

Abstract
The Ebola virus protein VP40 is a transformer protein that possesses an extraordinary ability to accomplish multiple functions by transforming into various oligomeric conformations. The disengagement of the C‐terminal domain (CTD) from the N‐terminal domain (NTD) is a crucial step in the conformational transformations of VP40 from the dimeric form to the hexameric form or octameric ring structure. Here, we use various molecular dynamics (MD) simulations to investigate the dynamics of the VP40 protein and the roles of interdomain interactions that are important for the domain–domain association and dissociation, and report on experimental results of the behavior of mutant variants of VP40. The MD studies find that various salt‐bridge interactions modulate the VP40 domain dynamics by providing conformational specificity through interdomain interactions. The MD simulations reveal a novel salt‐bridge between D45‐K326 when the CTD participates in a latch‐like interaction with the NTD. The D45‐K326 salt‐bridge interaction is proposed to help domain–domain association, whereas the E76‐K291 interaction is important for stabilizing the closed‐form structure. The effects of the removal of important VP40 salt‐bridges on plasma membrane (PM) localization, VP40 oligomerization, and virus like particle (VLP) budding assays were investigated experimentally by live cell imaging using an EGFP‐tagged VP40 system. It is found that the mutations K291E and D45K show enhanced PM localization but D45K significantly reduced VLP formation.
Keywords: Interdomain salt‐bridges, Ebola Virus, VP40, latch‐like interaction, PM localization, transformer protein
Introduction
The Ebola virus is a lipid‐enveloped virus that causes hemorrhagic fever with a high mortality rate. The virus uses its negative sense RNA genome to replicate its seven genes in the host cell. To overcome the limitations posed by its small genome, the Ebola virus utilizes proteins with astonishing abilities to change structures and perform multiple functions.1 The Ebola virus protein VP40 shows remarkable conformational plasticity in forming various oligomeric structures that perform different functions and is an example of a “transformer protein,” which is a recently identified class of multifunctional proteins.2, 3, 4 VP40 consists of a N‐terminal domain (NTD) that is involved in dimerization and a C‐terminal domain (CTD) that mediates the membrane binding and further oligomerization.5 VP40 exists in various conformations depending on the required function: a butterfly shaped dimer is involved in the transport of the protein to a membrane, a hexamer to form the viral matrix, and an octamer ring structure to bind to RNA and regulate viral transcription.5 As with other recently identified transformer proteins such as the RfaH transcription factor,2, 3, 6 the structural basis and mechanisms of large‐scale structural transformation in VP40 are not well understood.
Formation of larger oligomers from VP40 dimers requires the C‐terminal domain to disengage from the N‐terminal domain, though they remain connected by a linker segment. This is supported by the observation that the complete removal of the CTD, leaving only the NTD, facilitates hexamerization of VP40.7 Various studies have explored the role of CTD flexibility on the conformational changes that occur during VP40 oligomerization.8 Similarly, membrane insertion of the CTD has been shown to induce a conformational change of VP40 from dimeric to hexameric forms.9 It has been shown that removal of the CTD‐tail segment (residues 320–326) leads to spontaneous hexamerization of VP40 and membrane binding in vitro.9 Recently, experiments by Silva et al.10 suggested that the NTD‐CTD closed‐form structure is stabilized by latch‐like interactions between an unstructured region of the N‐terminal domain and the C‐terminal tail region. This stabilization of the closed‐form structure may, therefore, mean that this latch‐like interaction is important for regulating oligomerization of VP40 upon RNA binding or membrane insertion.11
Salt‐bridge ionic interactions are important for protein stability, conformational specificity, and for positioning key residues that are essential for protein folding and function.12, 13, 14 Salt‐bridge interactions can provide the structural specificity required for proper folding of individual domains15, 16, 17 as well as guiding domain–domain association. Bhaskara et al.18 studied interdomain interactions in multidomain proteins and discussed the importance of the specificity of salt‐bridges, and also emphasized the role played by hydrophobic interactions in stabilizing the domain interface. Similarly, several interdomain salt‐bridge interactions stabilize the structure of VP40. In addition, in VP40 strategically positioned charged residues in the CTD are critical for membrane binding and subsequent conformational changes leading to further oligomerization.19
In this article, we use molecular dynamics (MD) simulations to investigate the interdomain interactions in the VP40 protein. Although the first crystal structure of VP40 was characterized as a monomer,8 it was later shown that it exists as a dimer in solution and that the dimer structure is required for VP40 trafficking to the cell membrane.5 For investigating the CTD disengagement from the NTD, we considered one subunit protomer (chain A) from the dimeric form in the protein data bank (PDB) (PDB code 4ldb). Our simulations reveal an interdomain salt‐bridge interaction involving the CTD‐tail segment latch. We also investigate the dynamics of other salt‐bridges that stabilize the closed‐form structure. Live cell experimental mutational analyses were performed to investigate the role of the computationally identified salt‐bridge interactions on VP40 plasma membrane (PM) localization and VP40 oligomerization. VLP budding assays were also performed to determine if any of the salt‐bridge interactions were important in budding. Identification and detailed investigations of such key interactions are an important step in understanding the mechanisms of structural transformations and reorganizations of VP40 into various oligomeric structures.
Results
MD simulations on the wild‐type (WT) VP40 as well as on several mutants were performed to investigate the interdomain interactions that are important in stabilizing the closed‐form structure, the sequence of steps that allow the CTD to disengage from the NTD, and the interactions that facilitate domain–domain association into the closed form. The effects of the removal of some important salt‐bridges that were identified computationally were further investigated experimentally by live cell imaging.
