

Abstract
Cells use the post‐translational modification ADP‐ribosylation to control a host of biological activities. In some pathogenic bacteria, an operon‐encoded mono‐ADP‐ribosylation cycle mediates response to host‐induced oxidative stress. In this system, reversible mono ADP‐ribosylation of a lipoylated target protein represses oxidative stress response. An NAD+‐dependent sirtuin catalyzes the single ADP‐ribose (ADPr) addition, while a linked macrodomain‐containing protein removes the ADPr. Here we report the crystal structure of the sitruin‐linked macrodomain protein from Staphylococcus aureus, SauMacro (also known as SAV0325) to 1.75‐Å resolution. The monomeric SauMacro bears a previously unidentified Zn2+‐binding site that putatively aids in substrate recognition and catalysis. An amino‐terminal three‐helix bundle motif unique to this class of macrodomain proteins provides a structural scaffold for the Zn2+ site. Structural features of the enzyme further indicate a cleft proximal to the Zn2+ binding site appears well suited for ADPr binding, while a deep hydrophobic channel in the protein core is suitable for binding the lipoate of the lipoylated protein target.
Keywords: ADP‐ribose, SauMacro, SAV0325, lipoate, ADP‐ribosylation, macrodomain, adenosine diphosphate ribose, lipolyation, oxidative stress, Staphylococcus aureus, X‐ray crystallography
Introduction
The reversible post‐translational transfer of adenosine diphosphate ribose (ADPr) moieties to proteins enables cells to dynamically control response to external stimuli. In bacteria, ADP‐ribosyltransferases (ARTs) are the primary family of proteins responsible for ADP‐ribosylation of target proteins.1, 2 Sirtuin proteins also have established functions in diverse cellular processes such as aging, metabolic regulation and gene silencing.3 While they typically catalyze protein deacetylation reactions, members of the Sirtuin family including the macrodomain‐linked sirtuins (SirTMs) have been shown to exhibit ADP‐ribosylating activity.3, 4, 5 Both ARTs and SirTMs utilize a bound nicotamide adenosine dinucleotide (NAD+) cofactor, and in their reactions release nicotamide while covalently linking ADPr to a substrate.1, 2, 3, 4, 5 Substrates may be either a specific amino acid (usually a glutamate/aspartate or lysine) on the target protein, or in the case of some eukaryotic ARTs, another ADPr though a glycosidic ribose–ribose bond forming linear and branched polymers. Structurally conserved in all domains of life, ARTs are an ancient protein class with diverse modifying abilities and target proteins. Bacterial ARTs act as toxins, modifying host cell signaling proteins, resulting in cell death and disease.6, 7, 8, 9, 10 ARTs primarily transfer a single ADPr, a reaction referred to as mono‐ADPribosylation (MARylation). In eukaryotes, ARTs have evolved into two distinct classes, those that resemble bacterial, MARylating ARTs and those capable of labeling target proteins with structurally and functionally diverse ADPr polymeric chains.11, 12 The latter enzymes are often referred to as poly‐ADPr polymerases (PARPs), and control diverse cellular processes including transcription and DNA repair through poly‐ADPribosylation (PARlyation).11, 12, 13, 14
The existence and coordination of enzymes capable of both mono ADP‐ribosylation and poly ADP‐ribosylation of target proteins paints a picture of a complex and regulated network of ARTs that can quickly and specifically alter cellular physiology post‐translationally.2 A number of enzymes complete the ADPr modifying cycle by catalyzing the removal of ADPr from poly‐ADPr‐targeted proteins.15 Many of these proteins contain a macrodomain fold, which enables selective recognition of ADPr16 and discrimination between mono‐ and poly‐ADPr.15 Initially discovered associated with peptidases in viruses17 macrodomains have been found in all forms of life, but it was not until crystal structures of macrodomain proteins from the thermophile Archaeoglobus flulgidus 16, 18 and subsequently coronavirus19 were their ADPr binding functions understood. Possessing a central six or seven stranded mixed β‐sheet sandwiched by four to five α‐helices, the macrodomain presents an L‐shaped cleft that envelops the ADPr moiety largely through conserved protein backbone interactions, as well as aspartate‐N6 adenosine and asparagine‐O2′ ribose interactions. Macrodomain proteins also function as O‐acetyl‐ADPr deacetylases, removing the acetyl moiety from the common reaction product of sirtuins.20 The macrodomain‐containing family includes the Poly‐ADPr glycohydrolases (PARGs).20, 21, 22 The catalytic functions of two additional human macrodomain proteins capable of reversing the glutamate‐linked mono‐ADPr, MacroD2,23, 24 and TARG1 (Terminal ADP‐Ribose Glycohydrolase 1),25 have also been reported. Thus, the ADP‐ribosylation cycle, including MARylation and/or PARylation, can be completed via macrodomain‐mediated removal of the terminal ADPr.
