

Significance
The A2AR is a G protein-coupled receptor (GPCR) that plays important roles in cardiovascular physiology and immune function. The A2AR is also a target for the treatment of Parkinson’s disease, where A2AR antagonists have been shown to enhance signaling through the D2 dopamine receptor. Here we present the crystal structure of the A2AR bound to a novel bitopic antagonist. As a result of structural changes needed to accommodate the bound antagonist, crystals could not be grown in lipidic cubic phase. Instead, crystals were grown in detergent with a type II packing rarely observed in GPCR crystals. The structure revealed a potential allosteric pocket that that can be exploited to develop subtype-selective allosteric modulators.
Keywords: GPCR, A2A adenosine receptor, structure, allosteric, Parkinson’s disease
Abstract
The adenosine A2A receptor (A2AR) has long been implicated in cardiovascular disorders. As more selective A2AR ligands are being identified, its roles in other disorders, such as Parkinson’s disease, are starting to emerge, and A2AR antagonists are important drug candidates for nondopaminergic anti-Parkinson treatment. Here we report the crystal structure of A2A receptor bound to compound 1 (Cmpd-1), a novel A2AR/N-methyl d-aspartate receptor subtype 2B (NR2B) dual antagonist and potential anti-Parkinson candidate compound, at 3.5 Å resolution. The A2A receptor with a cytochrome b562-RIL (BRIL) fusion (A2AR–BRIL) in the intracellular loop 3 (ICL3) was crystallized in detergent micelles using vapor-phase diffusion. Whereas A2AR–BRIL bound to the antagonist ZM241385 has previously been crystallized in lipidic cubic phase (LCP), structural differences in the Cmpd-1–bound A2AR–BRIL prevented formation of the lattice observed with the ZM241385–bound receptor. The crystals grew with a type II crystal lattice in contrast to the typical type I packing seen from membrane protein structures crystallized in LCP. Cmpd-1 binds in a position that overlaps with the native ligand adenosine, but its methoxyphenyl group extends to an exosite not previously observed in other A2AR structures. Structural analysis revealed that Cmpd-1 binding results in the unique conformations of two tyrosine residues, Tyr91.35 and Tyr2717.36, which are critical for the formation of the exosite. The structure reveals insights into antagonist binding that are not observed in other A2AR structures, highlighting flexibility in the binding pocket that may facilitate the development of A2AR-selective compounds for the treatment of Parkinson’s disease.
The adenosine A2A receptor (A2AR) is one of the four subtypes of adenosine receptors (A1R, A2AR, A2BR, A3R). As a member of the G protein-coupled receptor (GPCR) family, the A2A receptor couples to stimulatory G protein Gs and elevates intracellular cAMP upon activation by endogenous adenosine. The A2AR has been an attractive drug target due to its role in cardiovascular and immune system function, as well as in the central nervous system as a potential therapeutic target for Parkinson’s disease (PD) (1–4). PD is a neurodegenerative disease that affects more than 1% of the population over 65 years old. Currently, major treatments target the restoration of dopamine signaling, which is impaired in PD patients, by dopamine-replacing agents. Although these treatments effectively address PD-related motor disturbances, the long-term use of dopamine-replacing agents is associated with the development of motor complications; therefore, there is a need for nondopaminergic drugs (2). It is known that A2AR signaling regulates dopaminergic neurotransmission, and A2AR antagonists have been shown to enhance D2 dopamine receptor signaling, which has been found to improve PD symptoms in animal models and patients, without the side effects common to dopamine-replacing agents, such as dyskinesia (1, 5). Therefore, A2AR antagonists are good candidates for nondopaminergic treatment of PD. A more recent study found combined administration of A2AR and N-methyl d-aspartate receptor subtype 2B (NR2B) antagonists significantly improved motor activity, and the effects were sustained for longer than when administered separately at the same dose in a rodent model of PD (6). In an effort to identify a dual compound displaying A2AR as well as NR2B receptor antagonist activities, Cmpd-1 was designed and synthesized. The compound binds to both receptors with high affinity (Table 1). To understand the structure–activity relationship around the chemical series for the A2AR and facilitate further optimization, we sought to obtain the crystal structure of the A2AR in complex with Cmpd-1. Although a number of crystal structures of A2AR bound to various antagonists and agonists have been reported (7–14) using lipidic cubic phase (LCP) methods (15, 16), we were unable to obtain high-quality crystals of A2AR bound to Cmpd-1. As an alternative approach we obtained a 3.5-Å resolution crystal structure of A2AR in complex with Cmpd-1 by using a BRIL fusion construct and performing the crystallization in vapor-phase diffusion in the detergent lauryl maltose neopentyl glycol (LMNG). This structure of a nonrhodopsin GPCR was obtained in a detergent environment without thermostabilizing mutations or antibody fragment. Structural analysis provides insight into the binding mode of Cmpd-1 and reveals a potential allosteric pocket that is not observed in previously reported A2AR structures. The structural information provides a rationale for the higher affinity of Cmpd-1 compared with its closely related analogs.
Table 1.
