

Abstract
Bilirubin is a potent antioxidant that is produced from the reduction of the heme degradation product biliverdin. In mammalian cells and Cyanobacteria, NADH/NADPH‐dependent biliverdin reductases (BVRs) of the Rossmann‐fold have been shown to catalyze this reaction. Here, we describe the characterization of Rv2074 from Mycobacterium tuberculosis, which belongs to a structurally and mechanistically distinct family of F420H2‐dependent BVRs (F‐BVRs) that are exclusively found in Actinobacteria. We have solved the crystal structure of Rv2074 bound to its cofactor, F420, and used this alongside molecular dynamics simulations, site‐directed mutagenesis and NMR spectroscopy to elucidate its catalytic mechanism. The production of bilirubin by Rv2074 could exploit the anti‐oxidative properties of bilirubin and contribute to the range of immuno‐evasive mechanisms that have evolved in M. tuberculosis to allow persistent infection.
Keywords: F420, biliverdin reductase, biliverdin, bilirubin, Mycobacterium tuberculosis, mycobacteria, enzyme catalysis, flavin/deazaflavin oxidoreductase
Short abstract
PDB Code: 5JAB
Abbreviations
- BVR
biliverdin reductase
- ESI‐MS
electrospray ionization mass spectrometry
- F420
factor 420
- F‐BVR
F420H2‐dependent biliverdin reductase
- FDOR
flavin/deazaflavin oxidoreductase
- Fgd
F420‐dependent glucose‐6‐phopshate dehydrogenase
- G6P
glucose‐6‐phosphate
- HO
heme oxygenase
- MD
molecular dynamics
- NO
nitric oxide
- RMSD
root mean square deviation
- RMSF
root mean square fluctuation
- WT
wild type
Introduction
Biliverdin is a naturally occurring linear tetrapyrrole produced from the oxidative degradation of heme by heme oxygenases (HOs) (Fig. 1).1 It is used for a variety of purposes, for instance as the precursor to light harvesting phycobilins in Cyanobacteria and green algae,2 and as pigmentation in birds, amphibians and reptiles.3 In mammals, biliverdin is reduced to bilirubin by biliverdin reductases (BVRs).4 Bilirubin is toxic in high concentrations and mostly excreted after solubilization by conjugation with glucuronic acid.4 However, it is also a strong antioxidant and lipophilic radical scavenger,5 capable of compensating for a 10,000‐fold increase in reactive oxygen species and quenching reactive nitrogen species like nitric oxide (NO) to protect against cellular oxidative damage.6, 7, 8, 9
Figure 1.
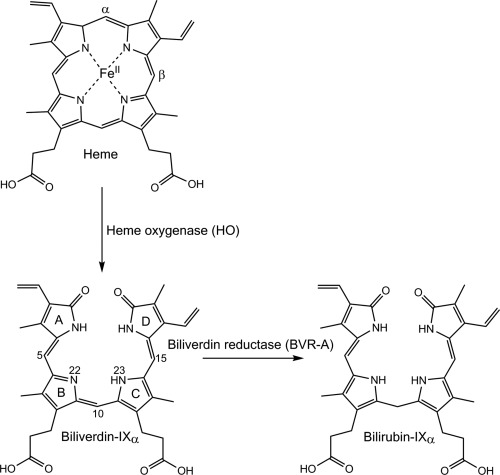
Structures of the heme degradation products biliverdin‐IXα and bilirubin IXα. Pyrrole rings (A–D) and atom positions discussed in the text are labeled on biliverdin‐IXα.
Two BVR isoforms have been identified from mammalian cells, BVR‐A and BVR‐B.6, 10, 11, 12 BVR‐A reduces biliverdin‐IXα (formed by the cleavage of heme at the α‐meso position) to bilirubin‐IXα (Fig. 1).10, 12 It is the predominant isoform found in adult mammalian tissue,13 with the highest expression levels found in the brain, lungs and pancreas.12, 14 BVR‐B is highly expressed in fetal tissue and reduces biliverdin‐IXβ (heme cleaved at the β‐meso position) to bilirubin‐IXβ.12 In adults, bilirubin‐IXβ constitutes just 3–5% of total bilirubin in bile while 95–97% is bilirubin‐IXα.15 Both mammalian BVR isoforms utilize a nicotinamide cofactor (NADH or NADPH) as an electron donor and have catalytic domains similar to short‐chain alcohol dehydrogenases with a core α/β dinucleotide binding motif (a “Rossmann” fold).16, 17 BVR‐A also contains an additional C‐terminal domain that forms a six stranded β‐sheet, containing additional motifs for regulatory serine/threonine kinase activity and DNA and transcription factor binding sites that allows participation in cellular signaling.11, 17 In mammals, BVR‐A expression is induced in response to stress resulting from reactive oxygen species, endotoxins and heavy metals,10, 18 and has been shown to confer resistance towards oxidative stress.19, 20 This is partly because of its involvement in signaling during inflammation,11, 21 for instance in the induction of anti‐inflammatory cytokines,22 modulation of downstream cytokine responses,23 and upregulation of oxidative stress response proteins like HO.24 However, the oxidative stress resistance conferred by BVR‐A can also be attributed to the antioxidative and cytoprotective nature of the bilirubin produced.6, 7, 8, 9
Although the antioxidative and anti‐inflammatory roles of BVR and bilirubin in mammalian cells have been widely described and studied,21 only one other BVR has been characterized; an NADH/NADPH‐dependent enzyme from the cyanobacterium Synechocystis sp. PCC 6803.25 In vivo production of bilirubin has been detected in Salmonella enterica serovar Typhimurium,26 but the protein that was responsible for this transformation was not identified. Although there has been relatively little focus on prokaryotic BVRs, bilirubin can have a similar antioxidative effect on bacterial cells.27 Recent work has shown that infection of macrophages by Mycobacterium tuberculosis, Mycobacterium avium, and Mycobacterium abscessus induces HO production,27, 28, 29 which produces biliverdin and subsequently bilirubin, and that these molecules have a protective effect on the bacteria during early infection.27 Bilirubin has also been shown to downregulate the production of inducible nitric oxide synthases,30, 31 that generate microbicidal NO in human and murine macrophages infected with M. tuberculosis.32, 33
The many strategies used by M. tuberculosis to evade the host immune response, including the involvement of BVRs, are not well understood, but are of critical importance because they allow the pathogen to persist within macrophages, leading to latent infection.34 Over a third of the world's population is infected by latent M. tuberculosis, which can convert to active tuberculosis (TB), especially in immune‐compromised patients, resulting in approximately 10 million cases of TB and 1.5 million deaths in 2015.35 Our recent functional assignment of a flavin/deazaflavin oxidoreductase (FDOR) superfamily in Actinobacteria,36 showed that the M. tuberculosis protein Rv2074 had significant sequence similarity to a novel BVR from Mycobacterium smegmatis (MSMEG_3880) that is structurally and mechanistically distinct to the previously described NADH/NADPH‐dependent BVRs, relying on the deazaflavin cofactor F420H2. 36 The crystal structure of Rv2074 was first solved as an apo‐protein 10 years ago,37 and annotated as an FMN‐dependent pyridoxine 5′‐phosphate oxidase. To better understand the function and physiological roles of F420H2‐dependent biliverdin reductases (F‐BVRs) in TB, we have performed a detailed mechanistic analysis of Rv2074, solving its X‐ray crystal structure in complex with F420 for the first time, and elucidating its catalytic mechanism using site‐directed mutagenesis, molecular dynamics simulations and NMR spectroscopy. We also show that this family of BVRs is exclusive to Actinobacteria and is abundant in pathogenic and commensal mycobacteria, where they are likely to produce bilirubin during infection as an antioxidant and cytoprotectant.
Results
Rv2074 reduces biliverdin‐IXα to bilirubin‐IXα
To further investigate the predictions from our bioinformatics analysis,36 specifically that the M. tuberculosis FDORs Rv2074 and Rv1155 are biliverdin reductases,36 we tested the ability of these enzymes to catalyze the reduction of biliverdin‐IXα to bilirubin‐IXα in vitro using purified, recombinant proteins. In the absence of either enzyme, no increase in absorbance was observed in the broad peak at 450 nm that is characteristic of bilirubin‐IXα production [Fig. 2(A)]. Similarly, the reaction did not proceed in the absence of glucose‐6‐phosphate (G6P) or F420‐dependent G6P dehydrogenase (Fgd) required for F420H2 production. In contrast, when Rv2074 and Rv1155 were included in the reaction, we observed a rapid change in the UV/Vis spectrum of the reaction, with a reduction in the absorbance of the peaks characteristic of biliverdin‐IXα at 390 nm and 690 nm. Bilirubin‐IXα formation is indicated by the increase in absorbance at 450 nm that includes a shoulder at 510 nm due to its low solubility in aqueous solutions at pH 7.5.38 The characteristic spectroscopic peaks of these compounds allowed us to monitor the reaction continuously and obtain kinetic parameters for the enzyme‐catalyzed reactions [Fig. 2(B), Table 1]. Rv2074 exhibited kinetic efficiency that is consistent with a native substrate, with a low K M value of 8.0 ± 1.0 µM and a k cat/K M of 9 × 103 M−1 s−1 that is comparable to other FDORs with their proposed native substrates like the menaquinone analogue menadione for the F420H2‐dependent quinone reductase Rv3547 (K M value of 3.4 ± 1.3 µM and k cat/K M of 9 × 104 M−1 s−1).39 It is also comparable to its homolog from M. smegmatis MSMEG_3880 (K M of 5.7 ± 0.2 and k cat/K M of 4 × 104 M−1 s−1) that has 85% amino acid identity with Rv2074.36 Mammalian and cyanobacterial BVR‐As have similar K M values ranging from 1 to 7 µM, and a range of specific activities reported at the maximum observed reaction rate (0.2–8 µmol min−1 mg−1)40, 41, 42, 43 that are comparable to Rv2074 (0.18 µmol min−1 mg−1). In contrast to Rv2074, the k cat/K M of Rv1155 for biliverdin‐IXα was 30‐fold lower (3 × 102 M−1 s−1), and the K M for the substrate was 10‐fold higher, at 82 ± 13 µM, which suggests biliverdin‐IXα is unlikely to be the native substrate of Rv1155, and that it is likely to have evolved to catalyze the reduction of an alternative molecule.
Figure 2.
