

Summary
Three-dimensional live cell imaging of the interaction of T cells with antigen presenting cells (APC) visualizes the subcellular distributions of signaling intermediates during T cell activation at thousands of resolved positions within a cell. These information-rich maps of local protein concentrations are a valuable resource in understanding T cell signaling. Here, we describe a protocol for the efficient acquisition of such imaging data and their computational processing to create four-dimensional maps of local concentrations. This protocol allows quantitative analysis of T cell signaling as it occurs inside live cells with resolution in time and space across thousands of cells.
Keywords: live cell imaging, computational image analysis, spatiotemporal distributions, immunological synapse, T cell activation
1. Introduction
T cell activation is regulated by the complex interactions of dozens of signaling intermediates. One of the great, general challenges in current biology is to understand how dozens of proteins function as an integrated system. As a key resource to elucidate such complexity, signaling intermediates are not evenly distributed through an activating T cell but enrich at particular locations at distinct times [1, 2]. Concurrent enrichment of two proteins increases their interaction probability and thus the local efficiency of the signaling step mediated by their interaction. Thus at the system scale uneven signaling distributions govern the information flow through signaling networks [3–6]. Supporting the importance of subcellular location for function in T cell activation, loss-of-function mutations of various signaling intermediates consistently yield diminished localization [7–11]. In addition, through the imaging of large numbers of signaling intermediates, subcellular structures mediating signal integration have emerged [12, 13]. Thus a quantitative determination of subcellular signaling distributions of a large number of T cell signaling intermediates is a powerful resource to discover functional signaling connectivity. Here we describe the methodology for such characterization across thousands of cells. Three-dimensional live cell imaging of the interaction of T cells with antigen presenting cells (APC) visualizes the subcellular distributions of signaling intermediates at thousands of resolved positions within a cell, i.e. it yields detailed three-dimensional maps of local concentrations. We describe a protocol for efficient acquisition of such data. Voxel-by-Voxel quantification of such imaging data provides full access to the three-dimensional maps of local concentrations. We describe a computational analysis routine to generate maps that are aligned across all T cells to be analyzed. Such maps form a powerful foundation for the computational analysis of signaling connectivity and the modeling of T cell signal transduction.
2. Materials
2.1. Retroviral transduction reagents
Phoenix-E (φNX-E) cell line (obtained from Nolan laboratory, Stanford University).
φNX-E incomplete media: DMEM with 4.5% glucose, 110mg/L sodium pyruvate and L-glutamine, 10% FBS, 100U/mL penicillin, 100µg/mL streptomycin, MEM non-essential amino acids.
φNX-E complete media: As 2.1.2 supplemented with 300µg/mL hygromycin and 1µg/mL diphtheria toxin.
Corning Primaria tissue culture plates 35mm or equivalent.
0.02% EDTA solution in PBS.
Chloroquine diphosphate: 4.1mg/mL chloroquine diphosphate solution in ddH2O and sterile filtered (e.g., Sigma).
2x Hepes buffered saline: 16g/L NaCl, 0.74g/L KCl, 0.27g/L NA2HPO42g/L D-glucose, 10g/L HEPES. Dissolve in ultrapure water, adjust pH to 7.05, and sterile filter.
Calcium chloride: 2M solution of calcium chloride in ddH2O. Sterile filtered.
Protamine sulfate: dissolved in PBS at 8mg/mL.
2.2. T cell culture components
Complete media: RPMI with L-glutamine, 10% FBS, 100U/mL penicillin, 100µg/mL streptomycin, 50µM 2-mercaptoethanol.
Interleukin-2 media: complete media supplemented with 50U/mL rhIL-2.
Agonist peptide for the TCR transgene (Moth cytochrome C for the 5C.C7 TCR): 5mM solution in ultrapure water.
2.3. Cell sorting and imaging components
Imaging buffer: PBS without calcium and magnesium, 10% FBS, 1mM calcium chloride, 500µM magnesium chloride.
Glass bottomed 348 well imaging plate (Brooks Life Science Systems) or equivalent.