Binding properties of Cmpd-1 to A2A, A1, and NR2B receptors
| Receptor | pKi* | n** | Radioligand |
| A2A | 7.95 ± 0.06 | 4 | [3H]-ZM241385 |
| A1 | 6.8 | 2 | [3H]-DPCPX |
| NR2B | 8.25 ± 0.17 | 4 | [3H]-UCB9352*** |
Negative logarithm of the equilibrium dissociation constant of Cmpd-1 determined by competitive binding assay.
Number of parallel tests for the binding assay.
Proprietary compound of UCB.
Results and Discussion
Cmpd-1, a Novel Potential Anti-Parkinson Candidate.
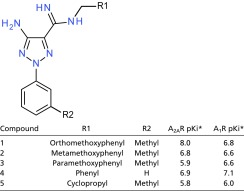
We identified a unique molecule, referred to as Cmpd-1 (Fig. 1A), that behaves as an A2AR/NR2B dual antagonist. Cmpd-1 has a 15-fold higher affinity for the A2AR than the A1R (with pKi values of 7.95 and 6.8, respectively; Table 1) and binds NR2B with high affinity (pKi = 8.25); it functions as an A2AR antagonist able to block CGS21680-induced A2A receptor activation with pKb 7.00 ± 0.32 (n = 4). Cmpd-1 contains three rings: methylphenyl, aminotriazole, and orthomethoxyphenyl (Fig. 1A). We synthesized and tested close analogs of Cmpd-1 where the orthomethoxyphenyl group was replaced by different R groups (Table 2). The results revealed that migration of the methoxy group around the ring, or removal of the methoxy group, reduced the affinity significantly for A2AR but not for A1R, whereas replacement of methoxyphenyl with a cyclopropyl ring reduced the affinity for both receptors. Our efforts to dock Cmpd-1 into previously reported A2AR structures could not help us to rationalize affinity-boosting effects associated with the orthomethoxyphenyl R group. Therefore, to help facilitate further optimization efforts around this chemotype we sought to obtain a crystal structure of A2AR in complex with Cmpd-1 to provide much-needed insight into the binding mode of this lead molecule.
Fig. 1.

Cmpd-1 chemical structure and A2AR–BRIL–Cmpd-1 crystal packing. (A) Chemical structure of several A2AR ligands, including Cmpd-1, ZM241385, and endogenous ligand adenosine. Cmpd-1 is predicted to be protonated under physiological pH. (B) Crystal packing of the A2AR–BRIL–Cmpd-1 structure represents an unusual type II crystal packing for a GPCR. The receptor is shown in green, and BRIL is shown in cyan. (C) Schemes of type I and type II packing for membrane protein crystals. Detergent and lipid molecules are shown in pink and gray, respectively. Transmembrane segments and polar region of the membrane protein are shown in green and cyan, respectively.
Table 2.
Binding properties of Cmpd-1 analogs to A2A and A1 receptors
 |
Negative logarithm of the equilibrium dissociation constant of compound x determined by competitive binding assay. The values are the means of 2–4 independent determinations.
Crystallization of A2AR in Complex with Cmpd-1.
The LCP method for membrane protein crystallogenesis (15, 16) has been used successfully to crystallize a number of GPCRs over the past several years and has become the method of choice; this is exemplified by the fact that 38 of the 39 GPCRs with structures reported have been successfully crystallized in LCP. Therefore, we initially attempted to crystallize A2AR in complex with Cmpd-1 in LCP. We started our trials in LCP using a construct with T4 lysozyme (T4L) inserted into the intracellular loop 3 (ICL3) of A2AR (A2AR–T4L) as previously reported (14). We were able to obtain crystals of A2AR–T4L bound to Cmpd-1 in LCP with comparable size and shape to those previously described (14). However, we were unable to optimize the crystal to diffract beyond 5 Å. As an alternative, we replaced T4L with a truncated modified T4L (mT4L), which has a deleted small lobe of T4L and has previously helped to improve diffraction of muscarinic M3 receptor crystals (17). We obtained slightly improved crystals and obtained a dataset at 4.6 Å resolution. Although the structure could be solved by molecular replacement, the electron density map showed no density to model the ligand (Fig. S1). We noted that the packing of this crystal lattice is not as tight as the packing in the reported A2AR–T4L or A2AR–BRIL crystals (Fig. S1), resulting in higher solvent content, and postulated that this may have contributed to the poorer diffraction. Insertion of thermostabilized apocytochrome b562RIL (BRIL) into ICL3 of A2AR was reported to improve the thermostability of the fusion protein (A2AR–BRIL), and crystals of A2AR–BRIL were obtained in LCP and diffracted to 1.8 Å (8, 18). However, despite extensive crystallization trials, we were not able to obtain any crystals of A2AR–BRIL bound to Cmpd-1 or its analogs in LCP. As a control experiment, we reproduced the crystals of A2AR–BRIL in complex with ZM241385 using previously reported conditions (8), and the crystals diffracted well beyond 2.0 Å resolution with one trial, indicating the protein preparation and methods used were adequate.
Fig. S1.