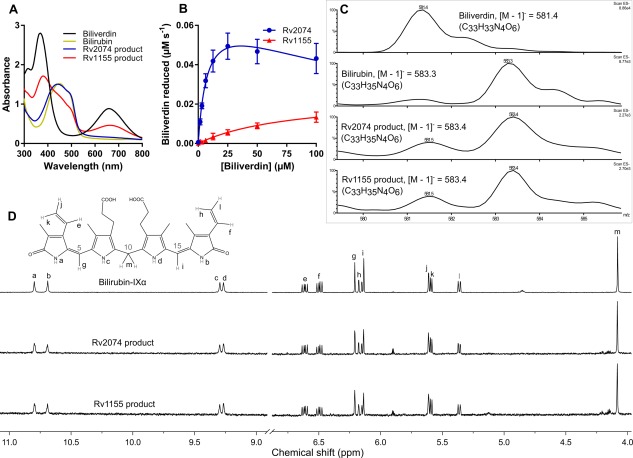
Characterization of F‐BVRs from M. tuberculosis and the reaction product formed. A. Absorbance spectra in aqueous solution of the reaction products formed by Rv2074 and Rv1155 in comparison to biliverdin‐IXα and bilirubin‐IXα. Differences between the Rv2074 and Rv1155 products to pure bilirubin‐IXα can be attributed to remaining unreacted biliverdin‐IXα. B. Activity of Rv2074 and Rv1155 (1 µM) with biliverdin‐IXα in the presence of reduced F420H2. For Rv2074, K M = 8.0 ± 1.0 µM, k cat = 7.5 × 10−2 ± 4.5 × 10−3 s−1 and k cat/K M = 8.9 × 103 M−1 s−1. For Rv1155, K M = 82.8 ± 12.7 µM, k cat = 2.4 × 10−2 ± 2.1 × 10−3 s−1, and k cat/K M = 2.9 × 102 M−1 s−1. C. Low resolution (ESI negative) mass spectra of the reaction products compared to biliverdin‐IXα and bilirubin‐IXα. D. Proton NMR spectra of the products formed compared with bilirubin‐IXα. Chemical shifts were assigned as previously reported,44 and peaks for the methyl and propionate substituents are not shown for clarity.
Table 1.
In vitro Activity of Rv2074, Rv1155, and Rv2074 Mutants with Biliverdin IXα
| Protein | k cat (s−1) | K M (µM) | k cat/K M (M−1 s−1) |
|---|---|---|---|
| Rv2074 | 7.2 × 10−2 ± 4.5 × 10−3 | 8.0 ± 1.0 | 8.9 × 103 |
| Rv1155 | 2.4 × 10−2 ± 2.1 × 10−3 | 82.8 ± 12.7 | 2.9 × 102 |
| R21A | 5.1 × 10−2 ± 5.5 × 10−3 | 15.4 ± 3.0 | 3.3 × 103 |
| R109A | 4.0 × 10−2 ± 2.3 × 10−3 | 21.2 ± 3.3 | 1.9 × 103 |
| R112A | 4.7 × 10−2 ± 3.3 × 10−3 | 13.8 ± 1.7 | 3.4 × 103 |
| R117A | 2.0 × 10−2 ± 1.4 × 10−3 | 19.7 ± 3.7 | 1.0 × 103 |
| Y104A | 0.7 × 10−2 ± 6.8 × 10−3 | 12.8 ± 3.9 | 0.5 × 103 |
| Y104F | 1.4 × 10−2 ± 1.4 × 10−3 | 14.8 ± 2.5 | 1.0 × 103 |
| Y108A | 0.4 × 10−2 ± 6.9 × 10−3 | 19.0 ± 8.4 | 0.2 × 103 |
| Y108F | 2.0 × 10−2 ± 5.6 × 10−3 | 7.6 ± 0.7 | 2.7 × 103 |
Our previous preliminary assignment of biliverdin reductase activity to this family of mycobacterial proteins was based on spectroscopic analysis, but did not explore the reaction mechanism in detail.36 To confirm whether reduction was taking place, and to identify the nature of the reaction, we performed mass spectrometry on the purified reaction products [Fig. 2(C)]. Electrospray ionization mass spectrometry (ESI‐MS) was used to confirm loss of a compound with an equivalent mass to biliverdin‐IXα (582.2 Da; observed [M−H] − m/z = 581.4) and accumulation of a product with an equivalent mass to bilirubin‐IXα (584.3 Da; [M−H] − m/z = 583.4). This was followed by proton NMR‐spectroscopy to identify the position at which biliverdin‐IXα was reduced [Fig. 2(D)]. The proton NMR spectra for the products (assigned as previously described)44 showed that the reaction products from the Rv2074 and Rv1155‐catalyzed reactions were essentially identical and displayed clear singlets at 4.09, 6.15 and 6.22 ppm corresponding to a methylene bridge at the C10 position and methine moieties at the C15 and C5 positions of bilirubin, respectively, thus confirming the specific reduction of biliverdin at the C10 position.
The structure of Rv2074 with its native cofactor, F420
The apo‐enzyme structure of Rv2074 has been reported previously,37 and was initially suggested to be an FMN‐dependent enzyme based on homology to the human and Escherichia coli pyridoxamine 5′‐phosphate oxidases.45, 46 However, our bioinformatics analysis suggested that it might instead function with F420 as a cofactor.36 The results presented in Figure 2 are the first demonstration of any catalytic activity with Rv2074, and notably occur in an F420H2 dependent‐fashion, suggesting that it is indeed an F420H2 dependent oxidoreductase. In order to better understand the catalytic mechanism involved in biliverdin‐IXα reduction, we have solved the structure of Rv2074 in complex with F420 at a resolution of 1.65 Å (Table 2). Omit electron density corresponding to one molecule of F420 with an oligoglutamate tail consisting of three residues is observed in the cofactor binding site of each of the four protein chains in the asymmetric unit. Each monomer adopts the conserved split β‐barrel protein fold characteristic of the FDORs, as previously described,36, 47, 48, 49 and the proteins are arranged as homodimers, which is supported by analysis from the Proteins, Interfaces, Structures and Assemblies (PISA) server [Fig. 3(A)].50 The F420 binding site is located at the dimer interface as observed in the Rv1155‐F420 complex,49 and also in complexes of FDORs with other flavin cofactors, including the FMN binding pyridoxamine‐5‐phosphate oxidases and the FAD binding MSMEG_4975 from M. smegmatis.36, 45, 46 In many respects, the binding of F420 to Rv2074 is similar to that observed in Rv1155 [Fig. 3(B)],49 particularly at the phosphate group, which is coordinated by a highly conserved lysine residue (Lys‐61) that is present in all members of the FDOR‐B superfamily.36 However, whereas the deazaflavin moiety in Rv1155 was observed to be present in a bent “butterfly” conformation, it is planar in Rv2074.
Table 2.
Data Collection and Refinement Statistics for Crystallography
| Data collection | |
| Space group | P 2 21 21 |
| Unit‐cell parameters | |
| a, b, c (Å) | 61.76, 88.62, 98.62 |
| α, β, γ (°) | 90, 90, 90 |
| Wavelength (Å) | 0.9537 |
| Resolution range (Å)a | 65.9–1.65 (1.68–1.65) |
| Unique reflectionsa | 65830 (3221) |
| Completeness (%)a | 100 (99.6) |
| Multiplicitya | 7.1 (6.7) |
| R merge a, b | 0.181 (1.261) |
| R pim a, c | 0.073 (0.527) |
| Mean <I/σ(I)>a | 9.3 (1.7) |
| CC1/2 a, d | 0.994 (0.581) |
| Molecules per asymmetric unit | 4 |
| Solvent content (%, v/v) | 43.8 |
| Refinement | |
| Reflections used | 62534 |
| Resolution range (Å)a | 65.92–1.65 (1.69–1.65) |
| R work/R free a, e | 0.178/0.217 (0.295/0.375) |
| Number of atoms (all) | 5032 |
| Water molecules | 654 |
| Average B‐factor (Å2) | |
| Main chains | 17.9 |
| Side chains | 22.3 |
| Water molecules | 30.5 |
| F420 molecules | 22.9 |
| R.M.S. deviations | |
| Bond lengths (Å) | 0.019 |
| Bond angles (°) | 1.962 |
| Ramachandran plot regions (%) | |
| Favored | 98.6 |
| Allowed | 1.4 |
| Outliers | 0 |
| PDB ID | 5JAB |
Values in parenthesis are for the highest‐resolution shell.
R merge = (Σh Σi|I h i – ⟨I h⟩|)/(Σh Σi ⟨I h⟩), where ⟨I h⟩ is the average intensity of i symmetry‐related observations of the unique refection h.
R pim = (Σh Σi (1/(n h – 1))1/2 |I h i – ⟨I h⟩|)/(Σh Σi ⟨I h⟩).
CC1/2 = linear correlation coefficient between intensities from random half‐datasets.
R work = Σh |F (obs) – F (calc)|/Σh |F (obs)| and 5% of the data that were excluded from the refinement were used to calculate R free.
Figure 3.
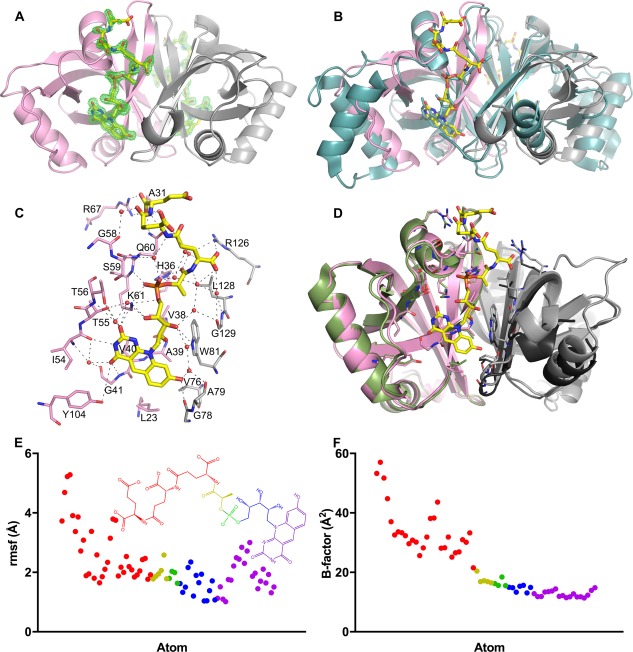
Structure of Rv2074 complexed with F420. A. Overall structure of the Rv2074:F420 complex at 1.65 Å resolution (chain A in pink and B in grey). Difference (mFo–DFc) density at 3 σ for the F420 molecules at each chain are shown in green. Clear density showed the placement of the alloxazine, ribityl and phospholactate moieties in all four monomers in the asymmetric unit, although the clarity of the density for the polyglutamate chain was variable. B. Structure shown in A overlaid with the homodimeric Rv1155:F420 complex in cyan (PDB ID: 4QVB).49 C. Residues involved in interacting with F420 in Rv2074. Pink and grey residues represent those from Chain A and Chain B of the homodimer, respectively. D. Comparison of the Rv2074:F420 complex in A with the structure of the apo‐protein (PDB ID: 2ASF).37 Chain A is shown in green and its crystallographic symmetry mate is in black, where key residues involved in F420 binding in C are shown as sticks. E. Plot of the RMSF values for the atoms of F420H2 showing the larger deviation of the oligo‐glutamate chain compared with the rest of the molecule over the course of the MD simulation. The atoms are color coded by group as represented in the inset structure. F. Plot of the temperature (B) factors in the crystal structure for the atoms in the F420 molecule in E, showing a similar trend.