Recently, an operon‐encoded reversible MARylation system was identified in the pathogenic bacteria Staphylococcus aureus and Streptococcus pyogenes.5 This system regulates the pathogen response to host‐initiated oxidative stress through lipoate protein ligase A (LplA2) transfer of a lipoate to a glycine cleavage system H‐like (GcvH‐L) target protein [Fig. 1(A)]. At the center of this regulation is a MARylation/deMARylation cycle, where a novel class of macrodomain‐linked sirtuins (SirTM) catalyzes the NAD+‐dependent transfer of a mono‐ADPr to GcvH‐L, while a macrodomain protein removes the moiety [Fig. 1(A)]. Rack et al. showed that only a lipoylated GcvH‐L could be MARylated by SirTM. Lipoylated GcvH‐L is also efficiently deMARylated by the macrodomain (SauMacro for S. aureus and SpyMacro for S. pyogenes). Altogether, the study points to the modification of a shuttle protein (GcvH‐L) with a redox‐active molecule (lipoate) under reversible control of a sirtuin/macrodomain‐catalyzed negative feedback mechanism, where MARlyated GcvH‐L represses and deMARlyated GcvH‐L activates pathogen redox defense.5 While the molecular basis of SirTM function has been established,5 the structure and function of the SauMacro protein remains less well defined. Here, we report the crystal structure of SauMacro (a.k.a. Sav0325) to 1.75‐Å resolution. Through sequence and structural analysis, we demonstrate that the SirTM‐linked macrodomain protein family consists of a core macrodmain fold decorated by an N‐terminal 3‐helix bundle which scaffolds a novel Zn2+ coordination loop proximal to the ADPr binding site. SauMacro is further characterized by a conspicuous hydrophobic protein core cavity that is suitable for engagement of the lipoylated target substrate.
Figure 1.
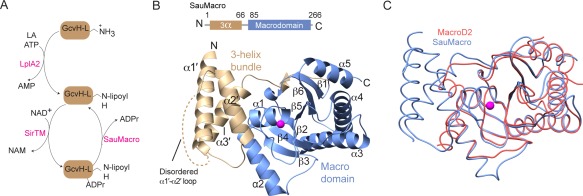
Overall structure of SauMacro. A. Lipoate/ADPr cycle. B. Overall fold of SauMacro. SauMacro is rendered as ribbons with secondary structures indicated, where amino‐terminal helices α1′‐α3′ are colored tan, carboxy‐terminal macrodomain fold is colored blue, and Zn2+ is rendered as a magenta sphere. C. Cα trace overlay of SauMacro (blue) with human MacroD2 (red). Structures were aligned using MatchMaker in Chimera.26 SauMacro Zn2+ is rendered as a sphere (magenta). All figures were prepared in Chimera26 unless otherwise indicated.
Results and Discussion
Overall structure of the SAV0325 macrodomain
Crystallization of recombinant SauMacro was facilitated by limited trypsin digestion of full‐length SauMacro immediately prior to crystallization. For structure solution, an unpublished structure of the Eschericia coli macrodomain fold protein (RCSB entry 1SPV) was successfully used as a molecular replacement search model. The asymmetric unit contains a single SauMacro molecule with continuous electron density in two segments corresponding to residues 1 to 22 (plus an N‐terminal non‐native “GSHMAS” sequence from the expression tag) and residues 30 to 263 of SauMacro [Fig. 1(B), Table 1]. We hypothesize that trypsin cleavage occurred in the arginine rich disordered α1′‐α2′ linker region (amino acids 23‐30), and that this aided in crystallization.
Table 1.
Crystallographic Data Collection and Refinement Statistics
| SauMacro‐Zn2+ complex | |
|---|---|
| PDB code | 5KIV |
| Data collection | |
| Wavelength (Å) | 1.0000 |
| Space group | P 21 21 21 |
| Cell dimensions | |
| a, b, c (Å) | 46.823, 47.853, 134.95 |
| α, β, γ (degrees) | 90, 90, 90 |
| Resolution (Å) | 50.0–1.75 (1.78–1.75) |
| Total reflections | 560,123 |
| R merge | 0.086 (0.345) |
| I/σI | 16.3 (3.3) |
| Completeness (%) | 98.83 (89.89) |
| Redundancy | 5.5 (2.8) |
| Refinement | |
| Resolution (Å) | 29.98–1.75 (1.812–1.75) |
| No. of reflections | 31085 (2784) |
| R work/R free | 0.157/0.194 (0.206/0.251) |
| No. of nonhydrogen atoms | 2398 |
| Protein | 2077 |
| Ligand | 14 |
| Water | 307 |
| Average B‐factors | 33.1 |
| Protein | 31.8 |
| Ligand | 37.7 |
| Water | 42.0 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (degrees) | 1.02 |
| Molprobity statistics | |
| All‐atom clashscore | 1.2 |
| Ramachandran favored (%) | 98 |
| Ramachandran allowed (%) | 2 |
| Ramachandran outliers (%) | 0 |
| Overall score | 0.93 |
Statistics for the highest‐resolution shell are shown in parentheses.