Dynamical analysis of the closed‐form structure: interdomain interactions and equilibrium dynamics
In many proteins, the CTD‐tail segment interactions affect the protein conformational dynamics and play important functional roles.20, 21, 22, 23 Though not resolved in the X‐ray crystal structure, the VP40 CTD‐tail segment (residues 320 − 326) has been shown to be important for regulating oligomerization. This is evidenced by the fact that the removal of the CTD‐tail segment leads to spontaneous oligomerization of VP40 and membrane binding in vitro.9 This is further supported by the suggestion that the VP40 in its closed‐form is stabilized by latch‐like interactions between an unstructured region of the N‐terminal domain and the C‐terminal tail region.10 In order to investigate the NTD‐CTD interactions involving the CTD‐tail segment, we modeled this segment by inserting the missing residues 320 − 326. The modeled CTD‐tail segment in the closed‐form structure was a random coil structure with no contacts with the NTD. Starting from this conformation, we performed eight independent MD simulations for 50 ns each (total of 400 ns) and monitored the CTD‐tail interactions with the NTD. We observed the latch‐like interactions with the NTD via a hydrophobic patch as well as the D45‐K326 salt‐bridge interaction, as shown in Figure 1, in four of the eight simulations. Figure S1 Supporting Information displays the representative trajectories that show the formation of the latch. The interdomain contacts involved in the latch are highlighted in Figure S2 Supporting Information. Once formed, the latch is stable for several nanoseconds (∼3 to 30 ns) before breaking. The flexibility of the CTD tail segment resulting from the transient stability allows the separation of the CTD from the NTD, as required for higher order oligomerization of the VP40 dimers. In addition, membrane association may make it easier for the latch to release10 to facilitate the VP40 hexamerization in the membrane.
In order to explore the interdomain as well as intra‐domain interactions and residue–residue contacts, we used 10 ns of the trajectory from the region with the intact latch and calculated a contact map for the VP40 structure using Carma.24 Figure 1(A) shows the resulting contact map obtained with a distance cut‐off of 5 Å. Residues 44 − 201 belong to the NTD and residues 202 − 326 belong to the CTD. The lower right block displays the interdomain contacts. This contact map allows us to identify all interdomain interactions and in this article, we focus on interdomain salt‐bridges and hydrophobic interactions. Salt‐bridge interactions were identified with a cut‐off distance of 4 Å and are shown in Figure 1(B). A total of five interdomain salt‐bridge interactions were obtained: D45‐K326, D56‐K256, E76‐K291, R148‐D312, and E160‐K212. As discussed later, the D45‐K326 salt‐bridge is important in guiding domain association in the open‐form monomer.
Figure 1.
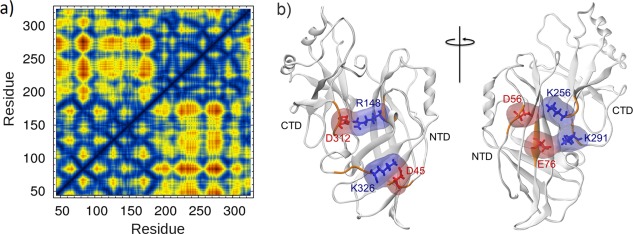
(a) Amino acid contact map of the VP40 protomer. The blue regions (<5 Å) in the lower right block display the NTD‐CTD interdomain contacts within 5 Å distance (b) VP40 inter‐domain salt‐bridges. Red are negatively charged amino acids and blue are positively charged.
Domain dissociation by forced pulling
We used Steered Molecular Dynamics (SMD) to investigate the relative strengths of interdomain interactions by pulling the center of mass of the C‐terminal domain (CTD) away from the harmonically restrained N‐terminal domain. For SMD simulations, the last frame of the 5‐ns equilibrium NVT simulation was taken as the initial structure and solvated again in a larger rectangular box to allow room for extension. In order to focus on the relative motion of the NTD and the CTD, translation and rotation was prevented by harmonically restraining the protein during heating and equilibration. After that, the NTD was kept harmonically restrained and the center of mass of the CTD was pulled away from the NTD using SMD. The protein was aligned so that the NTD‐CTD interface is approximately perpendicular to the pulling direction (−x). Stages in the resulting domain separation due to the SMD pulling are shown in Figure 2.
Figure 2.
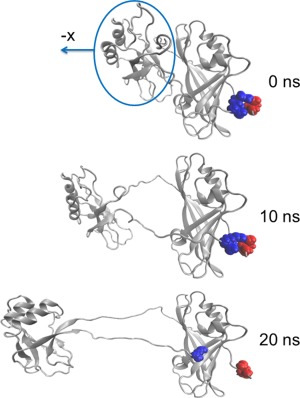
Domain separation during the SMD simulation in which the CTD (circled) is pulled away from the NTD. Residues D45 and K326 that participate in the interdomain salt‐bridge are highlighted. This salt‐bridge that participates in the latch remains intact for 20 ns and restricts CTD−NTD relative reorientation even when the domains are well separated.
We performed seven 25‐ns SMD simulations and averaged the force profiles for pulling the CTD center of mass away from the harmonically restrained NTD. During the initial stage of pulling, the SMD force continuously increase because all NTD−CTD interactions are intact and undergoing stress as shown in Figure S3 Supporting Information. We monitored the interdomain interactions during the pulling for a representative trajectory and the results are displayed in Figure 3. Figure 3(A) shows the SMD force necessary to move the center of mass of the CTD at a constant speed of 2 Å/ns as it disengages from the NTD. The SMD force curve initially increases and reaches a peak of 700 pN around 4 ns [region A in Fig. 3(A)] and then suddenly drops to approximately 300 pN. Just before this sharp drop in the SMD force, the solvent accessible surface area (SASA) at the interface of the NTD and CTD suddenly increases [Fig. 3(C)] by 800 Å2, suggesting the separation of the interdomain hydrophobic interface and the exposure of hydrophobic residues to water. This may be precipitated by the breaking of hydrogen bonds [Fig. 3(B)] and the breaking of the R148−D312 salt‐bridge [Fig. 3(D)] that occur at approximately 1.5 ns. In addition, two other salt‐bridges, D56‐K256 and E76‐K291, are each stretched and weakened before 1.5 ns. Thus, it appears that the weakening of specific salt‐bridges and hydrogen bonds permits enough flexibility to allow water to enter into the hydrophobic domain–domain interface.