Each F420 molecule is coordinated through an extensive network of intermolecular interactions [Fig. 3(C)]. Hydrogen bonds between the nitrogen and carboxyl groups of the deazaflavin moiety of F420 and residues from Chain A of the homodimer include the main chain amide group of Gly‐41 and the side chains of Thr‐55, Thr‐56, and Tyr‐104. In addition, the carbonyl group of Gly‐78 of Chain B hydrogen bonds to the hydroxyl group of the deazaflavin. Hydrophobic interactions from Leu‐23, Ala‐39, and Val‐40 in Chain A and Val‐76 in Chain B provide additional stabilization to the aromatic rings. The highly conserved base Lys‐61 from Chain A stabilizes the ribityl moiety and forms a salt bridge to the negatively charged phosphate group, along with His‐36. Trp‐81 from Chain B also hydrogen bonds with the ribityl moiety. Val‐38 of Chain A and Leu‐128 of Chain B form hydrophobic interactions with the methyl group of the lactyl moiety. A series of positively charged residues form salt bridges with the negatively charged glutamate side chains of the oligoglutamate tail (His‐36, Gln‐60, Arg‐67 of Chain A and Arg‐126 of Chain B).
In comparison with the apo‐enzyme,37 the overall topology of the F420 bound protein remains mostly unchanged, except that residues at the cofactor binding site undergo conformational change to optimize its interaction with F420 [Fig. 3(D)]. These include Lys‐61, Arg‐67, Gln‐60, and His‐36 from Chain A and Arg‐126 from Chain B that interact with the oligoglutamate chain of F420 and Leu‐23 from Chain A that has moved further away due to hydrophobic interactions with the deazaflavin moiety. In addition, Chain B of the F420 bound complex moves closer to Chain A, due to the association of Val‐76, Gly‐78, Ala‐79, Trp‐81, and Leu‐128 of Chain B with the cofactor at the dimer interface [Fig. 3(D)].
To investigate the contribution of the different regions of the protein to F420 binding, we performed molecular dynamics (MD) simulations with the cofactor bound homodimer for a total of 100 ns. These results show that the ribityl‐phospholactyl region maintains salt bridge interactions with Lys‐61 and His‐36 during the simulations, anchoring F420H2 in the binding pocket. The oligoglutamate tail and, to some extent, the deazaflavin group seem to be more mobile [Fig. 3(E)], consistent with the decreasing electron density and increasing temperature factors observed in this region of the oligoglutamate chain [Fig. 3(A,F)].
Biliverdin binding and reduction by Rv2074
Extensive co‐crystallization and crystal‐soaking experiments with Rv2074 and biliverdin‐IXα resulted in weak, ambiguous electron density within the active site that was not sufficient for accurate modeling of the substrate. To further investigate the mode of substrate coordination and the likely catalytic mechanism, we have used a combination of in silico docking and MD simulations to predict the most likely conformation of biliverdin in the active site of Rv2074 [Fig. 4(A)]. Using AUTODOCK VINA,51 we obtained initial binding poses that showed biliverdin with the pyrrole rings B and C on either side of the reactive C10 atom stacked on to the deazaflavin moiety with the propionate chains and the distal pyrrole rings A and D in variable positions. Through manual inspection and analysis of the docking scores, the most plausible binding orientation, with the propionate chains stabilized by arginine residues in the active site (Arg‐21, Arg‐109, Arg‐112 and Arg‐117) and the distal pyrrole rings A and D oriented such that biliverdin is in a porphyrin‐like conformation, was then subjected to two independent sets of 50 ns MD simulations for a combined total 100 ns [Fig. 4(A)]. These results show that the substrate is stable in this conformation for the length of the simulation and is stabilized by hydrogen bonding of the propionate side chains with Arg‐109 and Arg‐112, with intermittent binding contributions from Arg‐21 also being observed during the simulation. In addition, Arg‐117 forms cation–π interactions with pyrrole ring C and pyrrole ring B forms aromatic interactions with the deazaflavin of F420. The propionate side chains of rings B and C show significant flexibility [Fig. 4(B)], which is consistent with the flexibility of the multiple arginine residues (Arg‐21, Arg‐109, and Arg‐112) that interact with them. In contrast, the pyrrole rings B and C are less mobile, especially in comparison to the terminal pyrrole rings A and D, which are extremely flexible in the large open active site [Fig. 4(B)], with no specific interactions with the protein [Fig. 4(A)]. These observations suggest that the hydride transfer from F420H2 to biliverdin‐IXα relies upon correct alignment and stable binding of the B and C pyrrole groups that determine the position of the reactive C10 of biliverdin with respect to the cofactor.
Figure 4.
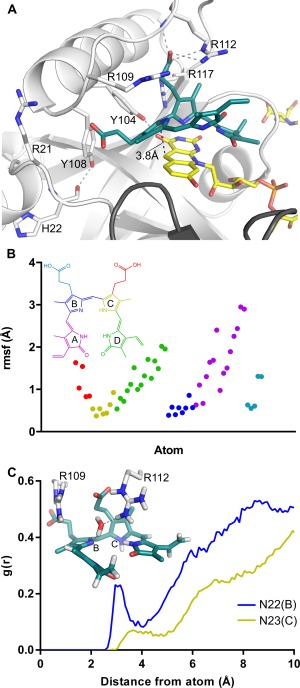
Biliverdin binding mode of Rv2074. A. Dominant biliverdin binding conformation simulated by MD, representing 59% of the structures at a cutoff of 2 Å. Residues chosen for mutagenesis are shown as sticks and dotted lines in grey represent hydrogen bonding interactions. B. Plot of the RMSF values for biliverdin demonstrating the mobility of the propionate groups and pyrrole rings A and D compared to pyrrole rings B and C. The atoms are color‐coded by group as represented in the inset structure. C. Radial distribution function g(r) of water plotted by distance from reference nitrogen atoms N22 on pyrrole ring B or N23 on pyrrole ring C to oxygen of water. The inset figure shows one of the frames from the simulation where a water molecule is hydrogen bonding to N22(B) and to R112 within sufficient distance for proton transfer.
The MD simulation results were analyzed to identify the most populated conformations of the active site in the presence of substrate using cluster analysis (see Methods). The analysis was performed on the subset of atoms that included the substrate and the surrounding active site residues Arg‐21, Tyr‐104, Tyr‐108, Arg‐109, Arg‐112, and Arg‐117. Structures from the individual frames of the simulation were clustered together if their RMSD was lower than the cutoff value, which was set to 2.0 Å. The most populated cluster consisted of 59% of the structures at an RMSD cutoff of 2.0 Å and the representative conformation of substrate and the residues is shown in Figure 4(A). The C–C distance between F420 and substrate in the representative conformation is 3.8 Å, but the average value of this distance measured during simulations is 3.9 ± 0.3 Å. In previously solved ternary structures of F420 binding proteins with their substrates this distance was 2.7 Å and 3.1 Å,52, 53 This is consistent with typical Michaelis complex structures that precede the formation a transition state, which is characterized with donor–acceptor distance of ∼2.7 Å for hydride transfer.54 The flexibility of the deazaflavin ring of F420H2 and the C10 and pyrrole rings B and C of biliverdin, as evident from the RMSF plots which show fluctuations of 0.5 Å and 2 Å, respectively [Figs. 3(D) and 4(B)], can allow for the donor–acceptor distance to reach optimum values for successful hydride transfer.
To identify the residues within the active site that are directly involved in biliverdin reduction, we generated eight point mutants of Rv2074: Arg‐21‐Ala, Arg‐109‐Ala, Arg‐112‐Ala, Arg‐117‐Ala, Tyr‐104‐Ala, Tyr‐104‐Phe, Tyr‐108‐Ala, and Tyr‐108‐Phe [Fig. 4(C), Table 1]. These residues were chosen as they are located in active site pocket and are highly conserved among the putative F‐BVR family [Figs. 4(A) and 5(A)], suggesting that they are under evolutionary selection and are likely to have a functional role. All of these mutations led to some reduction in the catalytic efficiency, particularly the mutations of the active site tyrosine residues. Tyr‐104 hydrogen bonds to the carbonyl group on the pyrimidine ring of F420H2 and mutation of this amino acid to either alanine or phenylalanine reduces both biliverdin affinity (increased K M) and catalytic rate (k cat), suggesting that a stabilized F420 molecule is important for the reaction [Figs. 3(C) and 4(A)]. Although Tyr‐108 is located on the same loop as Arg‐109 that bind the propionate side chains of biliverdin, it is directed away from both the substrate and cofactor, with minimal fluctuation over the course of the simulation. However, the Tyr‐108‐Ala mutant shows almost complete loss of catalytic activity (k cat), while the Tyr‐108‐Phe mutant retains 30% activity compared to wild‐type Rv2074. In the wild type protein, the hydroxyl group of Tyr‐108 hydrogen bonds to the carbonyl group of the His‐22 backbone. This hydrogen bond is important to hold the loop containing the important arginine residue Arg‐21 within the correct vicinity. In addition to this, the aromatic group of Tyr‐108 and the Tyr‐108‐Phe mutant could also provide a secondary effect towards the catalytic activity by desolvation of the F420H2‐biliverdin interaction region. Although the immediate area above biliverdin was solvent accessible in the MD simulations, the area immediately below where the hydride transfer takes place was devoid of water molecules. The catalytic advantages for desolvated protein cages have been highlighted,55 where charge transfer occurs between reactant and product as the desolvated microenvironment can aid charge reorganization through stabilization of charged transition states such as those found in our system.
Figure 5.