3. Methods
3.1. Generation of retrovirus
Stock plates of φNX-E cells are maintained in φNX-E complete media in a 37°C, 6% CO2 incubator. When splitting cells, aspirate media and add 1mL 0.02% EDTA per plate. Incubate cells at 37°C for approximately 3 minutes (see Note 1). Following incubation, tap each plate firmly with a finger to fully loosen the cells. Use 5mL DMEM to collect cells from the plate and transfer to a Falcon tube. Centrifuge for 3 minutes at 200×g, resuspend in 1mL of φNX-E media and count cells using a hemocytometer. Plate 600,000 cells into a fresh Falcon Primaria tissue culture plate in a total volume of 6mL media. φNX-E complete media should be used for general maintenance (see Note 2), whereas incomplete media should be used when cells will undergo transduction.
On day 3 of culture in incomplete media, φNX-E cells are ready for transduction via calcium phosphate precipitation. Make a fresh solution of chloroquine diphosphate and add 25µL to each plate. Swirl gently to mix and return cells to incubator.
For each plasmid, label two 15mL Falcon tubes as tube A and tube B. In tube A mix 0.5mL 2× HBS with 2µL 1M NaOH (see Note 3). In tube B mix 0.5mL sterile ddH20 with 10µg plasmid DNA. Add 62µL 2M CaCl2 drop by drop to tube B.
Using a 1mL tissue culture pipette, bubble air vigorously through tube A whilst adding the contents of tube B in a drop-by-drop fashion. Let sit at room temperature for a minute.
Gently add the entirety of this solution to the φNX-E cells, swirling gently to aid distribution. Return cells to the incubator overnight.
The following day check for the presence of the calcium phosphate precipitate as black puncta much smaller than the φNX-E cells. Gently aspirate the media from the φNX-E cells and replace with 3.2mL incomplete media (see Note 4). Incubate cells, without any media changes or splitting, for approximately another 48 hours. At this time the media will contain retrovirus carrying the DNA construct of interest.
3.2. Culture of T cells from lymph nodes
Cull a T cell receptor transgenic mouse over 6 weeks of age using a schedule one technique (see Note 5).
Dissect out lymph nodes (see Note 6). Collect them into a tube containing 5mL of complete media.
Place a 40µm cell strainer over a 50mL Falcon tube. Pour the lymph nodes into the strainer and gently disrupt them using the plunger of a 1mL syringe. Wash the strainer with 5mL complete media, and repeat the disruption process until the media passing through the strainer appears clear. Centrifuge the Falcon tube for 3 minutes at 200×g.
Discard supernatant and thoroughly resuspend the cell pellet in 1mL complete media. Count live cells and add complete media to the cells to bring a density to 4×106 cells/mL.
In a 24 well plate, add 0.65µL of 5mM agonist peptide to the bottom of each well you wish to use. Add 1mL of cell suspension to each well, so that each contains 4×106 cells. Incubate at 37°C, 6% CO2 overnight.
3.3. Retroviral transduction of T cells
After T cells have been activated overnight they are ready to be retrovirally transduced. Collect each well into a separate 15mL Falcon tube. If more than one well of cells will be transduced with the same construct they may be pooled.
Collect the media from the φNX-E cell plate into a separate 15mL Falcon tube. Gently add 1mL PBS to the φNX-E cells to prevent dehydration and set aside. Centrifuge both sets of tubes at 200×g for 3 minutes.
Discard the T cell supernatant. Take 2mL φNX-E cell media supernatant and use this to resuspend the T cells. Add 2µL of 8mg/mL protamine sulphate to the bottom of a 24 well plate well. Add the 2mL of cell suspension to this well.
Centrifuge the 24 well plate at 200×g32°C, for 2 hours (see Note 7).
In parallel on a wide field fluorescent microscope, use a 10× objective lens to focus on the φNX-E cells left in the plate. With an appropriate laser determine whether the φNX-E cells are expressing the fluorescent protein construct. If not, it is likely that the calcium phosphate precipitation has failed (see Note 8).
After centrifugation aspirate the media from each well of T cells, being careful not to disrupt the cells at the bottom of the well. Resuspend T cells in 2mL IL-2 media.
Incubate cells for a further 3 to 4 days to allow expression of the transduced protein and to expand cell numbers. On a daily basis check the cell density. Split each well into two when the cells become fully confluent and maintain in IL-2 media.
3.4. Fluorescent activated cell sorting of successfully transduced T cells
On the third or fourth day of culture following transduction collect the T cells into a Falcon tube and centrifuge at 200×g for 3 minutes. Discard the supernatant and resuspend the cell pellet in 150µL imaging buffer per well of cells.
Prepare a collection tube by adding 1.5mL IL-2 media to a 6mL FACS tube.