The SauMacro macrodomain structure is characterized by the canonical α/β macrodomain fold (six‐stranded mixed beta sheet (β1–β6) that is sandwiched by five alpha helices α1–α5), and an added N‐terminal helical domain. Overall, the macrodomain core of SauMacro resembles other macrodomain structures, and is most closely related to MacroD2 (RMSD of 1.6 Å) [Fig. 1(C), Table 2]. Three important differences distinguish SauMacro from other previously determined macrodomain structures. First, SauMacro contains an amino‐terminal extension comprised of an antiparallel 3‐helix bundle (α1′ ‐α3′, residues 1–66), which is structurally distinct from the two amino‐terminal helices found in MacroD2 and MacroD1 [Fig. 1(C), PDB: 2X47].20 Other macrodomain structures, including human TARG1 (PDB: 4J5S)25 and AF1521,16, 18 also lack these amino‐terminal helices. Secondly, we identify a Zn2+‐binding site, with Zn2+ coordination mediated by an insertion loop between β2 and α1. This Zn‐binding motif scaffolds the Zn2+ ion proximal to the presumed ADPr binding site. Lastly, the structure reveals a deep hydrophobic cavity in the SauMacro macrodomain that is proximal to the probable ADPr binding site.
Table 2.
Sequences and Crystal Structures Used in Multiple Sequence Alignments for This Study
| Abbreviation | Organism | Accession Codea | Homologyb |
|---|---|---|---|
| Sequence alignments | |||
| SauMacro | Staphylococcus aureus | 54042722 | — |
| SpyMacro | Streptococcus pyogenes | 134271958 | 88/47 |
| CboMacro | Clostridium bolteae | 524018385 | 96/39 |
| EclMacro | Enterobacter cloacae | 571249066 | 66/40 |
| YmdB | Escherichia coli | 26107583 | 67/39 |
| CalMfs1 | Candida albicans | 238883620 | 72/49 |
| AteMfs1 | Aspergillus terreus | 115391435 | 84/39 |
| Structure alignments | |||
| MacroD2 | Homo sapiens | 4IQY | 23.5 |
| MacroD1 | Homo sapiens | 2X47 | 23.1 |
| Phosphatase (putative) | Escherichia coli | 1SPV | 22.5 |
| PARP14 | Homo sapiens | 3Q6Z | 21.0 |
| MacroH2A.2 | Homo sapiens | 2XD7 | 20.4 |
| MacroH2A1.1 | Rattus norvegicus | 1YD9 | 20.1 |
| AF1521 | Archaeoglobus fulgidus | 2BFQ | 19.6 |
GenBank and PDB codes are given for sequence and structural alignments, respectively.
Percent coverage/identity and Dali reported Z scores with respect to SauMacro are reported for sequence and structural alignments, respectively.
A novel macrodomain Zn2+ binding site
The amino‐terminal three‐helix bundle is tightly packed against the β2‐α1 connecting loop containing the Zn2+ [Fig. 2(A–C)]. Conservation of the N‐terminal extension correlates with the conservation of the Zn2+ motif. A BLAST search of SauMacro related proteins (Table 2) reveals a distinct cutoff for conservation of the CxxxxHxC Zn2+‐binding motif, whereas the identity coverage drops below ∼70%, corresponding to a loss in the conservation of ∼80 amino‐terminal residues [Fig. 2(D), Table 2]. Notably, in the absence of ∼80 amino‐terminal sequence, proteins with high sequence homology to SauMacro also lack the Zn2+ binding motif. Thus the three‐helix bundle N‐terminal extension appears to provide an extended structural scaffold for the Zn2+‐binding β2‐α1 insertion loop.
Figure 2.
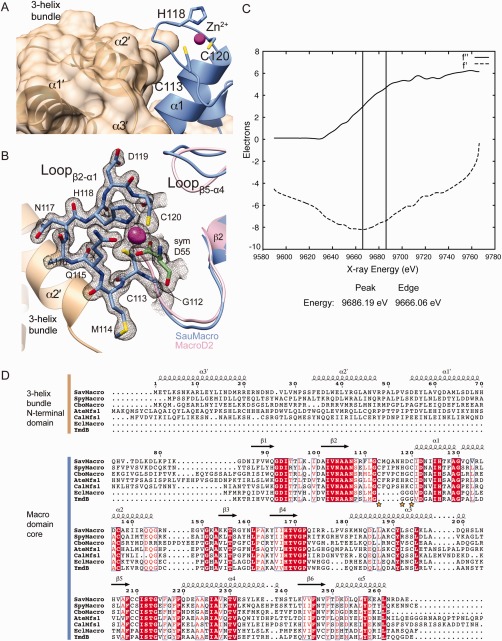
SauMacro Zn binding site. A. Packing of the three‐helix bundle with the Zn2+ binding site. Amino‐terminal helices α1′–α3′ are shown as ribbons and molecular surface (tan), carboxy‐terminal macrodomain is shown as ribbons (blue), with Zn2+ as a sphere (magenta). B. Close‐up view of Zn2+ binding site. SauMacro (blue) insertion loop residues are rendered as sticks, and the corresponding final 2mFo‐Dfc electron density map to 1.75‐Å resolution, contoured to 2σ is depicted as a mesh (gray). The location of the Zn2+ is revealed by the same 2mFo‐Dfc electron density map contoured at 10σ and depicted as a solid surface (magenta). SauMacro amino‐terminal helices (tan) and carboxy‐terminal helix α1 and Lβ5‐α4 (blue), as well as MacroD2 (PDB: 4IQY) α1 and Lβ5‐α4 are rendered as ribbons. C. X‐ray fluorescence scan at the Zn‐edge of purified, ammonium sulfate precipitated SauMacro protein. Anomalous scattering factors from X‐ray fluorescence f'' and derived from Kramers–Kronig (f') are consistent with the presence of Zn in our purified samples. D. Sequence comparison of SauMacro and related proteins (also see Table II). Sequence numbers and secondary structure assignments correspond to SauMacro. Red outlines indicate identical conservation, red letters indicate strong conservation (0.8 threashold). Yellow stars indicate Zn2+ binding residues in SauMacro. AteMfs1 and CalMfs1 contain additional carboxy‐terminal sequence corresponding to a fused sirtuin protein. EclMacro and YmdB lack amino‐terminal extensions and the conserved Zn2+ binding site.