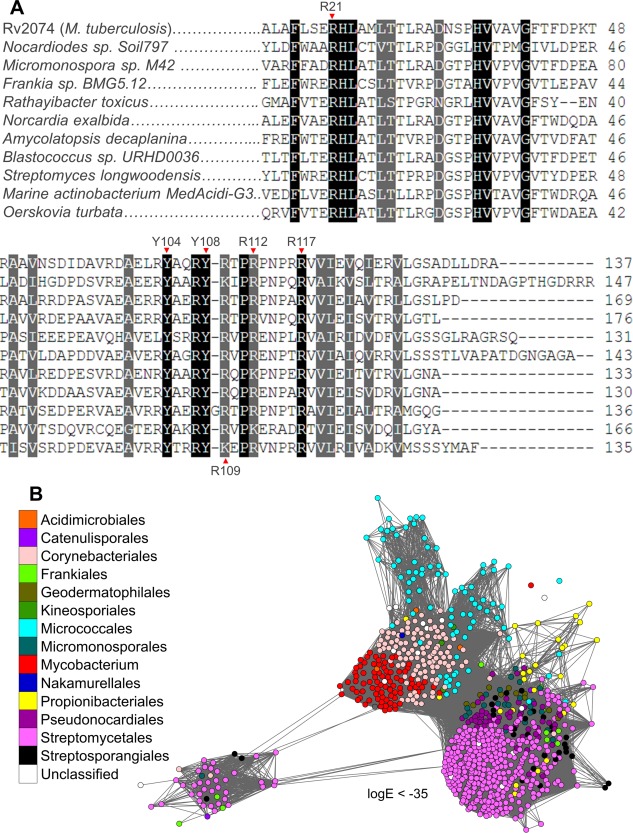
A. Sequence alignment of Rv2074 homologs from various Actinobacteria with ∼50% amino acid identity between them, showing conservation of active site residues chosen for site‐directed mutagenesis. B. SSN of all available F‐BVR homologs retrieved from the NCBI non‐redundant protein database using BLAST,59 showing their exclusive presence in species belonging to actinobacterial orders. Each node represents an individual protein, which are colored according to the taxonomic order of the originating organism, except for the genus Mycobacterium. Only edges with BLAST log E‐values less than −35 are shown.
All four arginine residues are involved in biliverdin binding by association with the propionate side chains (Arg‐21, Arg‐109, Arg‐112) and cation–π interactions with the pyrrole ring C (Arg‐117), and mutations of any of these residues to alanine negatively affects both k cat and K M (Table 1). Arg‐21 associates with biliverdin by hydrogen bonding with the propionate side chain of pyrrole ring B, although this was a transient interaction throughout the simulation, occurring intermittently when the biliverdin substrate drifted away from the proper cofactor alignment. Meanwhile Arg‐117 was consistently observed to form cation–π interactions with pyrrole ring C [Fig. 4(A)]. The Arg‐117‐Ala mutation has the highest influence on reaction rate, lowering the k cat to just 25% of the wild type, suggesting that the stabilization of pyrrole ring C by this residue is important in maintaining the correct biliverdin orientation for hydride transfer.
Since biliverdin features electronic delocalization across pyrrole rings B and C, either ring could be protonated at any one time. Both these protonated conformations were investigated in the MD simulations by manually protonating either N22 on ring B or N23 on ring C of biliverdin in separate simulations with otherwise identical parameters. The previously mentioned MD results were all performed with the N23 atom protonated since in the alternate conformation with N22 atom protonated, the enzyme‐substrate complex was observed to be unstable and the cofactor and substrate moved out of alignment for the length of the simulation. This further highlights the importance of the cation–π interactions involving Arg‐117, since it requires proper aromatic configuration of the biliverdin substrate to bind in a stable conformation. The very high sequence conservation of arginine at position 117 also lends credence to this as arginine residues are more often involved in cation–π bonding than other charged residues such as lysine [Fig. 5(A)].56 The complete reduction of biliverdin to bilirubin requires proton transfer to one of the pyrrole nitrogens in addition to hydride transfer to C10. In this study we have mutated every ionizable residue that forms significant interactions with the substrate near C10 to non‐ionizable residues, such as alanine or phenylalanine (Table 1). None of these mutations led to complete loss of activity, suggesting that the protein is unlikely to be the proton donor in the reaction. Computational analysis of biliverdin reduction by the human BVRs has suggested that the primary proton donor is likely to be a hydroxonium ion, generated via hydrogen bonding of a water molecule with a basic residue such as histidine or arginine.57, 58 Therefore, using MD simulations we investigated the permeability of solvent within the active site and the hydrogen bonding behavior near the pyrrole nitrogens of interest, N22 on ring B and N23 on ring C [Fig. 4(C)]. In statistical mechanics, the radial distribution function g(r) can be used to define the density distribution or the probability of finding particles as a function of a distance r from a central reference particle. Thus, in our case, the radial distribution function g(r) can be used to determine the probability of finding water molecules with respect to a distance r from the proton accepting nitrogen atoms which are used as our references. Water molecules were found to associate with N22, which was deprotonated in the simulation, with a clear peak in the probability distribution at a distance of 3.0 Å (from pyrrole nitrogen to the oxygen atom of water) before gradually extending into the bulk solvent, providing a sufficient window for hydrogen bonding. In contrast, the distribution with N23 (protonated) as a reference shows a much smaller probability peak at a longer distance of 3.6 Å suggesting that proton transfer to N22 during the reaction is more favorable in this biliverdin conformation due to easier solvent access. Furthermore, the presence of Arg‐109 and Arg‐112 in this vicinity [Fig. 4(C)], and the reduction of catalytic activity without these residues (Table 1), suggests that they could assist in generating a hydroxonium ion for this reaction (Fig. 6).
Figure 6.
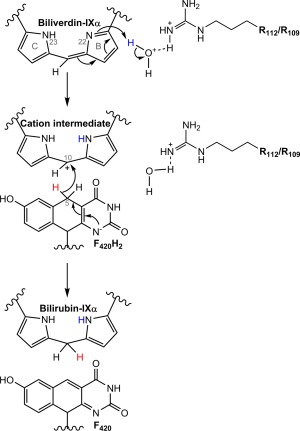
Proposed mechanism for biliverdin reduction by Rv2074. Proton donation to N22 on pyrrole ring B of biliverdin‐IXα by a hydroxonium ion generated by a nearby arginine residue occurs first, creating a cationic intermediate. This is then followed by hydride transfer from C5 of F420H2 to C10 of the intermediate, resulting in the formation of bilirubin‐IXα.
Abundance and distribution of F‐BVRs
Finally, we investigated the taxonomic distribution of the F‐BVR family, especially within pathogenic mycobacteria, and its presence in other Actinobacteria. We retrieved all sequences with more than 50% sequence identity to Rv2074 from the NCBI non‐redundant protein database using BLAST,59 and found that these proteins are exclusively found in Actinobacteria and are particularly abundant in mycobacteria (Fig. 5(B)]. In addition to M. tuberculosis, homologs were also identified from opportunistic human pathogens including the more commonly reported species Mycobacterium ulcerans, avium, marinum, intracellulare, abscessus, simiae, kansasii, haemophilum, xenopi, genavense, fortuitum, and chelonae (Table 3).60, 61, 62 It is absent from the genome of Mycobacterium leprae, which has undergone significant reductive evolution,63 and from the genome of the attenuated Mycobacterium bovis used for vaccination against M. tuberculosis.64 The presence of these proteins appears more common in obligate and opportunistic pathogens (86% of genomes) rather than in species that are not known to cause infection of which only 60% of the analyzed genomes encode an F‐BVR (Table 3). All other orders of Actinobacteria identified are predominantly soil bacteria and have previously been suggested to produce F420.65
Table 3.
Conservation of F‐BVRs in Mycobacteria
| Mycobacterium genome | Rv2074 homolog | common reservoir/isolation source59–61 | Genome accession |
|---|---|---|---|
| Pathogenic | |||
| Mycobacterium tuberculosis | Yes | Human infection | NC_000962.3 |
| Mycobacterium bovis | Yes | Human/animal infection | NZ_JKAL00000000.1 |
| Mycobacterium africanum | Yes | Human infection | NZ_JLAN00000000.1 |
| Mycobacterium canettii | Yes | Human infection | NC_019951.1 |
| Mycobacterium leprae | — | Human infection | NC_002677.1 |
| Opportunistic/commensal | |||
| Mycobacterium abscessus | Yes | Soil, water, infection | NC_010397.1 |
| Mycobacterium avium | Yes | Water, soil, infection | NC_002944.2 |
| Mycobacterium intracellulare | Yes | Water, soil, infection | NC_016946.1 |
| Mycobacterium marinum | Yes | Water, soil, infection | NC_010612.1 |
| Mycobacterium kansasii | Yes | Human infection | NC_022663.1 |
| Mycobacterium fortuitum | Yes | Soil, water, infection | NZ_CP011269.1 |
| Mycobacterium xenopi | Yes | Soil, water, infection | NZ_AJFI00000000.1 |
| Mycobacterium neoaurum | Yes | Soil, infection | NC_023036.2 |
| Mycobacterium ulcerans | Yes | Water, soil, infection | CP000325.1 |
| Mycobacterium chelonae | Yes | Water, soil, infection | NZ_CP010946.1 |
| Mycobacterium genavense | Yes | Human infection | NZ_JAGZ00000000.1 |
| Mycobacterium iranicum | Yes | Human infection | NZ_AUWT00000000.1 |
| Mycobacterium yongonense | Yes | Human infection | NC_021715.1 |
| Mycobacterium simiae | Yes | Rhesus monkey | NZ_CBMJ000000000.2 |
| Mycobacterium haemophilum | Yes | Human infection | NZ_CP011883.2 |
| Mycobacterium lepromatosis | — | Human infection | NZ_LAWX00000000.1 |
| Mycobacterium setense | Yes | Human infection | NZ_JTJW00000000.1 |
| Mycobacterium kyorinense | — | Human infection | NZ_BBKA00000000.1 |
| Mycobacterium vulneris | Yes | Human infection | NZ_CCBG000000000.1 |
| Mycobacterium celatum | — | Human infection | NZ_BBUN00000000.1 |
| Rarely pathogenic/commensal | |||
| Mycobacterium phlei | Yes | Soil | NZ_AJFJ00000000.1 |
| Mycobacterium smegmatis | yes | Soil | NC_008596.1 |
| Mycobacterium rhodesiae | Yes | Soil | NC_016604.1 |
| Non‐pathogenic | |||
| Mycobacterium rutilum | — | Soil | BBHF00000000.1 |
| Mycobacterium pallens | — | Soil | BBHE00000000.1 |
| Mycobacterium crocinum | — | Soil | BBHD00000000.1 |
| Mycobacterium rufum | Yes | Soil | NZ_JROA00000000.1 |
| Mycobacterium aromaticivorans | Yes | Soil | NZ_JALN00000000.2 |
| Mycobacterium indicus pranii | Yes | Soil | NC_018612.1 |
| Mycobacterium bovis (BCG vaccine) | — | Laboratory | NZ_JNAF00000000.1 |
| Mycobacterium vanbaalenii | Yes | Mineral oil, soil | NC_008726.1 |
| Mycobacterium gilvum | Yes | Water, soil | NC_014814.1 |
| Mycobacterium chubuense | Yes | Soil | NC_018027.1 |
Discussion
The catalytic mechanism of F‐BVRs
Proton NMR‐spectroscopy confirmed that Rv2074 catalyzes the reduction of biliverdin‐IXα at the C10 position in an F420H2‐dependent reaction. The crystal structure of the F420:Rv2074 complex allowed us to simulate biliverdin binding at the active site. Along with the site‐directed mutagenesis of key catalytic residues, we can now propose a plausible mechanism for this enzyme. Previous analysis of human BVR‐A and BVR‐B suggests that, in these enzymes, biliverdin reduction involves the use of NADPH as a cofactor and proceeds in two steps: (i) proton donation by a hydroxonium ion to a pyrrole nitrogen atom adjacent to C10 of biliverdin to make a cationic intermediate, followed by (ii) hydride transfer from NADPH to C10 to form bilirubin.57, 58 Based on our results, we also propose that a similar two‐step mechanism is plausible with F420H2 as the hydride donor (Fig. 6). This is also consistent with our recent work showing that F420 most likely exists in its deprotonated state when bound to FDORs,66 requiring the need for an alternate proton source during the reaction.