Figure 3.
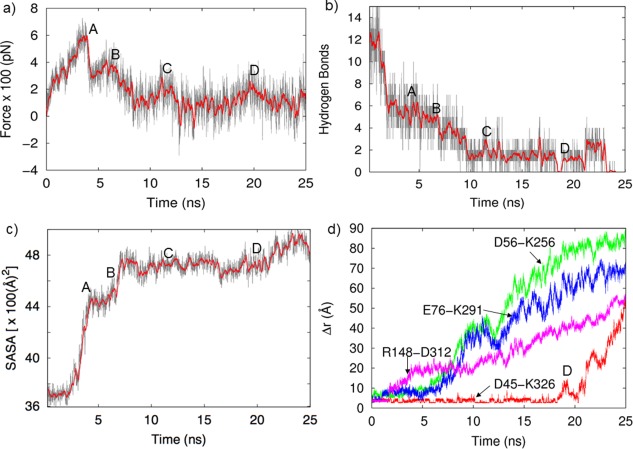
(a) SMD force necessary to keep the CTD moving at constant speed away from the NTD (gray curve: full data, red curve: time averaged data with 200 ps windows), (b) Time evolution of the number of hydrogen bonds in the interface, (c) solvent accessible surface area (SASA) of hydrophobic residues at the NTD‐CTD interface, and (d) various interfacial salt‐bridges distance.
Once the hydrophobic interactions begin to be disrupted, the SASA increases rapidly and the rest of the hydrophobic interactions are disrupted. This allows the domains to further separate, and additional hydrogen bonds are broken [region B in Fig. 3(B)]. Separation of the domains becomes easier, the SASA increases and the D56‐K256 and E76‐K291 salt‐bridges in the interface are fully broken by 8 ns. This sequence of bond disruption is consistent with the work of Bhaskara18 and Waldburger25 who emphasize the importance of hydrophobic interactions in stabilizing proteins. The NTD residues 72, 74, 95, 97, 125, 132, 156, 161, 162, 186, 187, 189, 191 and the CTD residues 283 − 286, 288 − 290, 293, 295, 317, 322, 323 form a strong hydrophobic interface. As shown in Figure 4(A), at 0 ns the interface is tightly packed and provides little access to water, By 4 ns [Fig. 4(B)], the domains have begun to separate and gaps open at the hydrophobic interface, exposing SASA as quantified in Figure 3(C). The SASA averaged over seven different trajectories in Figure S3 Supporting Information also shows the same general trend.
Figure 4.
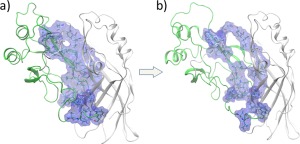
Hydrophobic residues at the domain–domain interface. (a) Initial closed‐form. (b) After the domains begin to separate (∼4 ns), gaps open at the hydrophobic interface, exposing SASA as quantified in Figure 3(c).
After 8 ns, the few remaining interdomain hydrogen bonds are broken [B→C in Fig. 3(B)]. Though the domains are always connected by the linker segment, after 10 ns, the only other interaction between the domains is the D45‐K326 salt‐bridge. Residue K326 is at the end of the flexible tail of the CTD. As the CTD separates from the NTD, this salt‐bridge remains fully intact and acts as a connecting latch until 20 ns [region D in Fig. 3(B)] as displayed in Figure 2. We will show later that the extended latch‐like D45‐K326 salt‐bridge facilitates association of the separated domains into the closed‐form structure.
Salt‐bridges and the interdomain stability
In order to understand the role of specific interaction in stabilizing the closed‐form structure, we mutated several important residues and pulled the CTD until the CTD is disengaged from the NTD. Figure 5 shows for different mutants, the force necessary to pull the CTD away from the NTD at constant speed. Since we are only interested in interdomain interactions, we pulled the CTD at a speed of 5 Å/ns for 4 ns and stopped after the interface begins to open. For each mutant, 10 similar runs were performed and the average force profile was calculated and displayed in Figure 5(A). Although the differences are small, the force profile for the WT generally shows more resistance to pulling compared to the mutants. The K291E mutation removes the 76–291 salt‐bridge that is present in the WT, and as seen in Figure 5(A), results in less force required to separate the domains. This shows that the K291 interaction is important in stabilizing the closed‐form structure. The comparison of hydrogen bonds in the WT and K291E as a function of time during SMD is displayed in Figure 5(B). The number of hydrogen bonds is reduced by half in the initial state of the mutant, also weakening the domain–domain interactions and contributing to the destabilization of the closed‐form structure.
Figure 5.
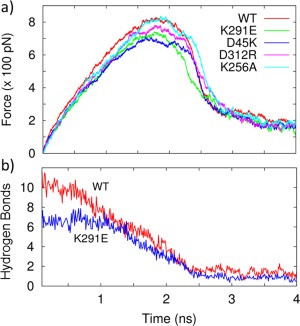
(a) Force required for separating the CTD away from the NTD at constant speed for the WT and different mutants. (b) The K291E mutation prevents formation of the 76–291 salt‐bridge and also decreases the number of interdomain hydrogen bonds, further destabilizing the closed‐form structure.
The D312R mutation removes the R148‐D312 salt‐bridge found in the WT. Removal of the 148–312 salt‐bridge weakens the domain interface and reduces the force necessary to separate the domains [Fig. 5(A)] compared to the WT. Another salt‐bridge, D56‐K256, is buried and is not a major contributor to interdomain stability.25 Upon alanine mutation K256A, which removes this salt‐bridge, there is little change compared to the WT in the amount of force necessary to separate the domains, as shown in Figure 5(A).