Electron density map and crystal packing of A2AR–mT4L–Cmpd-1 crystals grown in LCP. (A) Density map of A2AR–mT4L–Cmpd-1. 2Fo–Fc map is shown in gray mesh at 1.0 σ, and Fo–Fc map is shown in green (for positive peaks) and red (for negative peaks) mesh at 2.0 σ. (B) Close-up view of the potential ligand binding pocket. No density for side chains or the ligand was observed. (C) The crystal packing of A2AR–mT4L–Cmpd-1 (Left), A2AR–T4L–ZM241385 (Center), and A2AR–BRIL–ZM241385 (Right) shown in two orientations 90° apart. The center molecule is colored cyan, and surrounding molecules are colored green.
It is now widely understood that the key to successful crystallization of a GPCR is to maintain its conformational homogeneity. The bound ligand is critical in stabilizing a GPCR in a certain conformation, and if the ligand rapidly binds and dissociates due to its low affinity, the receptor will likely adopt multiple conformations that impair the crystallization process. The affinity of Cmpd-1 is ∼10- to 20-fold weaker than ZM241385 (14), and this difference could also be detrimental to crystallization efforts.
We finally obtained crystals of A2AR–BRIL in complex with Cmpd-1 by vapor diffusion with the receptor present in the detergent LMNG. This was surprising because all previously reported crystal structures of nonthermostabilized GPCRs with a fusion partner have been solved using LCP, and not vapor diffusion crystallization. Initial crystal hits were obtained through screening in commercial broad screens for membrane proteins and, after optimization, a 3.5-Å resolution data set was obtained by combining diffraction data from 20 crystals (Fig. S2). The structure was solved by molecular replacement and the quality of the electron density maps was very good for this resolution (statistics in Table S1). All residues and Cmpd-1 were well defined, with the exception of residues 1–3 at N terminus, residues 149–158 on extracellular loop 2 (ECL2), and residues 312–316 located at the C terminus that could not be modeled due to weak density. A2AR–BRIL is packed in the crystal lattice with a P41212 space group (Fig. 1B), representing type II crystal packing (Fig. 1C) (19), which is rarely observed for GPCRs. It is believed that type I packing is favored for GPCRs because the detergent micelle limits packing interactions that involve the transmembrane segments. In this lattice, with one molecule in the asymmetric unit, two adjacent receptors are packed against each other to form an antiparallel dimer through their transmembrane helix 1 (TM1), TM2, and Helix 8. Another adjacent A2AR–BRIL molecule is packed with its direction pointing 90° away along the crystallographic 41 screw axis in the lattice, mostly through extensive interaction among BRILs. BRIL also interacts with the Helix 8 and ECL3 of the receptor. Overall, most hydrophilic crystal contacts are mediated by BRIL, as is the case for all of the GPCRs crystallized with a fusion partner.
Fig. S2.

Crystals of A2AR–BRIL–Cmpd-1 obtained in vapor-phase diffusion. (A) Brightfield and UV images of the crystals grown in vapor-phase diffusion. (Scale bars: Left, 100 μm; Right, 50 μm.) (B) Diffraction pattern of the crystal.
Table S1.
Data collection and structural refinement statistics
| A2AR–BRIL–Cmpd-1 diffraction data | |
| No. of crystals | 20 |
| Space group | P41212 |
| Cell parameters | |
| a, b, c, Å | 131.2, 131.2, 89.7 |
| α, β, γ, ° | 90.0, 90.0, 90.0 |
| Resolution, Å | 50.0–3.50 (3.63–3.50)* |
| Average redundancy | 6.9 (2.6) |
| Average I/σ(I) | 11.4 (1.5) |
| Completeness, % | 95.9 (68.1) |
| Rmerge, % | 17.4 (45.0) |
| CC1/2 | 0.97 (0.75) |
| Refinement statistics | |
| No. of reflections | 10,167 |
| R factor, % | 27.6 |
| Free R factor, % | 29.9 |
| Free R percentage, % | 5.0 |
| Average B factors, Å2 | |
| Protein | 89.7 |
| Cmpd-1 | 93.9 |
| Rmsd from ideality | |
| Bond length, Å | 0.004 |
| Bond angles, o | 0.437 |
| Ramachandran favored | 91.4% |
| Ramachandran outliers | 0.3% |
Highest-shell statistics are in parentheses.
Structure of Cmpd-1–Bound A2AR–BRIL.
The overall structure of A2AR–Cmpd-1 is similar to other reported structures of A2AR in complex with antagonists (8–10, 12, 14). For example, our structure has an rmsd of 1.1 Å and 1.4 Å over 240 Cαs when aligned with A2AR–BRIL–ZM241385 structure (antagonist bound; PDB ID code 4EIY; Fig. 2A) and A2AR-T4L–UK-432097 (agonist bound, PDB ID code 3QAK) (11), respectively. Our structure represents an inactive state structure of the receptor based on the conformation of TM6, and conserved motifs such as E/DRY and NPXXY are virtually identical to that in A2AR–BRIL–ZM241385 structure (8). Nevertheless, our structure has notable differences compared with the reported structure of A2AR–BRIL–ZM241385, even though the crystals were generated from the same fusion protein. Some of these differences can be attributed to crystal packing, whereas others are most likely due to differences arising from interactions with the bound ligand.
Fig. 2.