- During FACS, use the following gating strategy to select for positively transduced lymphocytes (Fig. 1):
- Select live lymphocytes based on forward scatter versus side scatter.
- Select for singlets based on trigger pulse width versus side scatter.
Collect positive cells in multiples of 4×104 cells (see Note 10).
Return sorted cells to a 24-well plate in the incubator until ready for imaging. Image the same day as sorting to minimize loss of fluorescence (see Note 11).
Fig. 1. Gating strategy to sort GFP positive live lymphocytes.

CL4 T cells were retrovirally transduced to express Chronophin-GFP. i) The side scatter (SSC) versus forward scatter (FCS) gate to select live cells is given in red. ii) The trigger pulse width versus forward scatter (FCS) gate to select singlets is given in green. iii) A GFP positive sorting gate (green gate) is created around the GFP positive population which lies between 1 and 1.5 log shifts above the top of the GFP negative population (purple gate).
3.5. Live cell imaging of T cell:APC couple formation
For the 5C.C7 TCR transgene CH27 cells are used as APCs and cultured in complete media (see Note 12). To peptide load APCs collect approximately 1×106 cells into 1mL of complete media and add to a 24 well plate. Add 2µL of 5mM MCC peptide and mix. Incubate for a minimum of 4 hours.
Meanwhile, ensure a spinning disk microscope fitted with an environmental chamber and a 40× oil objective lens seated on a piezo motor is fully pre-heated to 37°C (see Note 13). Also pre-heat a glass bottomed 348 well imaging plate.
Collect the sorted T cells into a microcentrifuge tube. Collect 300µl of the peptide loaded APCs into a separate microcentrifuge tube. Centrifuge both at 300×g for 5 minutes in a tabletop centrifuge.
Carefully remove the supernatant from the T cell pellet. Resuspend the pellet in 5µL of pre-heated 37°C imaging buffer per 4×104 cells.
Repeat with the APCs, resuspending in 50µL of pre-heated imaging buffer.
Add 50µL imaging buffer to an empty well on the imaging plate. Add 5µL of T cells to this well and fit onto the microscope. Allow the plate temperature to equilibrate for 5 minutes.
Meanwhile set up the microscope so that a z-stack consisting of 21 images 1µm apart is taken at three time points per minute in the fluorescent channel. A single differential interference contrast (DIC) reference image should be taken mid-stack at each time point. Images should be taken for 15 minutes (see Note 14). We use 2×2 binning to improve the signal-to-noise ratio of the imaging data (see Note 15).
Once the plate has equilibrated, ensure the T cells are in focus. Firmly flick the microcentrifuge tube containing the APCs to generate a single cell suspension. Gently add 5µL APCs to the well so that the layer of T cells is not disturbed. Immediately begin monitoring the well through the eyepiece and wait for the APCs to begin settling amongst the T cells. Select a field of view with a good distribution of T cells and APCs and begin imaging (see Note 16).
Once imaging is completed separate the images into two different channels: DIC and fluorophore. Split the fluorescent channel into separate folders for each time point, with each folder containing the 21 z-stack images. Export all files as TIFFs (see Note 17).
3.6. Annotation of cell couples and synapse positions
For each movie, prepare an annotation file identifying the positions of the synapse between each T cell:APC couple as described below. First create a z-stack maximum projection for each time point, and then stack all the maximum projections so they can be scrolled through as a function of time. This can be accomplished with any of a number of basic image processing programs, such as ImageJ.
Remove any autoscaling from the images (see Note 18). Apply a pseudocolor lookup table to the fluorescence data to make differences in sensor intensity more readily apparent.
Use the DIC reference images to identify cell couples. Scroll through the time series until a T cell is observed forming an immune synapse with an APC, defined as a broad membrane interface between the T cells and APCs. Fluorescence images should only be used to confirm the identity of cells as either T cells or APCs (see Note 19). Start at the frame where initial contact between the T cell and APC is made and count forward a further two frames. Within these three frames, select the one where the membrane interface first reaches its widest point. Classify this frame as time point 0 or the earliest point at which the immune synapse is fully formed. For as many frames as desired record the coordinates for the two ends of the immune synapse and the frame number so that the cell couple can be found again for the automated analysis below (Fig. 2).