For the initial proton donation step, the most likely donor is a hydroxonium ion, as proposed for BVR‐A and BVR‐B,57, 58 considering the accessibility of the active site to water and the presence of Arg‐109 and Arg‐112 in the vicinity to generate the proton donating hydroxonium ion. The aromatic nature and steric bulk of the Tyr‐108 side chain most likely plays a role in desolvating the hydride transfer site and forming a nonpolar environment which helps enhance catalytic activity through strengthening polar interactions and stabilization of the charged transition state.55 This is corroborated by the MD simulations that show the Tyr‐108 residue does not form a direct association with either the cofactor or substrate, and instead has its hydroxyl group hydrogen bonded to the backbone carbonyl group of His‐22. This interaction helps align this loop within the correct distance for the neighboring Arg‐21 residue to interact with biliverdin. The alignment and distance between biliverdin and F420H2, which requires the presence of Arg‐21, Arg‐109, Arg‐112, and Arg‐117 for substrate stabilization, is essential for optimal hydride transfer to take place from C5 of F420H2 to C10 of biliverdin.
A physiological role for F‐BVRs
Of the two proteins with F‐BVR activity in M. tuberculosis, only Rv2074 appears to be functionally specialized in terms of its affinity for biliverdin IXα reduction and its catalytic efficiency. In contrast, Rv1155 does not bind biliverdin‐IXα with high affinity, suggesting that its biliverdin reductase activity is a promiscuous, and probably not physiologically relevant, function. Such substrate promiscuity appears to be common in the F420H2‐dependent FDORs, probably as a result of their common evolutionary origin.36 While homologs of Rv2074 are present in a number of pathogenic mycobacteria, we did not observe any closely related homolog (>50% amino acid identity) in the greatly reduced genome of M. leprae.63 However, M. leprae does have a homologue of Rv1155 (88% amino acid identity),36 that could at least in part compensate for the loss of the Rv2074 homolog through its promiscuous F‐BVR activity.
The abundance and conservation of F‐BVRs in Actinobacteria suggests that they are under evolutionary selection for an important physiological role. Heme is universally used as a prosthetic group in many proteins and, although it is synthesized in Actinobacteria,67 its acquisition from the environment and subsequent degradation by heme oxygenase (HO) liberates iron that is essential for bacterial growth and produces biliverdin.68 Little is known about the heme homeostasis and degradation process in Actinobacteria and it is likely that F‐BVRs contribute to this via metabolism and removal of the biliverdin produced by HO while simultaneously producing bilirubin. Recently, new classes of bacterial HOs have been discovered,69 which includes the mycobacterial MhuD that produces the biliverdin analogue mycobilin.70 Mycobilin differs from biliverdin by the presence of a ketone group instead of a carboxyl group at either of the terminal pyrrole rings A or D.70 The active site of Rv2074, which does not seem to have specific interactions for pyrrole rings A and D, could allow the reduction of mycobilin as well, which could have similar anti‐oxidative properties as bilirubin.
The over‐representation of F‐BVRs in pathogenic mycobacteria is notable, but consistent with the observation that their substrate, biliverdin‐IXα, is produced by mammalian cells. It has been observed that biliverdin‐IXα reduction produces a potent antioxidant system,6 making it possible that bilirubin‐IXα production by F‐BVRs could contribute to protecting pathogenic mycobacteria against oxidative stress. Proteomics studies have shown that Rv2074 is found in the membrane of M. tuberculosis,71, 72 and is also secreted.73 F420 is also known to be exported to the outer mycobacterial membrane.74 Therefore, it is likely that Rv2074 reduces the excess biliverdin produced in macrophages upon mycobacterial infection due to HO upregulation,27, 28, 29 producing bilirubin for its antioxidant activity.6, 7, 8, 9 HO upregulation has already been shown to promote M. tuberculosis and M. abscessus proliferation inside macrophages during early infection.27, 29 where bilirubin itself can also have a similar protective effect on intracellular M. abscessus.27 The bilirubin produced by F‐BVRs could form an “anti‐oxidative pocket” around the bacterial cell that can quench lethal nitrosative species like NO produced by the engulfing macrophage.8 NO production controls active mycobacterial infection inside macrophages,32, 33 and has multiple targets in M. tuberculosis.75, 76 It has already been shown that mutant M. tuberculosis strains lacking enzymes required for F420 biosynthesis are hypersusceptible to reactive nitrogen and oxygen species,39, 77 and bilirubin production through the activity of the F‐BVRs could be one of the contributing mechanisms through which F420 protects M. tuberculosis against oxidative stress. The potential involvement of bilirubin production by F‐BVRs in mycobacterial pathogenesis is further supported by comparative genomics indicating that Rv2074 is encoded in one of the regions of difference in the M. tuberculosis genome that are absent in the closely related attenuated M. bovis strains used for TB vaccination.64
Bilirubin produced by the F‐BVRs could also be involved in cell signaling as it can inhibit inducible nitric oxide synthase production,30, 31 which is required for the generation of NO in M. tuberculosis infected macrophages.32, 33 It is unlikely that the F‐BVRs with their different protein fold are able to mimic the direct regulatory functions of BVR‐A since mammalian BVR‐A contains an additional regulatory C‐terminal domain that comprises of multiple motifs involved in cellular signaling mechanisms which are absent in BVR‐B that has not yet been observed to have a similar regulatory role.11, 17 However, increased presence of biliverdin‐IXα has been shown to have anti‐inflammatory effects like the inhibition of pro‐inflammatory cytokine production,78, 79 and inhibition of antigen specific immune responses,80 that could be attributed to its fast conversion to bilirubin‐IXα by a BVR.21 Further work is required to confirm the exact physiological importance of F‐BVRs as part of an immune evasive strategy employed by mycobacteria to mediate oxidative stress and inflammation inside macrophages.
Summary
In this work, we describe the first comprehensive characterization of bacterial F420H2‐dependent biliverdin reductases, focusing on Rv2074 from M. tuberculsosis.36 Prior to this, biliverdin reductases had largely been studied from mammalian hosts. We have demonstrated that the F‐BVR family of BVRs is abundant in Actinobacteria, especially in mycobacterial pathogens, and characterized the mechanism by which Rv2074 from M. tuberculosis catalyzes the reduction of biliverdin‐IXα (the principle isomer produced in mammalian cells)13 to produce bilirubin‐IXα in an F420H2‐dependent manner, performing the same catalytic function as human BVR‐A. As previous work has already demonstrated the increased production of biliverdin by mycobacteria infected macrophages,27, 28, 29 anti‐oxidative properties of bilirubin,6, 7, 8, 9 and the presence of excreted Rv2074 in the extracellular medium,73 we suggests that Rv2074 may be involved in the pathogenesis of M. tuberculosis by conferring protection against oxidative and nitrosative species.
Materials and Methods
Materials
Biliverdin‐IXα, bilirubin‐IXα and G6P were purchased from Sigma‐Aldrich (Missouri, U. S. A.). The codon optimized sequences for rv2074 and rv1155 from M. tuberculosis (H37Rv) were purchased as GeneStrings from ThermoFisher Scientific (Massachusetts, U. S. A) and cloned into the expression vector pETMCSIII using Gibson assembly,81 as previously described for other FDORs.36 The rv2074 mutants were generated by Dr Ruhu Qi from the ANU Mutagenesis and Cloning service and validated by Sanger sequencing from the ANU Biomolecular Resource Facility. The plasmids used for F420 and Fgd overexpression (pYUBDuet‐fbiABC and pDEST17‐fgd, respectively) have been previously described.47, 82, 83 General molecular biology reagents and chemicals were purchased from Sigma‐Aldrich or Astral Scientific.
F420 purification
F420 was overexpressed and purified from M. smegmatis using a modified protocol based on previously published methods.83, 84 Briefly, M. smegmatis (mc24517) cells were transformed with pYUBDuet‐fbiABC by electroporation, plated on LB tween80 agar plates containing 50 µg/mL each of hygromycin B and kanamycin, and grown for 3 days at 37°C. Individual colonies were used to grow starter cultures for 5 days at 37°C in LB media containing 0.05% tween 80, 50 µg/mL hygromycin B and 50 µg/mL kanamycin C. This was then diluted into 500 mL of the same media and grown for a further 5 days at 37°C. Cells were harvested by centrifugation at 10,000g and resuspended in 20 mM tris(hydroxymethyl)aminomethane, pH 7.5, before being autoclaved at 121°C for 20 min. The insoluble fraction was removed by further centrifugation at 20,000g for 1 h. F420 was purified from the filtered lysate using a 60 mL Macro‐prep High‐Q ion‐exchange column (Bio‐Rad, California, U. S. A.). Fractions containing F420 (identified by the presence of an absorption peak at 420 nm) were further purified using a high‐capacity C‐18 Reversed‐Phase Extract‐Clean column (Alltech, Kentucky, U. S. A.). Fractions containing F420 (eluted using a methanol gradient from 0% to 100%) were freeze dried for storage at −80°C.
Protein expression and purification
Fgd for F420H2 generation was expressed and purified as described previously.82 Expression vectors for rv2074, its mutants and rv1155 were transformed into E. coli (BL21(DE3) from New England Biolabs, Massachusetts, U. S. A.) and grown overnight at 37°C on LB agar containing 100 µg/mL ampicillin and 0.5% glucose. Starter cultures were grown the following morning in Terrific Broth (TB) containing 100 µg/mL ampicillin and 0.5% glucose at 37°C for 5 h and transferred to 500 mL (or 1L for Rv2074 grown for protein crystallization) of modified auto‐induction media,36 for overnight growth at 30°C. Cells were harvested by centrifugation at 5000g for 15 min at 4°C, resuspended in lysis buffer (50 mM NaPO4, 300 mM NaCl, 25 mM imidazole at pH 8) and lysed by sonication using an Omni Sonicator Ruptor 400 (3 × 3 min at 60% power). The soluble fraction obtained from centrifugation at 13,000g for 1 hr at 4°C was passed over a gravity column containing 500 µL Ni‐NTA resin (Qiagen, Hilden, Germany). Columns were washed with 4 mL of lysis buffer followed by 4 mL of the same buffer containing 50 mM imidazole. Samples were eluted with 4 mL of elution buffer (same as lysis buffer but with 250 mM imidazole), dialyzed overnight and stored at −80°C in 50 mM Tris, 300 mM NaCl and 10% glycerol at pH 7.5 until further use.