Structure features of A2AR–BRIL in complex with Cmpd-1. (A) Overall structure comparison of A2AR–BRIL–Cmpd-1 (receptor and ligand in green; BRIL in cyan) and A2AR–BRIL–ZM241385 (receptor and ligand in orange; BRIL in gold). (B) Structural comparison of the intracellular part of TM5. In the A2AR–BRIL–Cmpd-1 structure, TM5 is displaced by 8 Å at the Cα of Gln2075.69 relative to the A2AR–BRIL–ZM241385 structure. (C) In the A2AR–BRIL–Cmpd-1 structure, Glu2286.30 forms a salt bridge with Arg1073.55, which is only possible because of the displacement of TM5 noted above. The map is 2Fo–Fc map contoured at 1.5 σ. (D) Structural comparison of the N-terminal aspect of the TM1 helices between the two structures where the Cα atoms of Met41.30 are ∼4 Å apart.
The first difference can be found at the cytoplasmic end of TM5, which showed an ∼8-Å outward movement comparing to its position in A2AR–BRIL–ZM241385 structure (Fig. 2B) at the Cα carbon of Gln2075.69 (superscripts indicate Ballesteros–Weinstein numbers) (20). We cannot attribute this difference to any difference in the ligand binding pocket, and the difference may result indirectly from crystal packing, because TM5 is fused to BRIL and/or it may reflect the intrinsic flexibility of the receptor. It is noteworthy that in a thermostabilized A2AR bound to the xanthine XAC (A2AR-TS–XAC), Glu2286.30 forms an ionic interaction with Arg1023.50 in A2AR (12) that was first observed in rhodopsin (21) and has also been observed in the D3 dopamine receptor (22). This interaction, called the ionic lock, is expected to stabilize the inactive state. In contrast, in our structure, Glu2286.30 forms an ionic interaction with Arg1073.55 with a distance of 4.0 Å (Fig. 2C), which would also stabilize the inactive conformation of TM6. This interaction between Glu2286.30 and Arg1073.55 cannot be formed in A2AR–BRIL–ZM241385 because of the position of TM5, and it is not possible in A2AR-TS–XAC because Arg1073.55 was mutated to Ala. The second difference relates to the conformation of Helix 8 (Fig. 2A). Unlike the straight helix observed in the A2AR–BRIL–ZM241385 structure, Helix 8 in our structure has a kink starting from Gln297. Because this part of Helix 8 is extensively involved in crystal contacts (Fig. 1B), this difference is likely caused by crystal packing. The third difference pertains to the location of the Cα atom of Met41.30 toward the extracellular end of TM1, which is ∼4 Å further from the TM core than in the A2AR–BRIL–ZM241385 structure (Fig. 2D). This difference, albeit more subtle in comparison with the first two, is most likely caused by the binding of Cmpd-1 and may explain the reason for not obtaining the same crystal packing as A2AR–BRIL–ZM241385 in LCP; this will be discussed in more detail below.
It should be noted that electron density provided evidence for an alternate conformation for ECL2 in our structure involving Phe168 and, to a lesser extent, Leu167 and Glu169. The side chain of Phe168 adopts two very different conformations (Fig. S3). According to the density map and refinement results, the conformation in which the side chain of Phe168 projects into the receptor core and makes π–π interaction with the ligand seems to be the dominant conformation at occupancy of 0.7, which is consistent with its conformation in all other reported A2AR structures (7–14). Further discussion of the binding pocket will be based on this single conformation.
Fig. S3.

Alternate conformation of Leu167 to Glu169. The involved residues and the ligand Cmpd-1 are shown in sticks. 2Fo–Fc map are shown in blue mesh contoured at 1.0 σ level. The major difference is with Phe168ECL2. The conformation that allow its aromatic ring to stack with Cmpd-1 is the dominant conformation with the occupancy of 0.7 (shown in green), and the occupancy of the other conformation is 0.3 (shown in gray).
Cmpd-1 Binding Pocket.
Cmpd-1 is well defined in the density map (Fig. 3A). The closely linked methylphenyl and aminotriazole rings occupy similar positions as the furan ring and triazolotriazine core of ZM241385, respectively (Fig. S4) (8, 10, 12, 14); in this deep pocket, they make extensive hydrophobic interactions and form hydrogen bonds with surrounding residues (Fig. 3B). Some interactions are common among various A2AR ligands: the amide nitrogen of Asn2536.55 makes a hydrogen bond with a nitrogen atom on the aminotriazole ring, and Phe168 on ECL2 makes aromatic stacking interaction with the aminotriazole ring. These two residues also make critical interactions with A2AR ligands in all reported A2AR structures. In addition, Glu169 on ECL2 forms a hydrogen bond to the amine group of the aminotriazole ring, and it also interacts with adenosine by forming a hydrogen bond. Overall, the methylphenyl and aminotriazole groups bind within the orthosteric pocket of A2AR, as defined by the space occupied by adenosine (Fig. S4) (13).
Fig. 3.

Binding interactions between the A2AR and Cmpd-1. (A) Cmpd-1 binding pocket. The side chains of residues within 4 Å of the ligand are shown in stick. Hydrogen bonding interactions are depicted by dashed lines. The Fo–Fc omit map around the ligand is shown as mesh contoured at 2.5 σ. (B) A schematic representation of Cmpd-1 binding interactions. The residues within 4 Å of Cmpd-1 are shown as spheres, with polar, hydrophobic, and negatively charged residues colored cyan, green, and red, respectively. The π−π stacking interaction of the aminotriazole ring with Phe168 is depicted with a solid green line. The hydrogen bonding interactions with the side chains of Asn253 and Glu169 are depicted with dotted pink lines. The protein pocket is displayed with a line around the ligand, colored according to the nearest residue. A gap in the line shows an opening in the pocket, and the minimum gap observed in this diagram indicates that Cmpd-1 is very well buried. The iminium of Cmpd-1 and His250 are predicted to be protonated due to the low pH (6.5) of crystallization conditions.