The proximity of the Zn2+‐binding motif to the predicted ADPr pocket implies that it may play roles in substrate binding and catalysis. The conserved residues C113, H118, and C120 mediate Zn2+‐coordination [Fig. 2(A)]. To validate the identity of the metal, we conducted X‐ray fluorescence measurements on purified SAV0325 protein [Fig. 2(C)]. This analysis indicates that Zn is abundant in our protein purifications, consistent with Zn2+ modeled in the crystal structure. We also note that binding site geometry is consistent with a Zn2+ binding as confirmed by the “checkmymetal” (CMM) binding site validation server27 (http://csgid.org/csgid/metal_sites/). The loop containing the 113‐CxxxxHxC‐120 Zn2+ binding motif appears to be an insertion relative to other known macrodomain structures, all of which replace this Zn2+‐binding sequence with a poly‐glycine linker. An inspection of the surface of SauMacro relative to other macrodomains containing ADPr reveals that the Zn2+‐binding motif does not occlude the canonical ADPr substrate‐binding cleft. Interestingly, Asp55 of a symmetry related SauMacro molecule completes the tetrahedral coordination shell of the bound Zn2+ [Fig. 2(B)]. In the case of SpyGcvH‐L, the MARlyated residue is expected to be either and Asp or Glu,5 so the observed Asp55‐Zn2+ interaction might reflect a possible substrate‐binding mode of the ADP‐ribosylated target. Thus, we hypothesize that SauMacro and related sirtuin‐linked macrodomains use this Zn2+ site to coordinate substrate binding and/or electron movements during catalysis.
ADPr binding site
ADPr was added to the crystallization solution, however no evidence for bound ligand was observed in our electron density maps. Notably, the loop between α4 and β6 is found in a conformation that partially occludes the predicted ADPr binding site, in a mode reminiscent of apo MacroD1. The CASTp server identifies a large contiguous solvent‐accessible surface pocket in proximity to the Zn2+ site [Fig. 3(A–D)]. This pocket comprises over 1400 Å2 of surface area, and can be divided into two main sections connected via a narrow tunnel: one adjacent to the Zn2+ and the others containing the conserved acidic residue D93. Mapping the ADPr from the MacroD2 structure (PDB: 4IQY) onto this surface places the terminal ribose (ribose") in the Zn2+ ‐adjacent pocket and the adenosine near D93. Electrostatic surface analysis implicates the surface around the Zn2+ pocket as electropositive, while the surface that would presumably contact the adenosine as electronegative [Fig. 3(B)]. Taken together, we infer that the identified pocket could accommodate the ADPr, consistent with SauMacro de‐ADP‐ribosylation function.5
Figure 3.
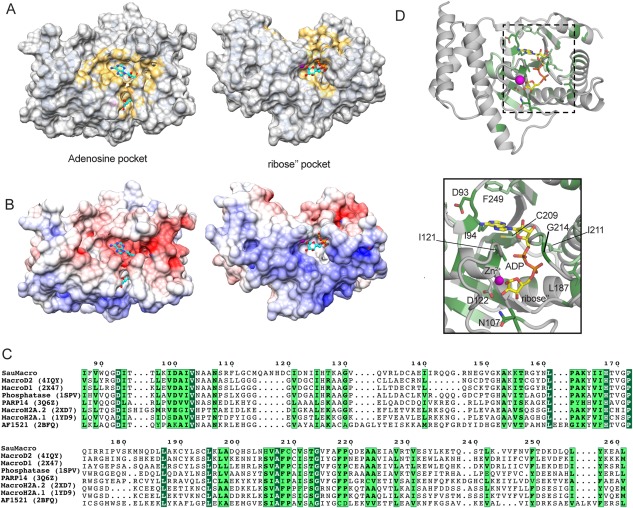
Predicted ADPr‐binding site. A. CASTp28 calculated solvent accessible surface pockets on SauMacro. SauMacro rendered as a molecular surface (gray) where yellow represents pocket atoms. ADPr from MacroD2 is shown in ball‐and‐stick (cyan). Adenosine (left) and ribose’’ (right) pocket views correspond in both panels A and B. B. SauMacro electrostatic surface. Poisson‐Boltzman electrostatics for SauMacro are rendered as surfaces, ranging from electropositive (blue), neutral (white) to electronegative (red). C. Structure‐specific sequence alignments of SauMacro with related macrodomains (see Table II). Dark green outlines indicate identical homology, while light green outlines indicate highly similar homology (0.8 threashold). Structures aligned with DALI server.29 D. Conserved residues (green) mapped onto SauMacro structure (gray). SauMacro rendered as ribbons with residues colored as in C. The position of ADPr from MacroD2 (yellow), as well as SauMacro residues predicted to interact with ADPr are rendered as sticks. Zn2+ (magenta) is shown as a sphere. Inset: Closeup view of ADPr binding site within the dashed box is presented.