For protein crystallization, Rv2074 was purified using a 5 mL GE HisTrap FF column using the same protocol as above but with 25 mL wash and elution steps instead. Purified protein was passed through a GE HiPrep 26/10 desalting column for buffer exchange into 50 mM Tris and 150 mM NaCl at pH 8.0. The histidine‐tag was then cleaved using TEV protease as described for other FDORs.36 The tag‐free protein was concentrated to ∼1 mM and incubated at 4°C overnight with a 1.5‐fold excess of F420, followed by size exclusion chromatography to remove excess unbound ligand on a GE Hiload 16/600 Superdex 75 pg column in buffer containing 20 mM HEPES and 150 mM NaCl at pH 7.5. The final sample was concentrated to 1.4 mM (21 mg/mL) and stored at 4°C in 20 mM HEPES and 50 mM NaCl at pH 7.5.
Enzyme activity assays and reaction product analysis
1 µM enzyme was added to reactions of 1 µM F420, 1 µM Fgd, 2.5 mM G6P, and 0–100 µM biliverdin in 100 µL buffer containing 50 mM Tris at pH 7.5. Enzyme activity was measured in 96‐well plates (path‐length 0.25 cm) by following the decrease in absorbance at 650 nm (ε = 25,000 M−1 cm−1)85 on an Epoch Microplate Spectrophotometer (BioTek, Vermont, U. S. A.). Average K M and V max values for three independent experiments were calculated from substrate inhibition curves of best fit ( on GraphPad Prism. The specific activity of Rv2074, calculated for comparison with previously characterized BVRs was measured at a concentration of 25 µM biliverdin IXα, which gave the highest reaction rate.
For the purification and analysis of reaction products, 5 mL reactions containing 100 µM biliverdin, 5 mM G6P, 1 µM Fgd, 1 µM F420, and 10 µM Rv2074 or Rv1155 were left incubating at room temperature for 2 h in 50 mM Tris at pH 7.5. A control reaction without enzyme, and another with 100 µM bilirubin instead of biliverdin were also set. Reaction products were purified in a similar manner to other studies of heme degradation products.86 Reactions were terminated with 1 mL each of 3M HCl and glacial acetic acid and the porphyrin products were extracted into 1 mL of chloroform. The aqueous layer was discarded; the chloroform layer was washed twice with 1 mL water and finally evaporated under a stream of nitrogen. For proton NMR, samples were resuspended in 500 µL deuterated chloroform and data were collected on a Bruker Ascend 700 MHz NMR spectrometer. For mass spectrometry, samples were resuspended in 500 µL methanol and spectra were collected on a Micromass ZMD mass spectrometer in the ESI negative mode.
Protein crystallization, data collection, and structure determination
The saturated Rv2074:F420 complex used was at a concentration of 1.4 mM (21 mg/mL) in 20 mM HEPES and 50 mM NaCl at pH 7.5, and was obtained after incubation of the pure protein with a 1.5‐fold excess of F420 followed by gel purification. SG1 (Molecular Dimensions, Newmarket, Suffolk, U. K.) and Index (Hampton Research, California, U. S. A.) high‐throughput crystallization screens were set to identify initial crystallization conditions for the Rv2074:F420 complex. The final crystal that was used for data collection was grown in 27% PEG‐3350, 0.2M MgCl2 and 0.1 M BisTris at pH 5.5. The cryobuffer used was the same as the crystallization conditions, with the addition of an additional 8% ethylene glycol. The crystal was flash‐cooled under a stream of nitrogen gas at 100 K. X‐ray diffraction data were collected at the Australian Synchrotron beamline MX2 with an ADSC Q315 detector and Blu‐Ice software.87 Diffraction data were integrated using XDS,88 and scaled using AIMLESS,89 in the CCP4 suite.90 Molecular replacement was performed with the previously solved apo‐structure of Rv2074 (PDB ID: 2ASF)37 using Phaser,91 and F420 was manually modeled into the difference density (mFo –DFc) at the binding site using COOT.92 Interspersed structural refinement was performed using Refmac and PHENIX,93, 94 with local non‐crystallographic symmetry restraints, and the final submitted structure was refined using Refmac.93 The Proteins, Interfaces, Structures and Assemblies server (http://www.ebi.ac.uk/pdbe/pisa/)50 that compares the interface between protein chains was used to analyze the most probable multimeric state of Rv2074 in solution.
Substrate docking and molecular dynamics simulations
Biliverdin‐IXα was docked to chain D of the Rv2074:F420 complex using Autodock Vina.51 Both protein and ligand were prepared using AutodockTools using default settings,95 where R21, Y104, Y108, R109, R112, and R117 were flexible. The MD simulations on the dimeric structure was performed with the GROMACS 4.6.5 suite,96 using the most plausible biliverdin conformation that was chosen based on the predicted free energy scores of binding and recurrence during manual inspection. Since there are delocalized electrons in the two central pyrrole rings, conformations with either ring protonated were used for separate simulations to investigate any preference in the binding modes and stability. The second chain in the homodimer included F420H2, but not biliverdin, to allow analysis of the cofactor stability in the enzyme complex. F420H2 and biliverdin were parameterized for simulation using the Automated Topology Builder (MolIDs: F420H2 for both chains; 29471 and 29452, Biliverdin N23‐protonated; 29436 and Biliverdin N22‐protonated; 29350) and the GROMOS 54A7 force field,96, 97. The simulations were performed under periodic boundary conditions with the system coordinates in a cubic box with 1.4 nm distance between the protein and the box wall. The van der Waals and Coulombic interactions were evaluated using a 1.4 nm cut‐off scheme for both. A dielectric constant of ε r = 78.5 was used to apply a corrective reaction‐field for long‐range electrostatic interactions beyond the cut‐off. Bond lengths were constrained with a time step of 2 fs using the LINCS algorithm.98 The simulations were performed at reference pressure and temperature of 1 bar and 300 K, respectively, which was achieved with the Berendsen weak coupling method,99 with time constants of 0.4 ps and 0.1 ps. VMD was used to visualize the simulation results.100
The dimeric structure in the box was solvated using SPC water.101 To ensure the overall charge neutrality of the complex, 14 Na+ ions were added to the solvent to counter the −14 e charge on the protein complex. The resulting system was minimized via steepest descent, followed by gradual relaxation in series of 4 simulations with position restraints placed on the protein‐cofactor‐substrate complex. Each simulation was 2 ns long and the force constants used were set to 1000 kJ mol−1 nm−1, 500 kJ mol−1 nm−1, 200 kJ mol−1 nm−1, and 50 kJ mol−1 nm−1, respectively. After the equilibration, two independent unrestrained runs of 50 ns were performed. The flexibility and motion of the substrate and cofactor in the active site were analyzed by calculating the root mean square fluctuation (RMSF) of their atoms, which were then grouped based on the chemical structure of the compounds. To investigate the accessibility of the solvent to the active site and probability of water molecules within sufficient distance to facilitate possible proton transfer, radial distribution function was calculated and graphed with the nitrogen atoms of interest as reference particles. The two trajectories were concatenated prior to cluster analysis, which was performed using the method by Daura et al.102 Briefly, all frames of the concatenated trajectory were aligned with respect to the reference (initial) structure by fitting the backbone root mean square deviation (RMSD). Cluster analysis was performed on the subset of atoms consisting of the substrate and active side residues Arg‐21, Tyr‐104, Tyr‐108, Arg‐109, Arg‐112, and Arg‐117. Conformations were considered to fall within the same cluster when their RMSD was less than the specified cutoff value of 2.0 Å.
Sequence similarity networks (SSNs)
Sequences of proteins homologous to Rv2074 were retrieved from the NCBI non‐redundant protein database using BLAST to an E‐value cut‐off of 1,103 and curated with CD‐hit,104 removing species level duplicates with more than 98% sequence identity. The SSN was generated using the Enzyme Function Initiative Enzyme Similarity Tool,105 where each node represents an individual protein and edges represent the BLAST logE‐value.59 A logE‐value cut‐off of −20 was used to identify the cluster of Rv2074 homologs of >50% amino acid identity (FDOR‐B4)36 from other distantly related proteins. The presence of F‐BVRs in pathogenic and non‐pathogenic mycobacteria were compared using 38 mycobacterial genomes from species that are documented to either cause infection or are only in the environment.60, 61, 62
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Author contributions
FHA performed most of the experiments. AEM proposed the catalytic mechanism and performed the molecular dynamics simulations with assistance from KCJ and MLO. PDC acquired crystallography data and finalized structural refinement. BML purified F420. FHA and CJJ designed the project and FHA, AEM, and CJJ wrote the paper.
Acknowledgments
We thank the ANU Cloning and Mutagenesis Facility and Dr Ruhu Qi for generating the Rv2074 mutants. We also thank Dr Matt Taylor at the Commonwealth Scientific and Industrial Research Organisation (CSIRO) for providing the facilities for F420 purification. We also thank Dr Stephen Watt and Anithahini Jeyasingham at the Australian National University Mass Spectrometry facility and Chris Blake at the Australian National University Nuclear Magnetic Resonance Centre for data collection on the purified biliverdin degradation products. This research was undertaken on the MX2 beamline at the Australian Synchrotron, Victoria, Australia.
Statement: Rv2074 from Mycobacterium tuberculosis, the causative agent of Tuberculosis, belongs to a novel class of F420H2‐dependent biliverdin reductases found in Actinobacteria. It reduces biliverdin‐IXα to bilirubin‐IXα, a potent antioxidant. As biliverdin‐IXα is produced in high amounts in macrophages infected with M. tuberculosis, its reduction by Rv2074 could play a role in protecting mycobacteria against oxidative stress, aiding the persistence of M. tuberculosis infection.