Fig. S4.

Comparison of various A2AR ligands binding positions. The structure of A2AR–BRIL–Cmpd-1 (green) is superimposed with A2AR–BRIL–ZM241385 (cyan; PDB ID code 4EIY) and A2AR-TS–adenosine (salmon; PDB ID code 2YDO). The ligands are shown in stick. The methylphenyl and aminotriazole rings of Cmpd-1 occupy positions that collectively are similar to that of adenosine in the orthosteric site.
The methoxyphenyl ring of Cmpd-1 extends to an adjacent and previously unobserved binding pocket formed by Tyr91.35, Ala632.61, Ile662.64, Ser672.65, Leu2677.32, Met2707.35, Tyr2717.36, and Ile2747.39, toward the extracellular aspect of TM1, TM2, and TM7 (Fig. 3); this is different from the binding mode of ZM241385 in A2AR–BRIL, which has its phenol ring extended toward solvent-exposed surface, but is somewhat reminiscent of its binding mode in thermostabilized A2AR (Fig. 4A) (8). Among the surrounding residues, Tyr91.35 and Tyr2717.36 make key aromatic interactions with Cmpd-1’s phenyl ring, and their positions and rotamers are unique compared with other reported A2AR structures (Fig. 4A and Fig. S5) (7–14). Specifically, Tyr91.35 moves 1.7 Å away from the TM core (Fig. 4A) compared with its position in A2AR–BRIL–ZM241385 structure, to enable the receptor to accommodate the methoxyphenyl moiety. The difference in the position of Tyr91.35 is amplified at the extracellular end of TM1 (Cα of Met41.30) to ∼4 Å. In the A2AR–BRIL–ZM241385 crystal lattice, the extracellular end of TM1 makes important crystal lattice contacts with an adjacent symmetry-related molecule, and the position of TM1 observed in our structure is incompatible with this packing (Fig. S6) (8), which may explain why we failed to crystallize A2AR–BRIL–Cmpd-1 in the same lattice form as observed for A2AR–BRIL–ZM241385 in LCP. The other aromatic residue Tyr2717.36 effectively covers the methoxyphenyl group and prevents it from becoming directly exposed to the solvent by adopting a rotamer very different from that found in the structures of A2AR–BRIL or thermostabilized A2AR bound to ZM241385 (Fig. 4A and Fig. S5) (12). The dramatic loss of potency observed upon replacement of the methoxyphenyl with cyclopropyl indicates the critical importance of aromatic interactions for Cmpd-1 binding (Table 2). In comparison, substitution of the phenol ring of ZM241385 with a cycloalkyl group did not alter the affinity for A2AR significantly (23), consistent with the reported structures that the phenol ring is primarily making hydrophobic interactions with A2AR (PDB ID code 3PWH), or primarily solvent exposed (PDB ID code 4EIY) (12, 14).
Fig. 4.

Potential allosteric site identified in A2AR–BRIL–Cmpd-1 structure. (A) Comparison of Tyr91.35 and Tyr2717.36 positions and rotamers viewed from extracellular side. The structure of A2AR–BRIL–Cmpd-1 (green) is superimposed with A2AR–BRIL–ZM241385 (orange; PDB ID code 4EIY) and A2AR-TS–ZM241385 (blue; PDB ID code 3PWH). Residues and ligand in A2AR–BRIL–Cmpd-1 are shown in stick, and they are shown in lines in other structures. (B) The potential allosteric site. The methoxyphenyl ring is located in a compact pocket formed by eight surrounding residues. Cmpd-1 is shown in spheres, and residues are shown in sticks, with the contact residues’ van der Waals radius depicted as dots. (C) Sequence alignment of human adenosine receptors. Residues forming the allosteric pocket are indicated by blue circles. Residues that are strictly conserved among all subtypes of adenosine receptors are highlighted by red shade, and partially conserved residues are highlighted in red characters.
Fig. S5.

Comparison of Tyr91.35 and Tyr2717.36 positions and rotamers with other A2AR structures. The structure of A2AR–BRIL–Cmpd-1 (green) is superimposed with A2AR-T4L–UK-432097 (magenta; PDB ID code 3QAK), A2AR-TS–adenosine (salmon; PDB ID code 2YDO), A2AR-TS–NECA (salmon; PDB ID code 2YDV) and A2ARTS–T4E (yellow; PDB ID code 3UZC). Tyrosine side chains and ligands are shown in stick for A2AR–BRIL–Cmpd-1 and in lines for the other structures.
Fig. S6.