VP40 mutant cellular localization analysis
The effects of some mutations in the salt‐bridge network were assessed in live HEK293 cells using an established EGFP‐tagged VP40 system.26 The mutants assessed included D45K, R148A, E160A, and K291E, which should abrogate the aforementioned salt‐bridges, as well as a control K90A. Lys90 was not shown to participate in interdomain salt‐bridges and previously was shown to form virus like particles (VLPs) and localize to the plasma membrane inner leaflet, similar to WT VP4027 in CHO‐K1 cells. EGFP‐VP40 localized to the PM with characteristic high intensity membrane protrusions [Fig. 6(A)].26, 27 Further quantification using a MATLAB script revealed that K291E increased the %PM localization of VP40 by ∼20% (P < 0.07) compared to the WT (Fig. 6). These results suggest that removing the 76–291 salt‐bridge may alter the time scale of VP40 structural transitions but without impairing the important interdomain associative forces likely provided by other salt‐bridge and hydrophobic interactions. The D45K had a similar PM localization as K291E. It is important to note that the P‐value for these measurements was >0.05. Because these experiments are done at high concentrations of VP40 at a significant timeframe post‐transfection, time‐dependent assays may be needed to detect the time dependent changes of the VP40 assembly to better assess the statistical significance of each mutation on plasma membrane localization and oligomerization.
Figure 6.
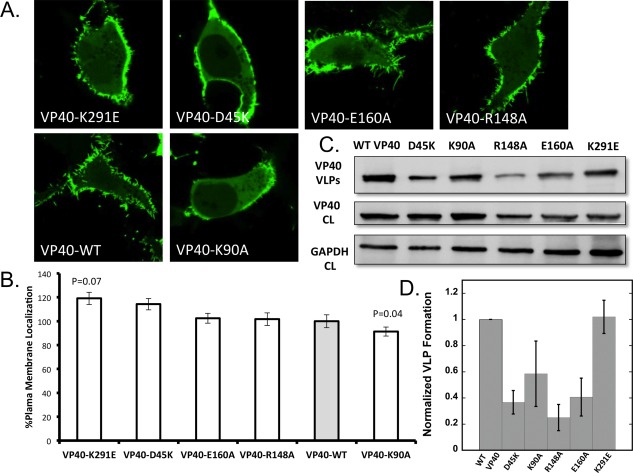
Cellular localization and VLP analysis of interdomain NTD−CTD salt‐bridge mutants. (a) Representative images of VP40‐EGFP constructs imaged 18–24 h post transfection into HEK293 cells. All images were acquired with a zoom of 8.0, therefore, the frame size is 20 μm and equivalent for all images. (b) Average plasma membrane localization for each construct. Image analysis was performed in MATLAB to determine the %PM localization for each. Error bars are ± the standard error of the mean. At least 28 cells were imaged per construct over three independent experiments on three different days. (c) Representative blots of VP40 and respective mutations in VLPs (top panel) or cell lysates (middle panel) or GADPH from cell lysates (bottom panel). (d) WT‐VP40 VLP formation was set at 1 and each mutant was compared to WT‐VP40 as fraction a of VLP formation. The bars represent the averages ± standard errors of the means for three independent experiments quantified using ImageJ. P‐values for mutations were 0.002 for D45K, 0.001 for R148A, and 0.004 for E160A. K90A and K291E P‐values were not statistically significant.
R148A or E160A did not contribute significantly to changes in PM localization. As R148‐D312 was the first salt‐bridge to break in the domain separation simulation, R148 likely plays a lesser role in mediating domain separation. As expected, K90A appeared similar to WT and exhibited plasma membrane localization albeit slightly less than WT. As a whole, this live cell mutational analysis illustrates different functions of salt‐bridge networks in VP40 conformational regulation and opens the door to large‐scale cellular and in vitro analysis of VP40 conformational dynamics.
PM localization alone is not necessarily indicative of the oligomerization state of VP40 where hexamers and larger oligomers have been detected.11, 26, 27, 28 Thus, we monitored VP40 oligomerization state of EGFP‐VP40 and respective mutations using N&B analysis as previously reported.11, 26, 27, 28 EGFP‐VP40 and all mutations employed exhibited a similar extent of oligomerization (see Fig. S7 Supporting Information) 18–24 h after transfection suggesting mutations still permit assembly of VP40 in some fashion. However, the extent and type of oligomerization is difficult to discern with this technique, as N&B measures the size of oligomers based upon the assembly of each additional EGFP containing monomer. Thus, N&B is not able to decipher between a VP40 filament that provides the structure for viral egress and a filament that may be slightly twisted or less rigid based upon salt‐bridge changes.
To determine if any of the VP40 salt‐bridge mutations identified here influenced VLP formation, we measured VLP formation for WT and respective mutations using a VP40 antibody. VLPs were collected and measured 48 h post‐transfection using cell lysate expression of VP40 and GAPDH (total protein/cell content) as a control for each construct. Notably, several salt‐bridge mutations had a statistically significant effect on VLP formation. D45K, R148A, and E160A all reduced VLP formation while K291E had a similar amount of VLPs as WT VP40. K90A showed a slight reduction in VLP formation but statistical analysis demonstrated this reduction was not statistically significant. Thus, despite a similar level of PM localization and oligomerization, disruption of the D45‐K326, R148‐D312, or E160‐K212 salt‐bridges reduced VLP formation suggesting these interactions have an important role in formation of VP40 structural assemblies that mediate efficient VLP formation. Notably, previous mutation of K212 reduced VLP formation29 by ∼40%, further supporting the conclusions in this study.