- These coordinates should be saved into a spreadsheet either directly from the image-processing package or manually. The spreadsheet should have sets of rows with one set per cell couple and one row within the set for each time point (frame) for that couple. The sets should be separate by a blank row. In the row for each frame, put
- in column 1, the name of the image file (depending on how the images are acquired, there may be one file per time point or multiple time points in a single file)
- in column 2, the number of the channel within that file that contains the GFP fluorescence for that time point (if each time point is in a separate file this would typically be channel 1; if multiple time points are in the same file this would typically be the frame number)
- in columns 3 to 7, the X coordinate of the left end point, the Y coordinate of the left end point, the X coordinate of right end point, and the Y coordinate of right end point for the synapse in that time point.
- In column 8, the time difference for that frame relative to time point 0 (Fig. 3).
Save the spreadsheet as a CSV (Comma-separate value) file with a name that is specific (matched) to the movie file name.
Fig. 2. Cell coupling and interface annotation.

The coupling of an Ezrin-GFP-transduced 5C.C7 T cell with a CH27 APC in the presence of 10µM MCC agonist peptide is shown as a green transparent overlay of a maximum projection of the three-dimensional Ezrin-GFP fluorescence data onto DIC bight field images. Time is given in minutes relative to tight cell coupling. Black lines indicate the T cell:APC interface and the coordinates of the two ends of these lines are recorded for use in the computational analysis.
Fig. 3. Format of T cell:APC couple annotations for entry into the computational image analysis pipeline.
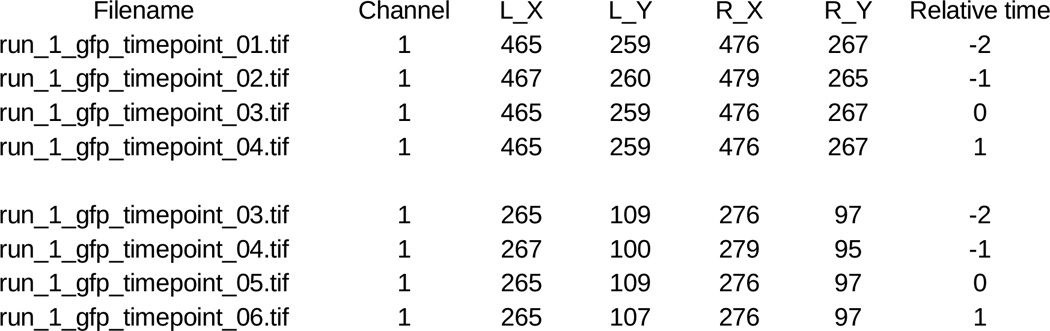
A representative spreadsheet is given.
3.7. Building models of four-dimensional protein distributions with CellOrganizer (see Note 20)
Install MATLAB if it is not already installed. Check http://cellorganizer.org to find out which versions of MATLAB are currently supported by CellOrganizer. Note that CellOrganizer is only supported for Mac OS and Linux systems.
Download the latest version of CellOrganizer from http://cellorganizer.org/Downloads, and extract it to some desired directory (i.e., a CellOrganizer directory (folder) in your home directory).
Launch MATLAB, and use the “cd” command to change your current working directory to the directory in which you put CellOrganizer. Type “setup” to initialize the environment for CellOrganizer.
If you want to test that the installation has occurred correctly, type “demo3Dtcell” which will process a small set of movies downloaded from the CellOrganizer website.
Type “copydemo(“demo3Dtcell”) and when prompted enter a name for the script it will create to analyze your movie or movies (e.g., “cofilin_analysis_May1”).
Type “edit” followed by a space and the name of the file you just created. Change the specification of the annotation file to match the movies that you want to analyze. For example, if your annotation file is called abc.csv, change “synapse_location=’annotation/*.csv’” to “synapse_location=’abc.csv’”. If you have more than one annotation file, they are in directory “mymovies”, and you want to process all of them, change “synapse_location=’annovation/*.csv’” to “synapse_location=’mymovies/*.csv’”
Change the path for where the results should be saved to the desired location by changing the value of “results_location”.
By default, the output models will have the same name as the base name of the annotation file with the number of the time point appended to it. For example, if the annotation file is “abc.csv”, the model for the first time point will be “abc_reltime_1.mat”. Also by default a model will be created for each time point in the annotation file. If a different name for the model is desired, set “options.model_prefix” to that name. If creation of models for only a subset of the time points is desired, edit “options.timepoints_to_include” to specify which time points to include (as a Matlab vector). For example, to analyze time points 1 to 7, set it to “[1:7]” or to analyze just time points 6 and 10, set it to “[6, 10]”.
