)
To check for the correctness of the standard library values of the guanidine moiety, we have analyzed its geometry in the contemporary Cambridge Structural Database9 and in the atomic resolution structures stored in the PDB. The geometry obtained for the crystal structure of L‐arginine phosphate monohydrate refined against high‐resolution X‐ray and neutron data,10 and arginine residues in trypsin refined using transferable aspherical atom model,11 showed significant difference from the EH libraries values. This study is an extension of our previous report on stereochemical restrains of histidine residues.12
Results and Discussion
The number of structures released by the PBD on March 23, 2016, is 218, of which 211 are obtained by X‐ray crystallography. The total number of disagreements above Z = 5 of various bond angles from the EH99 values in all 211 validation reports is 944, with 452 related to bond angles within the guanidinium group of arginines. After taking into account only these guanidinium groups that are in a single conformation, there are 222 NE‐CZ‐NH1 angles and 167 NE‐CZ‐NH2 angles classified as outliers. In several instances, the nomenclature of the terminal nitrogen atoms in the deposited structures is wrong, but all cases were inspected and the atomic names reverted, if necessary, to ensure that the NH1 atoms are cis and NH2 atoms are trans with respect to the CZ‐NE bond. Out of 222 NH1 outliers, 202 are larger than 122.8º and 20 smaller than 117.8º, and among 167 NH2 outliers 162 are smaller than 117.8º and 5 are larger than 123.0º. Thus the number of angular outliers within the guanidinium moieties in arginine residues is dramatically larger than any number of outliers in all remaining residue types. It is clear that the NE‐CZ‐NH1 angles tend to refine to values larger than the EH91 geometrical library target of 120.0º, and the NE‐CZ‐NH2 angles usually adopt values smaller than 120.0º.
Protein structures are usually refined even at atomic resolution with geometrical restraints applied to bond lengths, valence angles, planarity of certain groups, chiral volumes of asymmetric centers, and so forth. However, due to the large excess of measured reflections over the refined parameters, the effect of restraints on the “well‐behaving” groups, that is, fully occupied, low B‐factor fragments is very weak, and the restraints are in practice necessary only to preserve the stereochemical integrity of some disordered or highly flexible parts.13
In all structures deposited at a resolution 1 Å or higher in the PDB (on March 23, 2016) there are 916 guanidine moieties where all atoms are fully occupied and have B factors not exceeding 10 Å2. Statistics of their geometry is included in Table 1. The NE‐CZ‐NH1 and NE‐CZ‐NH2 angles show that the guanidinium group is not as symmetric as suggested by the EH values. It is somewhat surprising that even among 8183 instances of guanidinium groups with all atoms having B factors at most 20 Å2 among all PDB structures at resolution declared as 2.5 Å, the values of NE‐CZ‐NH1 and NE‐CZ‐NH2 angles are still clearly different, although their spread around the average values (RMSD ∼1.5º) is larger than for 1 Å resolution structures (RMSD ∼1.0º).
To validate these findings we performed a survey of the CSD that identified 148 organic structures containing 435 guanidinium moieties connected to the rest of the molecules through one aliphatic carbon atom and refined to an R factor lower than 0.075. The statistics of their geometry is presented in Table 1. The average bond lengths are in good agreement with the EH values, however the angles show significant differences, confirming the findings based on PDB structures.
Collectively, we recommend to correct the outdated libraries which are extensively utilized in macromolecular refinement and validating software to values based on the CSD survey presented here. This will diminish the number of reported geometric outliers for arginine that in reality reflect more correct values. Such a correction will also allow model refinement with proper restrains where their usage have stronger impact that is in flexible and disordered regions. Even though the difference between the NE‐CZ‐NH1 and NE‐CZ‐NH2 angles is small, it affects positions of hydrogen atoms, which can influence the results of further investigation, relying heavily on the crystal structures, for example docking of various ligands.
Acknowledgments
The content of this publication does not necessarily reflect the views or policies of the U.S. Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. MM acknowledges the Polish Ministry of Science and Higher Education for financial support through the “Mobility Plus” program.
References
- 1. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fitch CA, Platzer G, Okon M, Garcia‐Moreno EB, McIntosh LP (2015) Arginine: its pKa value revisited. Protein Sci 24:752–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Engh RA, Huber R (1991) Accurate bond and angle parameters for X‐ray protein structure refinement. Acta Cryst A47:392–400. [Google Scholar]
- 4. Engh RA, Huber R Structure quality and target parameters In: Rossmann MG, Arnold E, Eds. (2006) International tables for crystallography volume F: crystallography of biological macromolecules. Springer; Netherlands, pp 382–392. [Google Scholar]
- 5. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Cryst D67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ (1999) Efficient anisotropic refinement of macromolecular structures using FFT. Acta Cryst D55:247–255. [DOI] [PubMed] [Google Scholar]
- 7. Afonine PV, Grosse‐Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Cryst D68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Cryst D66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Allen FH (2002) The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Cryst B58:380–388. [DOI] [PubMed] [Google Scholar]
- 10. Espinosa E, Lecomte C, Molins E, Veintemillas S, Cousson A, Paulus W (1996) Electron density study of a new non‐linear optical material: l‐arginine phosphate monohydrate (LAP). Comparison between X–X and X–(X + N) refinements. Acta Cryst B52:519–534. [Google Scholar]
- 11. Malinska M, Dauter Z (2016) Transferable aspherical atom model refinement of protein and DNA structures against ultrahigh‐resolution X‐ray data. Acta Cryst D72:770–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Malinska M, Dauter M, Kowiel M, Jaskolski M, Dauter Z (2015) Protonation and geometry of histidine rings. Acta Cryst D71:1444–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dauter Z, Sieker LC, Wilson KS (1992) Refinement of rubredoxin from Desulfovibrio vulgaris at 1.0 A with and without restraints. Acta Cryst B48:42–59. [DOI] [PubMed] [Google Scholar]