Next, we structurally aligned a number of similar macrodomains predicted by the DALI server to have similar structural features (Z score > 19) [Table 2, Fig. 3(C)]. All of the compared structures contain an ADPr or ADPr‐like bound substrate molecule in a similar spatial orientation on the protein surface. In considering the structurally similar macrodomains, many of the residues involved in substrate recognition are conserved in SauMacro [Fig. 3(C,D)]. Important electrostatic interactions include Asp93, which could hydrogen bond to adenine N6 amino group, as well as N107 and D122, both of which would contact the O2′ hydroxyl of the terminal ribose. Substrate‐bound macrodomains contain an aromatic residue that provides π‐stacking interactions with the adenosine base; in SauMacro, F249 may perform this role [Fig. 3(D)]. C209, as well as the hydrophobic residues I94, I121, L187, and I211 line the pocket that could accommodate ADPr. G214 is a well‐conserved residue, and the presence of a bulky side chain at this position would likely occlude ADPr phosphate binding. Some backbone rearrangements of SauMacro would be necessary to prevent clashes with ADPr, especially the loop connecting β5 and α4. Overall, the surface properties of this cleft, strong sequence homology to known ADPr‐macrodomain complexes, and the previous report that SauMacro acts as an ADPr hydrolase5 strongly implicate that ADPr binds in the predicted pocket.
Predicted lipoate binding site
In addition to binding and hydrolyzing ADPr, SauMacro, and other related macrodomains occupying LpA2 and SirTM‐containing operons are expected to recognize lipoate. Efforts to co‐crystallize SauMacro with lipoate were unsuccessful and our structure does not contain lipoate. However, we identify a conspicuous deep hydrophobic pocket that both spatially and electrostatically may accommodate the redox‐active molecule [Fig. 4(A)]. This pocket is part of the surface identified by CASTp and resides in close proximity to the proposed ADPr binding site [Fig. 4(B)]. One face of the pocket contains residues 169‐TVGPQ‐173, which is highly conserved among macrodomains with high sequence homology to SauMacro [Fig. 2(D)]. The pocket is lined with highly conserved hydrophobic residues A105, I121, L187, F208, and I211, as well as Y191, which we predict would stack against the lipoate dithiolan ring. At the bottom of the pocket are two cysteines on the same face of helix α3, C190, and C194, which would lie within disulfide bonding distance to the two thiolates of the lipoate. Finally, the carboxyl face of the molecule, which would be covalently attached via an amide bond to a lysine of the target protein, extrudes out from the hydrophobic pocket, in potentially close contact to the conserved residue Q173.
Figure 4.
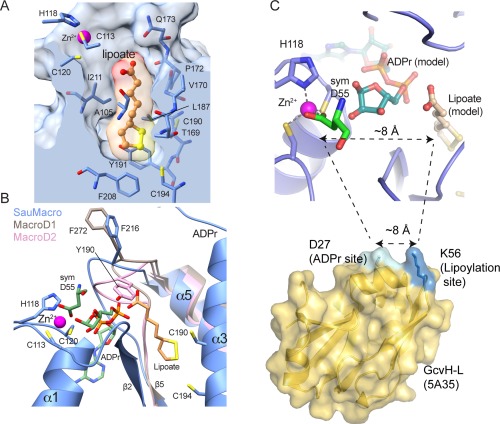
Predicted Lipoate binding site. A. Predicted lipoate binding pocket. A clipping plane through the SauMacro surface (blue) is placed at the center of a manually modeled lipoate molecule, which is rendered in ball‐and‐stick (orange) and molecular surface colored by atom. SauMacro residues forming the pocket are rendered as sticks, and the Zn2+ (magenta sphere) binding site is shown. B. Alternative conformations of the β5‐α4 Loop. SauMacro (blue), MacroD1 (brown) and MacroD2 (pink) are rendered as ribbons, where residues amino‐terminal of β5 are presented for SauMacro for clarity. The position of the β5‐α4 Loop is revealed by F216 (SauMacro), F272 (MacroD2) and Y190 (MacroD1), where residue numbers correspond to the respective protein. The position of ADPr from MacroD2 (green) and the SauMacro symmetry mate of D55 (green), a potential bound substrate analog, are rendered as sticks. C. Closeup view of the Zn2+, ADPr and lipoate binding site. The spacing between the Asp‐ADPr and lipoate‐binding pocket correlates with separation of the ADPr and lipoylation sites on GcvH‐L. Figure 4(C) was created with PYMOL.