Save the script and run it (i.e., click the Run button) and wait for the script to finish the analysis.
3.8. Analysis models and creating figures (see Note 21)
To generate a figure showing slices through the 3D map for each time point, use the function “ShowTcellModelMap”. To do so, include the full or relative path of the model file as the argument of the function, e.g., “ShowTcellModelMap(‘/path/to/model/your_model_reltime_1.mat’)” (a full path) or “ShowTcellModelMap(‘mymodels/your_model_reltime_1.mat’)” (a path relative to the current working directory).
To generate a movie showing protein intensity change through time points, use the function “GenerateTcellMovie”. The function can show up to three proteins in different colors, and it can therefore take up to three arguments. As before, each argument is a path to one of the models, e.g., “GenerateTcellMovie(‘models/protein1/*.mat’,’models/protein2/*.mat)” would create a two-color movie.
To compare the intensities of each voxel for different models, use the function “CompareTcellModels”. The function needs two paths, one for each model, e.g., CompareTcellModels(‘/path/to/models/model1.mat’, ‘/path/to/models/model2.mat’)”.
To show how much of the protein is enriched in the synapse region at various time points, use the function “ShowTcellEnrichment”. The input argument of the function are the filenames of the models to be compared, i.e. “ShowTcellEnrichment(‘/path/to/model/*.mat’)”.
Acknowledgments
The original research upon which these protocols are based was supported in part by National Institutes of Health grant P41 GM103712, by National Science Foundation grants MCB1121793 and MCB1121919, and by European Research Council grant PCIG11-GA-2012-321554.
Footnotes
The use of EDTA alone rather than the usual Trypsin/EDTA combination and limiting the time of EDTA on the φNX-E cells optimizes φNX-E cell health.
We find that maintaining the parental φNX-E stock under continuous selection ensures consistently high viral titers over months. Nevertheless, the stock is replaced from freezes about every 6 to 9 months.
For each batch of HBS the exact amount of NaOH to be added is determined in a dose response series of NaOH. The NaOH amount giving the smallest visible calcium phosphate precipitate is chosen.
By reducing the medium volume from 6 to 3.2 ml the concentration of virus in the supernatant is effectively doubled.
Our most widely used TCR transgene is the 5C.C7 TCR. However, this protocol has worked equally well with a number of other TCR transgenic mouse strains, including DO11.10, OTII, AND, Tg4, P14, CL4, and HY [14–18].
We commonly use combined inguinal, axillary, submandibular, and mesenteric lymph nodes. Spleen can be used instead, yet requires red blood cell lysis.
The centrifuge is heated by air friction. Therefore, pre-spin until 32°C are reached.
The LLMV long terminal repeat functions as a very strong promoter in the φNX-E cells and resulting viral mRNA is efficiently translated. The presence of imaging sensor in the φNX-E cells thus is an indirect yet convenient readout of the amount of viral mRNA produced.
The 1 to 1.5 log range above cellular autofluorescence background corresponds to the minimal fluorescence level detectable by sensitive microscope systems, a GFP concentration of 2.5µM [18]. The use of minimal sensor concentrations optimizes physiological relevance of the imaging data by minimizing interference of the imaging sensor with the signaling event to be studied. Using this sorting strategy across a range of actin regulators, the combined concentration of endogenous protein plus its retrovirally expressed GFP-tagged version as an imaging sensor was commonly within the 5 to 95 percentile of the endogenous protein concentration [19].
Transduction efficiencies vary as a function of (I) the TCR transgene and (II) the protein to be expressed. (I) The more vigorous the initial T cell proliferation in tissue culture is, the higher the transduction efficiency becomes. Therefore MHCI-restricted CD8 TCR transgenes generally yield higher efficiencies that can easily exceed 50 %. (II) The expression of a sensor is tightly linked to the endogenous concentration of the protein it is derived from. Thus Actin-GFP expresses substantially better than e.g. Itk-GFP. Sensors based on synthetic constructs or isolated protein domains, such as the F-tractin peptide of the PLCδ-PH domain, generally express well. Transduction efficiencies as low as 0.01 % can be used for subsequent imaging.
We find that homogeneously high sensor expression at the time of sorting becomes variable already on the time scale of days.