The outward displacement of TM1 in A2AR–BRIL–Cmpd-1 structure is incompatible with the crystal packing of A2AR–BRIL–ZM241385. A2AR–BRIL–Cmpd-1 (green) is aligned with A2AR–BRIL–ZM241385 (orange). A symmetry molecule of A2AR–BRIL–ZM241385 (gray) that packs through their TM1 and TM7 shows steric conflicts with TM1 in A2AR–BRIL–Cmpd-1 and therefore prevents crystallization in the same lattice as A2AR–BRIL–ZM241385 in LCP. Residues with steric conflicts are shown in sticks.
As a result of the unique conformations of these two aromatic residues, a relatively compact pocket is formed in our structure that accommodates the methoxyphenyl ring (Fig. 4B and Fig. S7), whereas the equivalent part in other reported A2AR structures is either very shallow (i.e., in A2AR–BRIL–ZM241385) (8) or more exposed to solvent as a cleft (i.e., in A2AR-TS–ZM241385) (12) (Fig. S7). The methoxyphenyl ring of Cmpd-1 has lower B-factors (averaged at 78.1 Å2) than the whole ligand (averaged at 93.9 Å2), suggesting that binding within this pocket stabilizes its position in the crystal. The eight residues that form this pocket tightly surround the methoxyphenyl R group of Cmpd-1 (Fig. 4B and Fig. S7), and leave no additional space for accommodating this ring with a methoxy group in either the meta or para position. Specifically, metamethoxyphenyl substitution will cause steric clash with Tyr2717.36, or Ala632.61, Tyr91.35, Ile662.64, whereas paramethoxyphenyl substitution will cause steric clash with Tyr91.35, and Ala632.61 or Tyr2717.36, depending on the rotamers of the methoxyphenyl ring. This binding mode is consistent with the results that meta- and paramethoxy analogs of the Cmpd-1 have significantly lower affinity for A2AR (Table 2).
Fig. S7.

Comparison of the allosteric pocket in A2AR–BRIL–Cmpd-1 structure to equivalent parts in other reported A2AR structures. The structures shown here include A2AR–BRIL–ZM241385 (cyan; PDB ID code 4EIY), A2AR-TS–ZM241385 (blue; PDB ID code 3PWH), A2AR-TS–1,2,4-triazine antagonist (A2AR-TS-–T4E, yellow; PDB ID code 3UZC), and A2AR-TS–adenosine (salmon; PDB ID code 2YDO). These structures are all aligned with the A2AR–BRIL–Cmpd-1 structure (green), and surfaces are all viewed and clipped at same position for comparison. Red arrows point to the equivalent part in each structure and the sites are either very shallow pockets or clefts exposed to solvent.
Because the native ligand adenosine does not extend to this pocket, it may represent a potential allosteric site that would not be predicted from other reported A2AR structures. Binding results also suggest that the binding interactions in this pocket are critical for the selectivity of Cmpd-1 for A2AR over A1R, because modifications of the orthomethoxyphenyl ring result in loss of selectivity between A2AR and A1R (Table 2). Sequence alignment showed that five of the eight residues forming this allosteric pocket are not conserved among adenosine receptors (Fig. 4C). For example, the equivalent residues of A2AR Ser672.65, Leu2677.32, and Met2707.35 are Asn702.65, Ser2677.32, and Thr2707.35 in A1R, respectively. Specifically, Leu2677.32 and Met2707.35 in A2AR make contacts with the methoxy group (Fig. 3), and the removal of the methoxy group results in decrease of affinity on A2AR and loss of selectivity between A2AR and A1R (Table 2). The variability of the residues forming this pocket suggests it may be a target for the development of novel selective A2AR antagonists and/or allosteric modulators.
Conclusion
We report here the crystal structure of A2AR in complex with a unique antagonist, Cmpd-1—a potential therapeutic candidate for PD; this is an example of a nonthermostabilized GPCR with fusion partner to be crystallized by vapor diffusion in detergent micelles, and demonstrates how subtle differences in a bound ligand can influence crystal packing in a GPCR. Although the A2AR is a well-studied receptor with a number of structures bound to various ligands reported, the structure we obtained with Cmpd-1 showed a difference in the ligand-binding mode that resulted in structural changes necessitating a different crystal packing. Structural analysis revealed a cryptic pocket not observed in previously reported structures, which could facilitate the development of novel A2AR subtype-selective compounds as therapeutic agents for PD and other disorders.
Experimental Procedures
Expression and Purification.
A2AR was cloned into pFastBac vector with HA signal sequence followed by FLAG epitope tag at its N terminus, and its C terminus is truncated at Ala316; ICL3 residues are replaced by fusion partners, T4L or BRIL, in the same positions as previously reported (8, 14). We also generated an mT4L fusion in the same position as a previously reported T4L fusion (17). To facilitate purification, an N-terminal Flag epitope was inserted after a signal peptide and a C-terminal six-histidine tag was introduced. These constructs were expressed in Sf9 cells using the pFastBac baculovirus system (Invitrogen). Cmpd-1 was added to the medium during expression at 100 nM. The cells were infected with baculovirus at 27 °C for 48 h before collection.