The loop connecting β5 and α4 adopts three different conformations among the MacroD2, MacroD1, and SauMacro structures [Fig. 4(B)]. MacroD2 (PDB: 4IQY) and MacroD1 (PDB: 2X47), which are both ADPr‐binding proteins, have very similar overall folds (RMSD of 0.85 Å over 210 Cα's), and this loop represents the most significant structural deviation between the two structures. Additionally, MacroD2 was determined in complex with ADPr, while MacroD1 is a ligand‐free structure. Jankevicius et al. noted that an apparent 13 Å shift (5.3 Å for Cα positions) would be required to bring F272 in MacroD1 from an “open” conformation to the closed conformation occupied by Y190, the structurally homologous residue in MacroD2, stacks against the distal ribose of ADPr.23 In SauMacro, the corresponding residue is likely F216, and its loop adopts an intermediate position, ∼10 Å (3.4 Å for Cα atoms) away from Y190 in MacroD2 [Fig. 4(B)]. SauMacro F216 is less likely to adopt the same closed conformation as Y190, as this position might occlude binding to the lipoate.
The close juxtaposition of the Zn‐binding site, ADP‐ribose pocket and the proposed hydrophobic lipoate‐binding cavity suggests that the ADPr and lipoylation sites on the target GcvH‐L proteins should be within close three‐dimensional proximity of one another. The distance between the aspartic acid ligand bound in our structure (the proposed Asp‐ADPr binding site) to the solvent accessible region of the lipoate binding site is ∼8 Å [Fig. 4(C)]. Overall this geometry closely correlates with the spacing of the known GcvH‐L ADPr modification (D27 of GcvH‐L) and lipoylation (K56 of GcvH‐L) sites [Fig. 4(C)].
Conclusions
The structure of SauMacro reported here reveals the first atomic details of a sirtuin‐linked macrodomain. As a recently discovered class of oxidative‐stress response regulators in pathogenic bacteria, the biochemical and biomedical relevance of these molecules are only beginning to be understood. Our structure suggests that SauMacro is a unique macrodomain protein with a novel Zn2+ binding insertion loop relative to other macrodomains. A three‐helix bundle provides the structural scaffolding for this site, and sequence homology implies that this amino‐terminal structure may be conserved for related macrodomains in bacteria and fungi. Through structural homology and analysis, we propose potential binding sites for ADPr and lipoate on SauMacro. Future co‐crystal structures of SauMacro in complex with these substrates, or with the GcvH‐L target protein, and in concert with biochemical analysis, will provide the foundation for potential novel antimicrobials targeting this stress response pathway.
Materials and Methods
Protein expression and purification
N‐terminally 6x His‐tagged SauMacro (SAV0325) was expressed in from pET28a in E. coli Rosetta 2 (DE3) (Novagen). Terrific broth media was inoculated with saturated cell cultures and allowed to grow at 37°C until OD260∼1.5, at which point the temperature was lowered to 15°C, induced with 0.2 mM IPTG and further incubated overnight. Cells were pelleted, resuspended in 20 mM Tris pH 7.5, 150 mM NaCl, 10 mM Imidazole, sonicated and clarified by centrifugation. Affinity chromatography was accomplished by flowing clarified cell lysate over Ni‐NTA resin. Bound protein was eluted by addition of imidazole and further purified by size‐exclusion chromatography using a Superdex 75 column (GE Healthcare) equilibrated in 20 mM Tris pH 7.5, 500 mM NaCl, and 0.1% βME. Prior to crystallization a purified SAV0325 was subjected to limited trypsin digest at room temperature for 1 hr at a ratio of 650:1 (w/w) SauMacro to trypsin. Trypsinized protein was resolved by additional size‐exclusion purification, followed by a final cation‐exchange purification eluted in 20 mM Tris pH 7.5, 175 mM NaCl, and 0.1% βME. The resulting fractions from the single peak from the ion‐exchange purification were pooled and concentrated to 25 mg/mL for crystallographic screening.
Crystallization and data collection
Crystals were grown at 4°C using the sitting‐drop vapor diffusion method, by mixing SauMacro (25 mg mL−1 protein plus 1 mM ADP‐ribose) with 0.1 M CHES pH 9.5, 15% (v/v) ethanol in a 1:1 ratio (250 nL: 250 nL) using a Mosquito robot (TTP Labtech, Hertfordshire, UK). The resulting rod shaped crystals were cryoprotected in a solution of mother liquor supplemented with 26% (v/v) ethylene glycol and flash frozen in liquid nitrogen for subsequent data collection. Diffraction data were collected to 1.75 Å on a MAR 225 CCD detector at Southeast Regional Collaborative Access team (SER‐CAT) 22‐BM beamline at the Advanced Photon Source, Argonne National Laboratory. Data were indexed, integrated, and scaled using HKL2000.30 The data were processed in P212121 space group, with unit cell dimensions of a = 46.823, b = 47.853, c = 134.950, α = β = γ = 90° (also see Table 1). X‐ray fluorescence scans [Fig. 2(C)] on ammonium sulfate precipitated SAV0325 were performed and analyzed at SER‐CAT 22‐BM.