Domain association
The various functions of VP40 require that the NTD and CTD domains be able to exist in both a closed, associated form as well as an open, dissociated form. In both the closed and open forms, a flexible segment consisting of residues 187 − 201 links the domains.8 In general, multidomain proteins fold to the functional states by association of already folded domains.30 The rate‐limiting step for the complete folding is often the correct matching of the domains at the interface, similar to protein‐protein association of structured proteins that is generally “diffusion‐limited.”31, 32, 33, 34 Dissociated domains that are connected only with a flexible linker will have both rotational and translational degrees of conformational freedom and the linker flexibility provides a large entropic barrier for correct interface matching. VP40 seems to have reduced this problem by introducing a second interdomain interaction to guide the domain association by using a handshake‐like latch interaction of the CTD tail with a NTD flexible segment via the D45‐K326 salt‐bridge. The flexible nature of both the long CTD tail ending in K326 and the unstructured NTD segment containing the residue D45 increases the probability that these residues will interact in the open‐form of VP40 and form a salt‐bridge. This interdomain salt‐bridge handshake, along with the linker segment, reduces the number of possible relative orientations of the two domains as they approach and provides specificity for domain association. This increases the probability for a productive encounter that results in successful association of the CTD and the NTD. This is akin to the binding mechanism of flexible, structurally disordered proteins.35, 36, 37 The contact of the CTD tail section with a loop in the NTD is stabilized by both the D45‐K326 salt‐bridge as well as hydrophobic interactions between the CTD‐tail amino acids and amino acids in the NTD loop.
The initial structure for the association runs, displayed in Figure 7(A) was taken from a frame in the SMD dissociation simulation in which the domains were separated. This dissociated structure contained the latch‐like CTD (tail)‐NTD (loop) hydrophobic interactions and the D45−K326 salt‐bridge as shown in Figure S2 Supporting Information. Even with the intact latch‐like interaction, the timescales for domain association may still be greater than microseconds. In order to achieve domain association in a reasonable computational time, the initial separated structure was carefully chosen so that the residues in the flexible linker segment were minimally perturbed. Furthermore, only the 15 residues in the linker segment (residues 187 − 201) and the last seven residues (320 − 326) in the CTD tail were allowed to be fully flexible and all other residues in both domains were harmonically restrained. Figure 7(B) displays the final frame of the association run at 100 ns, along with the superimposed native structure of the closed‐form as a reference to display the good similarity. The associated structure was within 2.6 Å rmsd compared to the native structure. The time course of the distance d cm between the center‐of‐mass (cm) of the NTD and the cm of the CTD in Figure 7(C) gives the approximate time of association. At approximately 12 ns, the distance is ∼25 Å, which is the NTD(cm)−CTD(cm) distance in the WT VP40.
Figure 7.
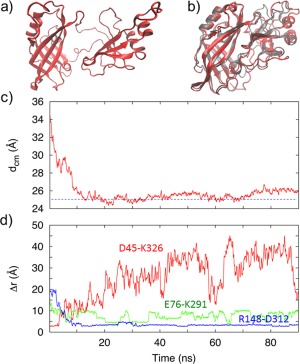
(a) Initial structure of the open‐form of the VP40 used in the association MD simulations. (b) The final closed‐formed structure at the end of the 100‐ns association simulation, along with the PDB structure of the closed‐form for reference. (c) Separation between the NTD center of mass (cm) and the CTD cm. The horizontal line at approximately 25 Å represents the cm separation in the WT. (d) Time course of the separation between the amino acids that make each of the three important salt‐bridges during domain–domain association.
We investigated the role played by the three salt‐bridges that we found to be especially important in domain association or in stabilizing the closed‐form: R148‐D312, E76‐K291 and D45‐K326. Figure S4 Supporting Information shows that initially in the dissociated form, only the D45‐K326 salt‐bridge is intact to guide the early stages of association. During the association, the D45−K326 salt‐bridge is broken, but the E76‐K291 and R148‐D312 salt‐bridges have formed to finalize association and stabilize the closed‐form structure.
More detailed information of the time course of these three salt‐bridges is given in Figure 7(D). The D45‐K326 salt‐bridge provides initial specificity for association when the domains are fully separated and starting to approach each other. This D45‐K326 salt‐bridge breaks at 4 ns, which from Figure 7(A) is at a stage in which the domains are almost in contact, but still separated by 4 Å compared to the fully associated conformation. At this stage, the domains are close enough that the other two salt‐bridges, E76‐K291 and R148−D312, have already started to form. These two salt‐bridges are critical for guiding the domains during the middle stage of the association process after the D45‐K326 salt‐bridge has broken, but before short range interdomain interactions are active. For the CTD, we computed the average Cα rmsf when the latch with the D45‐K326 salt‐bridge was intact, and compared it to the rmsf of the CTD in when the latch is broken. As can be seen in Figure S5 Supporting Information, the rmsf values of the CTD residues that are at the domain interface are smaller and, therefore, the CTD is more constrained when the CTD tail makes the salt‐bridge with the NTD loop.
The importance of other interactions in the association process was studied by comparing the SASA during dissociation and association as displayed in Figure S6 Supporting Information. The SASA during the association begins to significantly drop around 8 ns, which is well after the D45‐K326 guiding salt‐bridge has broken. At 8 ns, the domains are very near each other but not yet fully associated. Therefore, as expected for the short‐range hydrophobic interactions, they are helpful for guiding the final stages of association and are important for stabilizing the closed‐form structure. The guidance from hydrogen bonds and hydrophobic interactions only at the latter stages of association highlights the importance of salt‐bridges in the early and intermediate stages of the association process.
Methods
Molecular dynamics simulations
For the MD computational simulations, the initial structure of the VP40 protomer in the closed‐domain form was obtained from the dimeric VP40 structure (PDB code 4ldb). The CTD is composed of residues 202 − 326 and residues missing in the X‐ray crystallographic structure, such as the CTD tail residues 320 − 326, were inserted using Modeller.38 Since the unstructured N‐terminal domain segment composed of the first 43 amino acids was shown to have no effect on oligomerization,5, 39 we did not include this segment. All simulations were performed with all‐atom, explicit solvent MD simulations using NAMD 2.1040 with the CHARMM36 force field.41 The VMD software42 was used to set‐up and analyze the simulations. Each system was solvated and electrically neutralized by adding two Cl− counter ions. The dynamics were propagated using a 1 fs integration time step. Each system was heated by gradually increasing the temperature from 20 to 300 K in steps of 20 K/ps during a simulation time of 1 ns, followed by structural relaxation and equilibration for 2 ns of NPT simulation using a Langevin piston, along with a further 5 ns NVT relaxation run.