The purifications for all of the constructs were carried out through Ni-affinity and FLAG affinity chromatography and size-exclusion chromatography (SEC). Infected Sf9 cell pellets were lysed in 10 mM Tris, pH 7.6, 1 mM EDTA, 2 mg/mL iodoacetimide, 2.5 μg/mL leupeptin, 0.16 mg/mL benzamidine, and 5 μM of Cmpd-1. Membrane sample was collected by spinning at 37,800 × g. Extraction of A2A receptor from Sf9 membranes was done with a Dounce homogenizer in a solubilization buffer comprised of 1% n-dodecyl β-d-maltoside (DDM), 0.03% cholesteryl hemisuccinate (CHS), 30 mM Hepes, pH 7.8, 800 mM NaCl, 30% (vol/vol) glycerol, 2 mg/mL iodoacetimide, 2.5 μg/mL leupeptin, 0.16 mg/mL benzamidine, and 5 μM of Cmpd-1. Ni-affinity resin was added and mixing continued for another 2 h at 4 °C. The Ni-affinity resin was collected by spinning, and washed extensively with Ni wash buffer comprised of 0.1% DDM, 0.03% CHS, 30 mM Hepes, pH 7.8, 800 mM NaCl, 10% glycerol, 5 mM imidazole, and 5 μM of Cmpd-1. The protein was eluted from Ni-affinity resin by the Ni wash buffer with 200 mM imidazole, and loaded onto an anti-Flag M1 affinity column with the addition of 2.5 mM CaCl2. The FLAG column was then washed extensively with FLAG wash buffer comprised of 0.1% DDM, 0.03% CHS, 30 mM Hepes, pH 7.8, 800 mM NaCl, 5 μM of Cmpd-1 and 2.5 mM CaCl2. The detergent DDM was then gradually exchanged over 1 h into a buffer with 0.01% LMNG, 0.001% CHS, 30 mM Hepes, pH 7.8, 800 mM NaCl, and 5 μM of Cmpd-1. The receptor was eluted from the FLAG column using the same buffer but without CaCl2 and containing 200 μM FLAG peptide and 5 mM EDTA. The elution sample was concentrated and loaded onto a SEC column (Superdex 200) with the buffer 0.01% LMNG, 0.001% CHS, 30 mM Hepes, pH 7.8, 800 mM NaCl, and 5 μM of Cmpd-1. The major peak fractions were combined and concentrated for crystallization and the ligand was added up to 20 μM final concentration.
Crystallization.
We performed extensive LCP crystallization trials for A2AR–T4L, A2AR–mT4L, and A2AR–BRIL following similar conditions as previous reported (8, 14). No crystal or only poorly diffracting crystals were obtained even after extensive optimization. We obtained crystals that diffracted beyond 3.5 Å resolution by vapor-phase diffusion. The crystals grew in 0.045 M Mes, pH 6.5, 0.045 M MgCl2, 28% (vol/vol) PEG400, 5% (vol/vol) Jeffamine M-600 (pH 7.0) at 20 °C and were harvested by flash-freezing directly into liquid nitrogen. All of the LCP and vapor-phase diffusion crystallization trials were performed by Gryphon LCP robot (Art Robbins Instruments).
Data Collection and Structure Determination.
Data collection was performed using the beamline 23-ID of GM/CA@APS at the Advanced Photon Source. Microbeams of 10 or 20 μm diameter were used to acquire all diffraction data. Owing to radiation damage, only 5–20° of rotation data were collected from each crystal. All data were processed with the HKL2000 package (24). A 3.5-Å dataset was obtained by merging diffraction data from 20 crystals. The space group was determined to be P41212. Molecular replacement was performed using the program Phaser in the CCP4 package (25), with separate A2AR and BRIL chain from A2AR–BRIL–ZM241385 structure (PDB ID code 4EIY) as the search model. Iterative model-building and structural refinement were performed using COOT (26) and Refmac5 (27), respectively. The quality of the structure was assessed using MolProbity (28). Data processing and structure refinement statistics are shown in Table S1.
Ligand Optimization.
Using Percepta 2014 software, Cmpd-1 was predicted to have a pKa of ∼9 and thus under physiological conditions will be protonated (ACD/Percepta; Advanced Chemistry Development, Inc.). The Percepta software predicted that the NH moiety of the linker group that is closest to the aminotriazole ring is the most basic. This protonated form of the ligand in the RX structure was then submitted for a quantum mechanical (QM) optimization calculation using Jaguar version 8.4 (29). The QM calculation was performed using the B3LYP density functional theory (DFT) method with the 6-31G** basis set and the Poisson–Boltzmann Finite water solvation model, and XYZ Cartesian constraints were applied to all of the heavy atoms of the ligand with the exception of the linker region to help relieve apparent intramolecular strains within the linker region. To be noted, a methoxy group is usually found to be coplanar with the aromatic ring of methoxyphenyl, because this allows for overlap between the oxygen atom nonbonding orbital and the aromatic π system; this is not possible, however, for Cmpd-1, because steric clashes with the protein prevent it from assuming a coplanar configuration with the ring. Although unusual, examples of displacement of the methoxy group away from coplanarity have been observed previously (compare PDB ID codes 2bkt and 2bks). Further, a 5° incremental rigid-torsion QM scan of the methoxy group (using the same version of Jaguar, solvation model, DFT, and basis set as described above) revealed that the orientation of the methoxy group in our structure incurs less than +2.5 Kcal/mol energy penalty relative to the configuration where the methoxy is coplanar.
Binding assay.