We have used a wide variety of APCs, from transfected CHO cells, various B and T cell lymphoma lines, dendritic cells to tumor target cells. In general non-adherent cells allow for easier detection of cell coupling as the T cell:APC interface forms perpendicular to the cover slip and thus is readily detectable in the DIC bright field images.
We have used magnifications from 20× to 100×. Higher magnifications trade better resolution for dimmer images and lower number of cells and thus cell couples in the field. Therefore, the majority of our experiments are done with 40× magnification. For cell coupling efficiencies below 10 % even lower magnification is desirable to capture an adequate number of cell couples.
Biochemically detectable T cell signaling activity in T cell:APC couples peaks in the first five minutes of cell coupling [20, 21]. NFAT and NFκB translocate into the nucleus within about 3 and 7 min respectively [13, 18]. Therefore, 15 minutes imaging times captures the peak of T cell signaling activity. Nevertheless, we have imaged as long as 16 h. Any imaging beyond 30 min will require measures to minimize buffer evaporation.
Binning of camera pixels trades resolution in x and y against signal-to noise ratio. With the chosen 2×2 binning on the 40× objective resolution is roughly equivalent in all three dimensions.
In response to strong stimulus, e.g. a high concentration of agonist peptide in the presence of costimulation, cell coupling can be virtually instantaneous. As cell couples that form before the onset of data acquisition cannot be accurately timed relative to the time of cell couple formation and thus have to be excluded from analysis, a rapid decision on the imaging field is essential.
Data can be saved in any format. However, archiving images as .tiff files ensure great compatibility with various analysis packages and ready exchange with colleagues.
Autoscaling, the process where the dimmest pixel in an image is set to black and the brightest one is set to white, is helpful in image acquisition to get an immediate first impression of the data. However in image analysis, in particular in the comparison of images across a time series, we display all images on the same scale, as thus apparent differences in brightness reflect actual differences in fluorescence intensity rather than potential differences in scaling.
We don’t use fluorescence data in the determination of cell coupling. This would set up a circular argument where fluorescence data would be used to determine time of cell coupling to then be analyzed relative to this time.
When imaging data are acquired with a 40× objective and 2×2 binning each T cell is represented by >5,000 voxels. This paragraph describes how to generate voxel-by-voxel-resolved models of these distributions. Importantly, the MATLAB script creates these models in a cell shape-normalized fashion such that each position in one T cell model can directly be compared to an equivalent position in any other model [19].
Once cell shape-normalized models of the three-dimensional protein distributions have been generated they can be computationally processed in whatever form desired. In this paragraph we list four examples for visualization and quantitative analysis.
Reference
- 1.Monks CR, Kupfer H, Tamir I, Barlow A, Kupfer A. Selective modulation of protein kinase C-theta during T-cell activation. Nature. 1997;385(6611):83–86. doi: 10.1038/385083a0. [DOI] [PubMed] [Google Scholar]
- 2.Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse: A molecular machinery controlling T cell activation. Science. 1999;285:221–226. [Google Scholar]
- 3.Schmick M, Bastiaens PI. The interdependence of membrane shape and cellular signal processing. Cell. 2014;156(6):1132–1138. doi: 10.1016/j.cell.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 4.Angermann BR, Klauschen F, Garcia AD, Prustel T, Zhang F, Germain RN, Meier-Schellersheim M. Computational modeling of cellular signaling processes embedded into dynamic spatial contexts. Nat Methods. 2012;9(3):283–289. doi: 10.1038/nmeth.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Purvis JE, Lahav G. Encoding and decoding cellular information through signaling dynamics. Cell. 2013;152(5):945–956. doi: 10.1016/j.cell.