References
- 1. Tenhunen R, Marver HS, Schmid R (1968) The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci USA 61:748–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beale SI, Cornejo J (1983) Biosynthesis of phycocyanobilin from exogenous labeled biliverdin in Cyanidium caldarium . Arch Biochem Biophys 227:279–286. [DOI] [PubMed] [Google Scholar]
- 3. Cornelius CE (1991) Bile pigments in fishes: a review. Vet Clin Pathol 20:106–115. [DOI] [PubMed] [Google Scholar]
- 4. Mantle TJ (2002) Haem degradation in animals and plants. Biochem Soc Trans 30:630–633. [DOI] [PubMed] [Google Scholar]
- 5. Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN (1987) Bilirubin is an antioxidant of possible physiological importance. Science 235:1043–1046. [DOI] [PubMed] [Google Scholar]
- 6. Barañano DE, Rao M, Ferris CD, Snyder SH (2002) Biliverdin reductase: a major physiologic cytoprotectant. Proc Natl Acad Sci USA 99:16093–16098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jansen T, Daiber A (2012) Direct antioxidant properties of bilirubin and biliverdin. Is there a role for biliverdin reductase? Front Pharmacol 3:30. doi: 10.3389/fphar.2012.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kaur H, Hughes MN, Green CJ, Naughton P, Foresti R, Motterlini R (2003) Interaction of bilirubin and biliverdin with reactive nitrogen species. FEBS Lett 543:113–119. [DOI] [PubMed] [Google Scholar]
- 9. Minetti M, Mallozzi C, Di Stasi AM, Pietraforte D (1998) Bilirubin is an effective antioxidant of peroxynitrite‐mediated protein oxidation in human blood plasma. Arch Biochem Biophys 352:165–174. [DOI] [PubMed] [Google Scholar]
- 10. Maines MD (2005) New insights into biliverdin reductase functions: linking heme metabolism to cell signaling. Physiology 20:382–389. [DOI] [PubMed] [Google Scholar]
- 11. Kapitulnik J, Maines MD (2009) Pleiotropic functions of biliverdin reductase: cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol Sci 30:129–137. [DOI] [PubMed] [Google Scholar]
- 12. Yamaguchi T, Komoda Y, Nakajima H (1994) Biliverdin‐IX alpha reductase and biliverdin‐IX beta reductase from human liver. Purification and characterization. J Biol Chem 269:24343–24348. [PubMed] [Google Scholar]
- 13. Maines MD (1988) Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J 2:2557–2568. [PubMed] [Google Scholar]
- 14. Komuro A, Tobe T, Nakano Y, Yamaguchi T, Tomita M (1996) Cloning and characterization of the cDNA encoding human biliverdin‐IXα reductase. Biochim Biophys Acta 1309:89–99. [DOI] [PubMed] [Google Scholar]
- 15. Yamaguchi T, Yamaguchi N, Komoda Y, Nakajima H, Ishikawa M (1979) Studies on bilirubin metabolism. Proc Jpn Acad Ser B Phys Biol Sci 55:84–88. [Google Scholar]
- 16. Pereira PJ, Macedo‐Ribeiro S, Párraga A, Pérez‐Luque R, Cunningham O, Darcy K, Mantle TJ, Coll M (2001) Structure of human biliverdin IXbeta reductase, an early fetal bilirubin IXbeta producing enzyme. Nat Struct Biol 8:215–220. [DOI] [PubMed] [Google Scholar]
- 17. Whitby FG, Phillips JD, Hill CP, McCoubrey W, Maines MD (2002) Crystal structure of a biliverdin IXalpha reductase enzyme–cofactor complex. J Mol Biol 319:1199–1210. [DOI] [PubMed] [Google Scholar]
- 18. Maines MD, Ewing JF, Huang TJ, Panahian N (2001) Nuclear localization of biliverdin reductase in the rat kidney: response to nephrotoxins that induce heme oxygenase‐1. J Pharmacol Exp Ther 296:1091–1097. [PubMed] [Google Scholar]
- 19. Young SC, Storm MV, Speed JS, Kelsen S, Tiller CV, Vera T, Drummond HA, Stec DE (2009) Inhibition of biliverdin reductase increases ANG II‐dependent superoxide levels in cultured renal tubular epithelial cells. Am J Physiol Regul Integr Comp Physiol 297:R1546–R1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sedlak TW, Saleh M, Higginson DS, Paul BD, Juluri KR, Snyder SH (2009) Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc Natl Acad Sci USA 106:5171–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wegiel B, Otterbein LE (2012) Go green: the anti‐inflammatory effects of biliverdin reductase. Front Pharmacol 3:47. doi: 10.3389/fphar.2012.00047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hu Z, Pei G, Wang P, Yang J, Zhu F, Guo Y, Wang M, Yao Y, Zeng R, Liao W, Xu G. (2015) Biliverdin reductase A (BVRA) mediates macrophage expression of interleukin‐10 in injured kidney. Int J Mol Sci 16:22621–22635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lerner‐Marmarosh N, Miralem T, Gibbs PEM, Maines MD (2007) Regulation of TNF‐alpha‐activated PKC‐zeta signaling by the human biliverdin reductase: identification of activating and inhibitory domains of the reductase. FASEB J 21:3949–3962. [DOI] [PubMed] [Google Scholar]
- 24. Tudor C, Lerner‐Marmarosh N, Engelborghs Y, Gibbs PEM, Maines MD (2008) Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO‐1 gene expression by haematin. Biochem J 413:405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schluchter WM, Glazer AN (1997) Characterization of cyanobacterial biliverdin reductase. Conversion of biliverdin to bilirubin is important for normal phycobiliprotein biosynthesis. J Biol Chem 272:13562–13569. [DOI] [PubMed] [Google Scholar]
- 26. Mölzer C, Huber H, Diem K, Wallner M, Bulmer AC, Wagner K‐H (2013) Extracellular and intracellular anti‐mutagenic effects of bile pigments in the Salmonella typhimurium reverse mutation assay. Toxicol In Vitro 27:433–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abdalla MY, Ahmad IM, Switzer B, Britigan BE (2015) Induction of heme oxygenase‐1 contributes to survival of Mycobacterium abscessus in human macrophages‐like THP‐1 cells. Redox Biol 4:328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Silva‐Gomes S, Appelberg R, Larsen R, Soares MP, Gomes MS (2013) Heme catabolism by heme oxygenase‐1 confers host resistance to Mycobacterium infection. Infect Immun 81:2536–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scharn CR, Collins AC, Nair VR, Stamm CE, Marciano DK, Graviss EA, Shiloh MU (2016) Heme oxygenase‐1 regulates inflammation and mycobacterial survival in human macrophages during Mycobacterium tuberculosis infection. J Immunol 196:4641–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang WW, Smith DLH, Zucker SD (2004) Bilirubin inhibits iNOS expression and NO production in response to endotoxin in rats. Hepatology 40:424–433. [DOI] [PubMed] [Google Scholar]
- 31. Lanone S, Bloc S, Foresti R, Almolki A, Taillé C, Callebert J, Conti M, Goven D, Aubier M, Dureuil B, El‐Benna J, Motterlini R, Boczkowski J. (2005) Bilirubin decreases nos2 expression via inhibition of NAD(P)H oxidase: implications for protection against endotoxic shock in rats. Faseb J 19:1890–1892. [DOI] [PubMed] [Google Scholar]
- 32. Chan J, Xing Y, Magliozzo RS, Bloom BR (1992) Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J Exp Med 175:1111–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kröncke K‐D, Fehsel K, Kolb‐Bachofen V (1998) Inducible nitric oxide synthase in human diseases. Clin Exp Immunol 113:147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gengenbacher M, Kaufmann SHE (2012) Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol Rev 36:514–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. World Heath Organization (2015) Global tuberculosis report. WHO Press, Geneva.
- 36. Ahmed FH, Carr PD, Lee BM, Afriat‐Jurnou L, Mohamed AE, Hong N‐S, Flanagan J, Taylor MC, Greening C, Jackson CJ (2015) Sequence‐structure‐function classification of a catalytically diverse oxidoreductase superfamily in mycobacteria. J Mol Biol 427:3554–3571. [DOI] [PubMed] [Google Scholar]
- 37. Biswal BK, Au K, Cherney MM, Garen C, James MNG (2006) The molecular structure of Rv2074, a probable pyridoxine 5′‐phosphate oxidase from Mycobacterium tuberculosis, at 1.6 A resolution. Acta Crystallogr 62:735–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee KS, Gartner LM (1976) Spectrophotometric characteristics of bilirubin. Pediatr Res 10:782–788. [DOI] [PubMed] [Google Scholar]
- 39. Gurumurthy M, Rao M, Mukherjee T, Rao SPS, Boshoff HI, Dick T, Barry CE, Manjunatha UH (2013) A novel F420‐dependent anti‐oxidant mechanism protects Mycobacterium tuberculosis against oxidative stress and bactericidal agents. Mol Microbiol 8:744–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Phillips O, Mantle TJ (1981) Some kinetic and physical properties of biliverdin reductase. Biochem Soc Trans 9:275–278. [DOI] [PubMed] [Google Scholar]
- 41. Kutty RK, Maines MD (1981) Purification and characterization of biliverdin reductase from rat liver. J Biol Chem 256:3956–3962. [PubMed] [Google Scholar]
- 42. Cunningham O (2000) Studies on the specificity of the tetrapyrrole substrate for human biliverdin‐IXalpha reductase and biliverdin‐IXbeta reductase. Structure–activity relationships define models for both active sites. J Biol Chem 275:19009–19017. [DOI] [PubMed] [Google Scholar]
- 43. Hayes JM, Mantle TJ (2009) The effect of pH on the initial rate kinetics of the dimeric biliverdin‐IXalpha reductase from the Cyanobacterium synechocystis PCC6803. FEBS J 276:4414–4425. [DOI] [PubMed] [Google Scholar]
- 44. Kaplan D, Navon G (1981) Nuclear magnetic resonance studies of the conformation of bilirubin and its derivatives in solution. J Chem Soc Perkin Trans 2:1374–1383. [Google Scholar]
- 45. Safo MK, Mathews I, Musayev FN, di Salvo ML, Thiel DJ, Abraham DJ, Schirch V (2000) X‐ray structure of Escherichia coli pyridoxine 5′‐phosphate oxidase complexed with FMN at 1.8 Å resolution. Structure 8:751–762. [DOI] [PubMed] [Google Scholar]
- 46. Musayev FN, Di Salvo ML, Ko T‐P, Schirch V, Safo MK (2003) Structure and properties of recombinant human pyridoxine 5′‐phosphate oxidase. Protein Sci 12:1455–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Taylor MC, Jackson CJ, Tattersall DB, French N, Peat TS, Newman J, Briggs LJ, Lapalikar GV, Campbell PM, Scott C, Russell RJ, Oakeshott JG. (2010) Identification and characterization of two families of F420H2‐dependent reductases from mycobacteria that catalyse aflatoxin degradation. Mol Microbiol 78:561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cellitti SE, Shaffer J, Jones DH, Mukherjee T, Gurumurthy M, Bursulaya B, Boshoff HI, Choi I, Nayyar A, Lee YS, Cherian J, Niyomrattanakit P, Dick T, Manjunatha UH, Barry CE 3rd, Spraggon G, Geierstanger BH. (2012) Structure of Ddn, the deazaflavin‐dependent nitroreductase from Mycobacterium tuberculosis involved in bioreductive activation of PA‐824. Structure 20:101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mashalidis EH, Gittis AG, Tomczak A, Abell C, Barry CE, Garboczi DN (2015) Molecular insights into the binding of coenzyme F420 to the conserved protein Rv1155 from Mycobacterium tuberculosis . Protein Sci 24: 279–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774–797. [DOI] [PubMed] [Google Scholar]