For structural relaxation of the protein in the closed‐form, we used a solvated water‐box with a padding distance of 10 Å that resulted in box size of 69 × 87 × 81 Å3. A 100 ns NVT production run of the closed‐form was performed and analyzed.
Steered molecular dynamics (SMD) simulations were performed by pulling on the center‐of‐mass of the CTD along the negative x‐axis. The system consisted of 61,320 atoms in a box of size 117 × 84 × 66 Å3. All Cα atoms were harmonically restrained during heating and equilibration. The dummy atom attached to the center‐of‐mass of the CTD via a virtual spring (k = 3 kcal/(mol/Å2)) was pulled at a constant speed of 2 Å/ns with respect to the harmonically restrained NTD (residues 44 − 194).
For the simulations of domain association, the initial open‐form structure was taken from a frame of the SMD simulations in which the domains are disengaged. The system consists of 44,250 atoms in a box of 90 × 80 × 60 Å3. The association dynamics were then investigated with a 20 ns simulation. During this simulation, the Cα of the NTD were harmonically restrained.
Molecular biology
Mutations were generated in a pcDNA3.1 construct containing an EGFP‐tagged VP4026 using a Quikchange XL mutagenesis kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer's instructions and were verified by automated DNA sequencing.
Cell culture and cell imaging
HEK293 cells were cultured and maintained in T‐25 flasks at 37°C and 5% CO2 in DMEM media containing 10% FBS and 1% Pen/Strep. Cells were seeded into an 8‐well Lab‐Tek IITM chambered coverglass (Fisher Scientific) to a final confluency of 50–70% and transfected the following day with 300 ng/well of DNA using Lipofectamine® LTX and PLUSTM reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.
Image acquisition for number and brightness (N&B) analysis was as previously described.28 Cells were imaged 22 − 24 h post‐transfection with an Olympus FV2000 confocal microscope using a Plan Apochromat 40× 1.3 NA oil objective and a 488 nm Argon laser to excite EGFP. The zoom was set to 8 for images acquired for %plasma membrane (PM) localization. At least 28 cells were imaged per mutant from a total of three independent transfections. Hundred images were acquired with raster scanning with photon count mode on, and zoom was set to 16.4 so each pixel is 0.5 μm.
Image analysis
The intensity of EGFP at the PM and in the cytosol was calculated using a script in MATLAB (Mathworks, Natick, MA) as previously described.28 The percent PM localization for each construct was calculated as PM intensity divided by PM + Cytosol intensity times 100.
N&B Analysis was performed in SimFCS (Globals Software, Irvine, CA) as previously described.28 First, EGFP was used to determine the true brightness of a monomer in HEK293 cells 18–24 h post transfection; the value from 8 cells over two independent experiments gave an average brightness of 0.168. 0.168 was used as the oligomeric step size in analysis of all VP40 constructs where 1–2.01 represent a monomer to a hexamer, 2.01–3.02 represent a hexamer to a 12 mer, and 3.03 to 5.03 represent a 12 mer to a 24 mer (indicated as 12 mer+). At least 8 cells imaged over three independent experiments were used in each calculation. The average monomer to a hexamer, hexamer to a 12 mer, and12 mer+ were determined and the percent of each population was determined for each cell. The average percent was then calculated for each construct.
VLP budding assays
HEK293 cells were maintained in DMEM supplemented with 10% FBS and 1% Pen/Strep at 37°C and 5% CO2. At 80% confluency, 2.5 × 106 cells were seeded into 100 mm tissue culture dishes. Cells were transfected 24 h later with 21.25 μg of the indicated endotoxin free expression plasmids using Lipofectamine LTX and PLUS reagent according to the manufacturers protocol (Invitrogen, Carlsbad, CA). Cells were transfected in serum‐free OPTI‐MEM media for 8 h, and then the media was exchanged for DMEM (containing no FBS or Pen/Strep). Culture medium was harvested at 48 h post transfection, each plate was washed with 1×‐PBS and DMEM supplemented with 10% FBS and 1% Pen/Strep was added back to the 100 mm dishes and placed back in to the incubator. The PBS wash, and HaltTM Protease Inhibitor cocktail (ThermoFisher, Waltham, MA) were added to the harvested culture medium and centrifuged at 1500 rpm for 10 min at 4° to remove cellular debris. The supernatant was then slowly loaded on top of a 20% sucrose cushion in STE buffer and centrifuged at 100,000g for 2 h at 4°C. The VLP‐containing pellet was resuspended in 200 μL of fresh 150 mM ammonium bicarbonate and stored at −80°C. During the 2 h centrifugation for VLP collection, cells were collected with a cell scraper from each 100 mm dish and centrifuged at 900 rpm for 6 min at 25°C. The cellular pellet was resuspended in 1×‐PBS and centrifuged again at 900 rpm for 6 min at 25°C. The supernatant was discarded and the cellular pellet was lysed using RIPA buffer. Cells were incubated in the RIPA buffer on ice for 1 h, and were vortexed every 15 min. Cell lysate samples were centrifuged at 2500g for 17 min at 4°C. The supernatant was collected, protein concentration was measured using a BCA assay (Pierce, Dallas, TX) and the supernatant was stored at −80°C.