Ligand-binding studies for the adenosine receptors were done in membranes pretreated with adenosine deaminase prepared from HEK293 cells. For inhibition of binding to the human A2AR, membranes were incubated for 60 min with [3H]-ZM241385 (0.4 nM) at 25 °C (30). The reaction was terminated by rapid filtration over polyethyleneimine-treated glass-fiber filters and nonspecific binding determined by 10 µM 5′-N-ethylcarboxamido adenosine (NECA). For inhibition of binding to the human A1R, membranes were incubated as above with 0.15 nM [3H]-DPCPX, and nonspecific binding was defined with 100 µM 2-chloroadenosine.
Functional assay.
The ability to inhibit A2AR function was determined in CHO cells expressing human A2A receptor. Cells were preincubated for 60 min with the compound, and then incubated for a further 60 min in the presence of a submaximal concentration (100 nM) of the agonist CGS21680 in buffer containing 10 µM rolipram. Changes in cAMP levels were determined using the CisBio HTRF Kit.
Footnotes
Conflict of interest statement: The research was performed by ConfometRx, Inc., cofounded by T.S.K. and B.K.K., in collaboration with UCB Pharma.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 5UIG).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1621423114/-/DCSupplemental.
References
- 1.de Lera Ruiz M, Lim YH, Zheng J. Adenosine A2A receptor as a drug discovery target. J Med Chem. 2014;57(9):3623–3650. doi: 10.1021/jm4011669. [DOI] [PubMed] [Google Scholar]
- 2.Shook BC, Jackson PF. Adenosine A(2A) receptor antagonists and Parkinson’s disease. ACS Chem Neurosci. 2011;2(10):555–567. doi: 10.1021/cn2000537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pinna A, Wardas J, Simola N, Morelli M. New therapies for the treatment of Parkinson’s disease: Adenosine A2A receptor antagonists. Life Sci. 2005;77(26):3259–3267. doi: 10.1016/j.lfs.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 4.Milne GR, Palmer TM. Anti-inflammatory and immunosuppressive effects of the A2A adenosine receptor. Sci World J. 2011;11:320–339. doi: 10.1100/tsw.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bara-Jimenez W, et al. Adenosine A(2A) receptor antagonist treatment of Parkinson’s disease. Neurology. 2003;61(3):293–296. doi: 10.1212/01.wnl.0000073136.00548.d4. [DOI] [PubMed] [Google Scholar]
- 6.Michel A, Downey P, Nicolas JM, Scheller D. Unprecedented therapeutic potential with a combination of A2A/NR2B receptor antagonists as observed in the 6-OHDA lesioned rat model of Parkinson’s disease. PLoS One. 2014;9(12):e114086. doi: 10.1371/journal.pone.0114086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lebon G, Edwards PC, Leslie AG, Tate CG. Molecular determinants of CGS21680 binding to the human adenosine A2A receptor. Mol Pharmacol. 2015;87(6):907–915. doi: 10.1124/mol.114.097360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu W, et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012;337(6091):232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Congreve M, et al. Discovery of 1,2,4-triazine derivatives as adenosine A(2A) antagonists using structure based drug design. J Med Chem. 2012;55(5):1898–1903. doi: 10.1021/jm201376w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hino T, et al. G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature. 2012;482(7384):237–240. doi: 10.1038/nature10750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu F, et al. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332(6027):322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doré AS, et al. Structure of the adenosine A(2A) receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure. 2011;19(9):1283–1293. doi: 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lebon G, et al. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474(7352):521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaakola VP, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322(5905):1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caffrey M. A comprehensive review of the lipid cubic phase or in meso method for crystallizing membrane and soluble proteins and complexes. Acta Crystallogr F Struct Biol Commun. 2015;71(Pt 1):3–18. doi: 10.1107/S2053230X14026843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4(5):706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thorsen TS, Matt R, Weis WI, Kobilka BK. Modified T4 lysozyme fusion proteins facilitate G protein-coupled receptor crystallogenesis. Structure. 2014;22(11):1657–1664. doi: 10.1016/j.str.2014.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chun E, et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure. 2012;20(6):967–976. doi: 10.1016/j.str.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Russo Krauss I, Merlino A, Vergara A, Sica F. An overview of biological macromolecule crystallization. Int J Mol Sci. 2013;14(6):11643–11691. doi: 10.3390/ijms140611643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 21.Palczewski K, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289(5480):739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 22.Chien EY, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330(6007):1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Zwart M, et al. Potent antagonists for the human adenosine A2B receptor. Derivatives of the triazolotriazine adenosine receptor antagonist ZM241385 with high affinity. Drug Dev Res. 1999;48:95–103. [Google Scholar]
- 24.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 25.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 28.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bochevarov AD, et al. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int J Quantum Chem. 2013;113(18):2110–2142. [Google Scholar]
- 30.Palmer TM, Poucher SM, Jacobson KA, Stiles GL. 125I-4-(2-[7-amino-2-[2-furyl][1,2,4]triazolo[2,3-a][1,3,5] triazin-5-yl-amino]ethyl)phenol, a high affinity antagonist radioligand selective for the A2a adenosine receptor. Mol Pharmacol. 1995;48(6):970–974. [PMC free article] [PubMed] [Google Scholar]