- 51. Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Warkentin E, Mamat B, Sordel‐Klippert M, Wicke M, Thauer RK, Iwata M, Iwata S, Ermler U, Shima S (2001) Structures of F420H2:NADP+ oxidoreductase with and without its substrates bound. EMBO J 20:6561–6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ceh K, Demmer U, Warkentin E, Moll J, Thauer RK, Shima S, Ermler U (2009) Structural basis of the hydride transfer mechanism in F420‐dependent methylenetetrahydromethanopterin dehydrogenase. Biochemistry 48:10098–10105. [DOI] [PubMed] [Google Scholar]
- 54. Hammes‐Schiffer S, Watney JB (2006) Hydride transfer catalysed by Escherichia coli and Bacillus subtilis dihydrofolate reductase: coupled motions and distal mutations. Philos Trans R Soc Lond B Biol Sci 361:1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Richard JP, Amyes TL, Goryanova B, Zhai X (2014) Enzyme architecture: on the importance of being in a protein cage. Curr Opin Chem Biol 21:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gallivan JP, Dougherty DA (1999) Cation–pi interactions in structural biology. Proc Natl Acad Sci USA 96:9459–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Smith LJ, Browne S, Mulholland AJ, Mantle TJ (2008) Computational and experimental studies on the catalytic mechanism of biliverdin‐IXβ reductase. Biochem J 411:475–484. [DOI] [PubMed] [Google Scholar]
- 58. Fu G, Liu H, Doerksen RJ (2012) Molecular modeling to provide insight into the substrate binding and catalytic mechanism of human biliverdin‐IXα reductase. J Phys Chem B 116:9580–9594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rastogi N, Legrand E, Sola C (2001) The mycobacteria: an introduction to nomenclature and pathogenesis. Rev Sci Tech 20:21–54. [DOI] [PubMed] [Google Scholar]
- 61. Tortoli E (2014) Microbiological features and clinical relevance of new species of the genus Mycobacterium . Clin Microbiol Rev 27:727–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. van Ingen J, Boeree MJ, Dekhuijzen PNR, van Soolingen D (2009) Environmental sources of rapid growing nontuberculous mycobacteria causing disease in humans. Clin Microbiol Infect 15:888–893. [DOI] [PubMed] [Google Scholar]
- 63. Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, Honore N, Garnier T, Churcher C, Harris D, Mungall K, Basham D, Brown D, Chillingworth T, Connor R, Davies RM, Devlin K, Duthoy S, Feltwell T, Fraser A, Hamlin N, Holroyd S, Hornsby T, Jagels K, Lacroix C, Maclean J, Moule S, Murphy L, Oliver K, Quail MA, Rajandream MA, Rutherford KM, Rutter S, Seeger K, Simon S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Taylor K, Whitehead S, Woodward JR, Barrell BG. (2001) Massive gene decay in the leprosy bacillus. Nature 409:1007–1011. [DOI] [PubMed] [Google Scholar]
- 64. Behr MA, Wilson MA, Gill WP, Salamon H, Schoolnik GK, Rane S, Small PM (1999) Comparative genomics of BCG vaccines by whole‐genome DNA microarray. Science 284:1520–1523. [DOI] [PubMed] [Google Scholar]
- 65. Selengut JD, Haft DH (2010) Unexpected abundance of coenzyme F420‐dependent enzymes in Mycobacterium tuberculosis and other actinobacteria. J Bacteriol 192:5788–5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mohamed AE, Ahmed FH, Arulmozhiraja S, Lin CY, Taylor MC, Krausz ER, Jackson CJ, Coote ML (2016) Protonation state of F420H2 in the prodrug‐activating deazaflavin dependent nitroreductase (Ddn) from Mycobacterium tuberculosis . Mol Biosyst 12:1110–1113. [DOI] [PubMed] [Google Scholar]
- 67. Dailey HA, Gerdes S, Dailey TA, Burch JS, Phillips JD (2015) Noncanonical coproporphyrin‐dependent bacterial heme biosynthesis pathway that does not use protoporphyrin. Proc Natl Acad Sci USA 112:2210–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li C, Stocker R (2009) Heme oxygenase and iron: from bacteria to humans. Redox Rep 14:95–101. [DOI] [PubMed] [Google Scholar]
- 69. Wilks A, Ikeda‐Saito M (2014) Heme utilization by pathogenic bacteria: not all pathways lead to biliverdin. Acc Chem Res 47:2291–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nambu S, Matsui T, Goulding CW, Takahashi S, Ikeda‐Saito M (2013) A new way to degrade heme: the mycobacterium tuberculosis enzyme MhuD catalyzes heme degradation without generating CO. J Biol Chem 288:10101–10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Målen H, Pathak S, Søfteland T, de Souza GA, Wiker HG (2010) Definition of novel cell envelope associated proteins in Triton X‐114 extracts of Mycobacterium tuberculosis H37Rv. BMC Microbiol 10:132. DOI: 10.1186/1471‐2180‐10‐132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. De Souza GA, Leversen NA, Målen H, Wiker HG (2011) Bacterial proteins with cleaved or uncleaved signal peptides of the general secretory pathway. J Proteomics 75:502–510. [DOI] [PubMed] [Google Scholar]
- 73. Målen H, Berven FS, Fladmark KE, Wiker HG (2007) Comprehensive analysis of exported proteins from Mycobacterium tuberculosis H37Rv. Proteomics 7:1702–1718. [DOI] [PubMed] [Google Scholar]
- 74. Bashiri G, Perkowski EF, Turner AP, Feltcher ME, Braunstein M, Baker EN (2012) Tat–dependent translocation of an F(420)‐binding protein of Mycobacterium tuberculosis . PLoS One 7:e45003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rhee KY, Erdjument‐Bromage H, Tempst P, Nathan CF (2005) S‐nitroso proteome of Mycobacterium tuberculosis: enzymes of intermediary metabolism and antioxidant defense. Proc Natl Acad Sci USA 102:467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Buck ZH, De J (2010) Cell wall proteome analysis of Mycobacterium smegmatis strain MC2 155. BMC Microbiol 10:121. doi: 10.1186/1471‐2180‐10‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Greening C, Ahmed FH, Mohamed AE, Lee BM, Pandey G, Warden AC, Scott C, Oakeshott JG, Taylor MC, Jackson CJ (2016) F420‐ and Fo‐dependent redox reactions: physiology, biochemistry, and applications. Microbiol Mol Biol Rev 80:451–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bellner L, Wolstein J, Patil KA, Dunn MW, Laniado‐Schwartzman M (2011) Biliverdin rescues the HO‐2 null mouse phenotype of unresolved chronic inflammation following corneal epithelial injury. Invest Ophthalmol Vis Sci 52:3246–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bisht K, Wegiel B, Tampe J, Neubauer O, Wagner K‐H, Otterbein LE, Bulmer AC (2014) Biliverdin modulates the expression of C5aR in response to endotoxin in part via mTOR signaling. Biochem Biophys Res Commun 449:94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yamashita K, McDaid J, Ollinger R, Tsui T‐Y, Berberat PO, Usheva A, Csizmadia E, Smith RN, Soares MP, Bach FH (2004) Biliverdin, a natural product of heme catabolism, induces tolerance to cardiac allografts. FASEB J 18:765–767. [DOI] [PubMed] [Google Scholar]
- 81. Gibson DG, Young L, Chuang R‐Y, Venter JC, Hutchison CA, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Meth 6:343–345. [DOI] [PubMed] [Google Scholar]
- 82. Bashiri G, Squire CJ, Moreland NJ, Baker EN (2008) Crystal structures of F420‐dependent glucose‐6‐phosphate dehydrogenase FGD1 involved in the activation of the anti‐tuberculosis drug candidate PA‐824 reveal the basis of coenzyme and substrate binding. J Biol Chem 283:17531–17541. [DOI] [PubMed] [Google Scholar]
- 83. Bashiri G, Rehan AM, Greenwood DR, Dickson JMJ, Baker EN (2010) Metabolic engineering of cofactor F420 production in Mycobacterium smegmatis . PLoS One 5:e15803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Isabelle D, Simpson DR, Daniels L (2002) Large‐scale production of coenzyme F420−5,6 by using Mycobacterium smegmatis . Appl Environ Microbiol 68:5750–5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chen Jason DDB, Kawasaki Y, Bommer J, Takemoto JY (2012) Scalable production of biliverdin IXα by Escherichia coli . BMC Biochem 12:89. DOI: 10.1186/1472‐6750‐12‐89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhu W, Wilks A, Stojiljkovic I (2000) Degradation of heme in Gram‐negative bacteria: the product of the HemO gene of Neisseriae is a heme oxygenase. J Bacteriol 182:6783–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. McPhillips TM, McPhillips SE, Chiu H‐J, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. (2002) Blu‐Ice and the distributed control system: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat 9:401–406. [DOI] [PubMed] [Google Scholar]
- 88. Kabsch W (2010) XDS. Acta Crystallogr D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Evans PR, Murshudov GN (2013) How good are my data and what is the resolution? Acta Crystallogr D69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Collaborative Computational Project N 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D50:760–763. [DOI] [PubMed] [Google Scholar]
- 91. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr 66:486–501. D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L‐W, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hess B, Kutzner C, van der Spoel D, Lindahl E (2008) GROMACS 4: algorithms for highly efficient, load‐balanced, and scalable molecular simulation. J Chem Theory Comput 4:435–447. [DOI] [PubMed] [Google Scholar]
- 97. Koziara KB, Stroet M, Malde AK, Mark AE (2014) Testing and validation of the Automated Topology Builder (ATB) version 2.0: prediction of hydration free enthalpies. J Comput Aided Mol Des 28:221–233. [DOI] [PubMed] [Google Scholar]
- 98. Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1997) LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18:1463–1472. [Google Scholar]
- 99. Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684–3690. [Google Scholar]
- 100. Humphrey W, Dalke A, Schulten K (1996) VMD – visual molecular dynamics. J Mol Graph 14:33–38. [DOI] [PubMed] [Google Scholar]
- 101. Smith PE, van Gunsteren WF (1993) The viscosity of SPC and SPC/E water at 277 and 300 K. Chem Phys Lett 215:315–318. [Google Scholar]
- 102. Daura X, Gademann K, Jaun B, Seebach D, van Gunsteren WF, Mark AE (1999) Peptide folding: when simulation meets experiment. Angew Chem Int Ed 38:236–240. [Google Scholar]
- 103. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410. [DOI] [PubMed] [Google Scholar]
- 104. Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD‐HIT: accelerated for clustering the next‐generation sequencing data. Bioinformatics 28:3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Gerlt JA, Bouvier JT, Davidson DB, Imker HJ, Sadkhin B, Slater DR, Whalen KL (2015) Enzyme Function Initiative‐Enzyme Similarity Tool (EFI‐EST): a web tool for generating protein sequence similarity networks. Biochim Biophys Acta 1854:1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]