Structure determination and analysis
Crystallographic phases were determined by molecular replacement using 1SPV as a search model in Phaser.31 Subsequent rounds of automated model building in PHENIX and manual model building in Coot32 with refinement in PHENIX yielded the final model. Water molecules were placed automatically using PHENIX and checked manually in Coot. Significant positive density was found in between residues C113, H118, and C120, indicative of a metal ion coordination site. Based on ionic distances and coordination geometry, the metal was assigned as Zn2+. Although the protein crystallization solution was supplemented with ADPr, no definitive difference electron density corresponding to this ligand was observed. Model validation was carried out automatically with Molprobity.33 Data collection and refinement statistics are detailed in Table 1. PyMOL was used for structure‐sequence conservation figures, while all other molecular graphics were produced with UCSF Chimera.26 Solvent accessible surface pockets were calculated using the CASTp server.28 To visualize electrostatic surfaces, the SauMacro coordinates were prepared for Poisson‐Boltzman calculations using PDB2PQR,34 and the calculations were performed with APBS.35
Sequence and structural alignments
Homologous sequences were manually selected from a DELTA‐BLAST search of the SauMacro protein sequence. These sequences were aligned using the MultAlign server36 and visualized with ESPript.37 Proteins with similar structural homology to SauMacro were identified with the Dali server,29 and subsequent sequence alignments and figures were prepared with ENDscript.37 All sequences used in this study are listed in Table 2.
PDB accession codes
Coordinates and structure factors have been deposited in the Protein Data Bank as PDB entry 5KIV.
Acknowledgments
C.D.A. expressed and purified recombinant SauMacro, as well as performed the trypsinolysis and crystallization. R.S.W. collected diffraction data and determined the SauMacro crystal structure; G.K.F., B.D.W. and R.S.W. refined and interpreted the structure and homology data. G.K.F., C.D.A, and R.S.W. wrote the manuscript. The authors thank A. Moon and S. Andres for comments and I. Ahel for the gift of the SauMacro expression vector. The authors declare no competing financial interest. We thank L. Pedersen of the NIEHS collaborative crystallography core for help with data collection, and M. Schellenberg for conducting fluorescence scans. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W‐31‐109‐Eng‐38. Our studies are supported by the US National Institute of Health Intramural Program: US National Institute of Environmental Health Sciences (NIEHS), 1Z01ES102765 (R.S.W.).
References
- 1. Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch‐Nolte F (2010) Toward a unified nomenclature for mammalian ADP‐ribosyltransferases. Trends Biochem Sci 35:208–219. [DOI] [PubMed] [Google Scholar]
- 2. Barkauskaite E, Jankevicius G, Ladurner AG, Ahel I, Timinszky G (2013) The recognition and removal of cellular poly(ADP‐ribose) signals. FEBS J 280:3491–3507. [DOI] [PubMed] [Google Scholar]
- 3. Choi JE, Mostoslavsky R (2014) Sirtuins, metabolism, and DNA repair. Curr Opin Genet Dev 26:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kowieski TM, Lee S, Denu JM (2008) Acetylation‐dependent ADP‐ribosylation by Trypanosoma brucei Sir2. J Biol Chem 283:5317–5326. [DOI] [PubMed] [Google Scholar]
- 5. Rack JG, Morra R, Barkauskaite E, Kraehenbuehl R, Ariza A, Qu Y, Ortmayer M, Leidecker O, Cameron DR, Matic I, Peleg AY, Leys D, Traven A, Ahel I (2015) Identification of a class of protein ADP‐ribosylating sirtuins in microbial pathogens. Mol Cell 59:309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Honjo T, Nishizuka Y, Hayaishi O (1968) Diphtheria toxin‐dependent adenosine diphosphate ribosylation of aminoacyl transferase II and inhibition of protein synthesis. J Biol Chem 243:3553–3555. [PubMed] [Google Scholar]
- 7. Collier RJ, Kandel J (1971) Structure and activity of diphtheria toxin. I. Thiol‐dependent dissociation of a fraction of toxin into enzymically active and inactive fragments. J Biol Chem 246:1496–1503. [PubMed] [Google Scholar]
- 8. Han S, Craig JA, Putnam CD, Carozzi NB, Tainer JA (1999) Evolution and mechanism from structures of an ADP‐ribosylating toxin and NAD complex. Nat Struct Biol 6:932–936. [DOI] [PubMed] [Google Scholar]
- 9. Holbourn KP, Shone CC, Acharya KR (2006) A family of killer toxins. Exploring the mechanism of ADP‐ribosylating toxins. FEBS J 273:4579–4593. [DOI] [PubMed] [Google Scholar]
- 10. Simon NC, Aktories K, Barbieri JT (2014) Novel bacterial ADP‐ribosylating toxins: structure and function. Nat Rev Microbiol 12:599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gibson BA, Kraus WL (2012) New insights into the molecular and cellular functions of poly(ADP‐ribose) and PARPs. Nat Rev Mol Cell Biol 13:411–424. [DOI] [PubMed] [Google Scholar]
- 12. Barkauskaite E, Jankevicius G, Ahel I (2015) Structures and mechanisms of enzymes employed in the synthesis and degradation of PARP‐dependent protein ADP‐ribosylation. Mol Cell 58:935–946. [DOI] [PubMed] [Google Scholar]