Western blotting
Cell lysates and VLP samples were mixed with 5× SDS loading buffer, boiled for 5 min and loaded onto a 12% SDS‐PAGE gel. The gels were then transferred to a nitrocellulose membrane (Bio‐Rad, Hercules, CA). Each membrane was blocked in 5% BSA‐TBST for 90 min at room temperature. To detect VP40 expression in the VLPs and cell lysates, each membrane was incubated with a primary rabbit polyclonal anti‐EBOV VP40 antibody (IBT Bioservices, Gaithersburg, MD) (10 ng/mL) for 2 h at room temperature. After washing with TBST, the membranes were incubated with goat anti‐Rabbit HRP conjugated antibody (Abcam, ab6806)(1:2000 dilution) for 30 min at room temperature. For visualization of protein bands, ECL Western Blotting Substrate was added (ThermoFisher, Walktham, MA) and blots were exposed using an ImageQuant LAS 4000 (GE Healthcare). The membranes were then reblocked with 5% BSA‐TBST for 90 min at room temperature. To detect the loading control GAPDH, each membrane was incubated with a primary mouse monoclonal anti‐GAPDH antibody (ab8245‐1:3000 dilution) (Abcam, Cambridge, UK) for 1 h at room temperature. After washing with TBST, the membranes were incubated with sheep anti‐Mouse HRP conjugated antibody (ab205718‐1:5000 dilution) (Abcam, Cambridge, UK) for 30 min at room temperature. Blots were exposed as described above. In two experiments, nitrocellulose membranes from cell lysate samples were cut horizontally using Precision Plus Protein ™ Kaleidoscope™ Prestained Protein Standards (Bio‐Rad, Hercules, CA) to isolate VP40 and GAPDH on separate membranes. Membranes were then blocked, incubated with the antibody targeting their respective protein and imaged as described above. To quantify expression of VP40 for each cell lysates and VLPs, ImageJ was used first to normalize VP40 band density with respect to wild‐type VP40, and then to the their respective GAPDH loading control. All values are represented as mean ± S.E.M.
Conclusions
Using various molecular dynamics simulations, we investigated the dynamics of the association and the dissociation processes of the NTD and CTD domains in the Ebola virus VP40 transformer protein. MD simulations reveal a novel interdomain salt‐bridge interaction between D45‐K326 as part of the NTD‐CTD latch that was shown to be important for regulating VP40 oligomerization. The results suggest that the salt‐bridge interaction between D45‐K326 is helpful in guiding domain–domain association, whereas the E76‐K291 interaction stabilizes the closed‐form structure. VLP analysis demonstrated that some of these salt‐bridges may be significant in forming the correct VP40 assembly structure for significant scission and VLP formation. For instance, mutation of the D45‐K326, R148‐D312, and E160‐K212 salt‐bridges led to a statistically significant reduction in VLPs. Live cell imaging using an EGFP‐tagged VP40 system revealed that the mutation K291E shows an enhanced PM localization. This may be because the K291E mutation that abrogates the salt‐bridge allows easier domain dissociation, which is an important step in VP40 hexamerization at the plasma membrane leading to VLP formation. These computational and experimental live cell mutational analyses illustrate the different functions of salt‐bridge networks in VP40 conformational regulation and open the door to large‐scale molecular, membrane, cellular and in vitro analysis, and computational studies of VP40 conformational dynamics. Further experiments are necessary to investigate the time‐dependency of plasma membrane localization and the biophysics of assembly of VP40. For instance, VP40 localization, trafficking and oligomerization may need to be studied from initial stages of VP40 protein expression to scission of mature VLPs detectable at early stages of infection.
Supporting information
Supporting Information
Acknowledgment
The Tesla K40 gpu used for this research was donated by the NVIDIA Corporation.
References
- 1. Radzimanowski J, Effantin G, Weissenhorn W (2014) Conformational plasticity of the Ebola virus matrix protein. Protein Sci 23:1519–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Knauer SH, Artsimovitch I, Rosch P (2012) Transformer proteins. Cell Cycle 11:4289–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burmann BM, Knauer SH, Sevostyanova A, Schweimer K, Mooney RA, Landick R, Artsimovitch I, Rosch P (2012) An alpha helix to beta barrel domain switch transforms the transcription factor RfaH into a translation factor. Cell 150:291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tomar SK, Knauer SH, Nandymazumdar M, Rösch P, Artsimovitch I (2013) Interdomain contacts control folding of transcription factor RfaH. Nucleic Acids Res 41:10077–10085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bornholdt ZA, Noda T, Abelson DM, Halfmann P, Wood MR, Kawaoka Y, Saphire EO (2013) Structural rearrangement of Ebola virus VP40 begets multiple functions in the virus life cycle. Cell 154:763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gc JB, Gerstman BS, Chapagain PP (2015) The role of the interdomain interactions on RfaH dynamics and conformational transformation. J Phys Chem B 119:12750–12759. [DOI] [PubMed] [Google Scholar]
- 7. Ruigrok RW, Schoehn G, Dessen A, Forest E, Volchkov V, Dolnik O, Klenk HD, Weissenhorn W (2000) Structural characterization and membrane binding properties of the matrix protein VP40 of Ebola virus. J Mol Biol 300:103–112. [DOI] [PubMed] [Google Scholar]
- 8. Dessen A, Volchkov V, Dolnik O, Klenk HD, Weissenhorn W (2000) Crystal structure of the matrix protein VP40 from Ebola virus. EMBO J 19:4228–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Scianimanico S, Schoehn G, Timmins J, Ruigrok RH, Klenk HD, Weissenhorn W (2000) Membrane association induces a conformational change in the Ebola virus matrix Protein. EMBO J 19:6732–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Silva LP, Vanzile M, Bavari S, Aman JM, Schriemer DC (2012) Assembly of Ebola virus matrix protein VP40 is regulated by latch‐like properties of N and C terminal tails. PLoS One 7:e39978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Adu‐Gyamfi E, Soni SP, Xue Y, Digman MA, Gratton E, Stahelin RV (2013) The Ebola virus matrix protein penetrates into the plasma membrane: a key step in viral protein 40 (VP40) oligomerization and viral egress. J Biol Chem 288:5779–5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Donald JE, Kulp DW, DeGrado WF (2011) Salt bridges: geometrically specific, designable interactions. Proteins 79:898–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vijayakumar M, Zhou H‐X (2001) Salt bridges stabilize the folded structure of barnase. J Phys Chem B 105:7334–7340. [Google Scholar]