- 13. Langelier MF, Planck JL, Roy S, Pascal JM (2012) Structural basis for DNA damage‐dependent poly(ADP‐ribosyl)ation by human PARP‐1. Science 336:728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pascal JM, Ellenberger T (2015) The rise and fall of poly(ADP‐ribose): an enzymatic perspective. DNA Repair 32:10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Forst AH, Karlberg T, Herzog N, Thorsell AG, Gross A, Feijs KL, Verheugd P, Kursula P, Nijmeijer B, Kremmer E, Kleine H, Ladurner AG, Schuler H, Luscher B (2013) Recognition of mono‐ADP‐ribosylated ARTD10 substrates by ARTD8 macrodomains. Structure 21:462–475. [DOI] [PubMed] [Google Scholar]
- 16. Karras GI, Kustatscher G, Buhecha HR, Allen MD, Pugieux C, Sait F, Bycroft M, Ladurner AG (2005) The macro domain is an ADP‐ribose binding module. EMBO J 24:1911–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gorbalenya AE, Koonin EV, Lai MM (1991) Putative papain‐related thiol proteases of positive‐strand RNA viruses. Identification of rubi‐ and aphthovirus proteases and delineation of a novel conserved domain associated with proteases of rubi‐, alpha‐ and coronaviruses. FEBS Lett 288:201–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Allen MD, Buckle AM, Cordell SC, Lowe J, Bycroft M (2003) The crystal structure of AF1521 a protein from Archaeoglobus fulgidus with homology to the non‐histone domain of macroH2A. J Mol Biol 330:503–511. [DOI] [PubMed] [Google Scholar]
- 19. Xu Y, Cong L, Chen C, Wei L, Zhao Q, Xu X, Ma Y, Bartlam M, Rao Z (2009) Crystal structures of two coronavirus ADP‐ribose‐1''‐monophosphatases and their complexes with ADP‐Ribose: a systematic structural analysis of the viral ADRP domain. J Virol 83:1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen D, Vollmar M, Rossi MN, Phillips C, Kraehenbuehl R, Slade D, Mehrotra PV, von Delft F, Crosthwaite SK, Gileadi O, Denu JM, Ahel I (2011) Identification of macrodomain proteins as novel O‐acetyl‐ADP‐ribose deacetylases. J Biol Chem 286:13261–13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kleine H, Poreba E, Lesniewicz K, Hassa PO, Hottiger MO, Litchfield DW, Shilton BH, Luscher B (2008) Substrate‐assisted catalysis by PARP10 limits its activity to mono‐ADP‐ribosylation. Mol Cell 32:57–69. [DOI] [PubMed] [Google Scholar]
- 22. Kim IK, Kiefer JR, Ho CM, Stegeman RA, Classen S, Tainer JA, Ellenberger T (2012) Structure of mammalian poly(ADP‐ribose) glycohydrolase reveals a flexible tyrosine clasp as a substrate‐binding element. Nat Struct Mol Biol 19:653–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jankevicius G, Hassler M, Golia B, Rybin V, Zacharias M, Timinszky G, Ladurner AG (2013) A family of macrodomain proteins reverses cellular mono‐ADP‐ribosylation. Nat Struct Mol Biol 20:508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rosenthal F, Feijs KL, Frugier E, Bonalli M, Forst AH, Imhof R, Winkler HC, Fischer D, Caflisch A, Hassa PO, Luscher B, Hottiger MO (2013) Macrodomain‐containing proteins are new mono‐ADP‐ribosylhydrolases. Nat Struct Mol Biol 20:502–507. [DOI] [PubMed] [Google Scholar]
- 25. Sharifi R, Morra R, Appel CD, Tallis M, Chioza B, Jankevicius G, Simpson MA, Matic I, Ozkan E, Golia B, Schellenberg MJ, Weston R, Williams JG, Rossi MN, Galehdari H, Krahn J, Wan A, Trembath RC, Crosby AH, Ahel D, Hay R, Ladurner AG, Timinszky G, Williams RS, Ahel I (2013) Deficiency of terminal ADP‐ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. EMBO J 32:1225–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 27. Zheng H, Chordia MD, Cooper DR, Chruszcz M, Muller P, Sheldrick GM, Minor W (2014) Validation of metal‐binding sites in macromolecular structures with the CheckMyMetal web server. Nat Protoc 9:156–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res 34:W116–W118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Holm L, Rosenstrom P (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res 38:W545–W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Otwinowski Z, Minor W, Processing of X‐ray diffraction data collected in oscillation mode In: Carter JCW, Sweet R, Eds. (1997) Methods in enzymology. New York, Academic Press, pp 307–326. [DOI] [PubMed] [Google Scholar]
- 31. Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L‐W, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive python‐based system for macromolecular structure solution. Acta Cryst D66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Cryst D66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen VB, Arendall WB, III , Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Cryst D66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA (2004) PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res 32:W665–W667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98:10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Corpet F (1988) Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 16:10881–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Robert X, Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]