- 14. Menon S, Sengupta N (2015) Perturbations in inter‐domain associations may trigger the onset of pathogenic transformations in PrP(C): insights from atomistic simulations. Mol Biosyst 11:1443–1453. [DOI] [PubMed] [Google Scholar]
- 15. Packer LE, Song B, Raleigh DP, McKnight CJ (2011) Competition between intradomain and interdomain interactions: a buried salt bridge is essential for villin headpiece folding and actin binding. Biochemistry 50:3706–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bolon DN, Mayo SL (2001) Polar residues in the protein core of Escherichia coli thioredoxin are important for fold specificity. Biochemistry 40:10047–10053. [DOI] [PubMed] [Google Scholar]
- 17. Hendsch ZS, Tidor B (1994) Do salt bridges stabilize proteins? A continuum electrostatic analysis. Protein Sci 3:211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bhaskara RM, Srinivasan N (2011) Stability of domain structures in multi‐domain proteins. Sci Rep 1:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stahelin RV (2014) Membrane binding and bending in Ebola VP40 assembly and egress. Front Microbiol 5:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Govindjee R, Ohno K, Chang CH, Ebrey TG (1984) The C‐terminal tail of bacteriorhodopsin–its conformation and role in proton pumping. Prog Clin Biol Res 164:13–25. [PubMed] [Google Scholar]
- 21. Wang Q, Li S, Li C, Liang J, Fang Z, Xie L, Zhang R (2008) The extra C‐terminal tail is involved in the conformation, stability changes and the N/C‐domain interactions of the calmodulin‐like protein from pearl oyster Pinctada fucata. Biochim Biophys Acta 1784:1514–1523. [DOI] [PubMed] [Google Scholar]
- 22. Bandyopadhyay S, Banik U, Bhattacharyya B, Mandal NC, Roy S (1995) Role of the C‐terminal tail region in the self‐assembly of lambda‐repressor. Biochemistry 34:5090–5097. [DOI] [PubMed] [Google Scholar]
- 23. Zhu Y, Yang S, Qi R, Zou Y, Ma B, Nussinov R, Zhang Q (2015) Effects of the C‐terminal tail on the conformational dynamics of human neuronal calcium sensor‐1 protein. J Phys Chem B 119:14236–14244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Glykos NM (2006) Software news and updates. Carma: a molecular dynamics analysis program. J Comput Chem 27:1765–1768. [DOI] [PubMed] [Google Scholar]
- 25. Waldburger CD, Schildbach JF, Sauer RT (1995) Are buried salt bridges important for protein stability and conformational specificity? Nat Struct Biol 2:122–128. [DOI] [PubMed] [Google Scholar]
- 26. Adu‐Gyamfi E, Johnson KA, Fraser ME, Scott JL, Soni SP, Jones KR, Digman MA, Gratton E, Tessier CR, Stahelin RV (2015) Host cell plasma membrane phosphatidylserine regulates the assembly and budding of Ebola virus. J Virol 89:9440–9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Adu‐Gyamfi E, Soni SP, Jee CS, Digman MA, Gratton E, Stahelin RV (2014) A loop region in the N‐terminal domain of Ebola virus VP40 is important in viral assembly, budding, and egress. Viruses 6:3837–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Johnson KA, Taghon GJ, Scott JL, Stahelin RV (2016) The Ebola virus matrix protein, VP40, requires phosphatidylinositol 4,5‐bisphosphate (PI(4,5)P2) for extensive oligomerization at the plasma membrane and viral egress. Sci Rep 6:19125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McCarthy SE, Johnson RF, Zhang YA, Sunyer JO, Harty RN (2007) Role for amino acids 212KLR214 of Ebola virus VP40 in assembly and budding. J Virol 81:11452–11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Crieghton T (1992) Proteins: structures and molecular properties. New York, NY: W. H. Freeman. [Google Scholar]
- 31. Sugase K, Dyson HJ, Wright PE (2007) Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 447:1021–1025. [DOI] [PubMed] [Google Scholar]
- 32. Schreiber G, Keating AE (2011) Protein binding specificity versus promiscuity. Curr Opin Struct Biol 21:50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rogers JM, Steward A, Clarke J (2013) Folding and binding of an intrinsically disordered protein: fast, but not 'diffusion‐Limited'. J Am Chem Soc 135:1415–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arai M, Ferreon JC, Wright PE (2012) Quantitative analysis of multisite protein‐ligand interactions by NMR: binding of intrinsically disordered p53 transactivation subdomains with the TAZ2 domain of CBP. J Am Chem Soc 134:3792–3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Babu MM, van der Lee R, de Groot NS, Gsponer J (2011) Intrinsically disordered proteins: regulation and disease. Curr Opin Struct Biol 21:432–440. [DOI] [PubMed] [Google Scholar]
- 36. Shaw AS, Filbert EL (2009) Scaffold proteins and immune‐cell signalling. Nat Rev Immunol 9:47–56. [DOI] [PubMed] [Google Scholar]
- 37. Munz M, Hein J, Biggin PC (2012) The role of flexibility and conformational selection in the binding promiscuity of PDZ domains. PLoS Comput Biol 8:e1002749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eswar N, Webb B, Marti‐Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A (2006) Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics Chapter 5:Unit 5.6. [DOI] [PMC free article] [PubMed]
- 39. Yamayoshi S, Kawaoka Y (2007) Mapping of a region of Ebola virus VP40 that is important in the production of virus‐like particles. J Infect Dis 196:S291–S295. [DOI] [PubMed] [Google Scholar]
- 40. Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD, Jr (2010) CHARMM general force field: a force field for drug‐like molecules compatible with the CHARMM all‐atom additive biological force fields. J Comput Chem 31:671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information