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mast FD, Ratushny AV, Aitchison JD. Systems cell biology. J Cell Biol. 2014;206(6):695–706. doi: 10.1083/jcb.201405027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeFord-Watts LM, Dougall DS, Belkaya S, Johnson BA, Eitson JL, Roybal KT, Barylko B, Albanesi JP, Wulfing C, van Oers NS. The CD3 zeta subunit contains a phosphoinositide-binding motif that is required for the stable accumulation of TCR-CD3 complex at the immunological synapse. J Immunol. 2011;186(12):6839–6847. doi: 10.4049/jimmunol.1002721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ksionda O, Saveliev A, Kochl R, Rapley J, Faroudi M, Smith-Garvin JE, Wulfing C, Rittinger K, Carter T, Tybulewicz VL. Mechanism and function of Vav1 localisation in TCR signalling. J Cell Sci. 2012;125(Pt 22):5302–5314. doi: 10.1242/jcs.105148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang Y, Cucchetti M, Roncagalli R, Yokosuka T, Malzac A, Bertosio E, Imbert J, Nijman IJ, Suchanek M, Saito T, Wulfing C, Malissen B, Malissen M. The lymphoid lineage-specific actin-uncapping protein Rltpr is essential for costimulation via CD28 and the development of regulatory T cells. Nat Immunol. 2013;14(8):858–866. doi: 10.1038/ni.2634. [DOI] [PubMed] [Google Scholar]
- 10.Paster W, Brockmeyer C, Fu G, Simister PC, de Wet B, Martinez-Riano A, Hoerter JA, Feller SM, Wulfing C, Gascoigne NR, Acuto O. GRB2-Mediated Recruitment of THEMIS to LAT Is Essential for Thymocyte Development. J Immunol. 2013;190(7):3749–3756. doi: 10.4049/jimmunol.1203389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singleton KL, Gosh M, Dandekar RD, Au-Yeung BB, Ksionda O, Tybulewicz VL, Altman A, Fowell DJ, Wülfing C. Itk controls the spatiotemporal organization of T cell activation. Sci Signal. 2011;4(193):ra66. doi: 10.1126/scisignal.2001821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roybal KT, Sinai P, Verkade P, Murphy RF, Wülfing C. The actin-driven spatiotemporal organization of T-cell signaling at the system scale. Immunol Rev. 2013;256(1):133–147. doi: 10.1111/imr.12103. [DOI] [PubMed] [Google Scholar]
- 13.Roybal KT, Mace EM, Mantell JM, Verkade P, Orange JS, Wulfing C. Early Signaling in Primary T Cells Activated by Antigen Presenting Cells Is Associated with a Deep and Transient Lamellal Actin Network. PLoS One. 2015;10(8):e0133299. doi: 10.1371/journal.pone.0133299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Purtic B, Pitcher LA, van Oers NS, Wülfing C. T cell receptor (TCR) clustering in the immunological synapse integrates TCR and costimulatory signaling in selected T cells. Proc Natl Acad Sci USA. 2005;102(8):2904–2909. doi: 10.1073/pnas.0406867102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sinai P, Dozmorov IM, Song R, Schwartzberg PL, Wakeland EK, Wulfing C. T/B-cell interactions are more transient in response to weak stimuli in SLE-prone mice. Eur J Immunol. 2014;44(12):3522–3531. doi: 10.1002/eji.201444602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sinai P, Nguyen C, Schatzle JD, Wülfing C. Transience in polarization of cytolytic effectors is required for efficient killing and controlled by Cdc42. Proc Natl Acad Sci U S A. 2010;107(26):11912–11917. doi: 10.1073/pnas.0913422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singleton KL, Parvaze N, Dama KR, Chen KS, Jennings P, Purtic B, Sjaastad MD, Gilpin C, Davis MM, Wülfing C. A large T cell invagination with CD2 enrichment resets receptor engagement in the immunological synapse. J Immunol. 2006;177(7):4402–4413. doi: 10.4049/jimmunol.177.7.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singleton KL, Roybal KT, Sun Y, Fu G, Gascoigne NR, van Oers NS, Wülfing C. Spatiotemporal patterning during T cell activation is highly diverse. Sci Signal. 2009;2(65):ra15. doi: 10.1126/scisignal.2000199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roybal KT, Buck TE, Ruan X, Cho BH, Clark DJ, Ambler R, Tunbridge HM, Zhang J, Verkade P, Wulfing C, Murphy RF. Computational spatiotemporal analysis identifies WAVE2 and cofilin as joint regulators of costimulation-mediated T cell actin dynamics. Sci Signal. 2016;9(424):rs3. doi: 10.1126/scisignal.aad4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Negulescu PA, Krasieva TB, Khan A, Kerschbaum HH, Cahalan MD. Polarity of T cell shape, motility, and sensitivity to antigen. Immunity. 1996;4(5):421–430. doi: 10.1016/s1074-7613(00)80409-4. [DOI] [PubMed] [Google Scholar]
- 21.Mustelin T, Tasken K. Positive and negative regulation of T-cell activation through kinases and phosphatases. Biochem J. 2003;371(Pt 1):15–27. doi: 10.1042/BJ20021637. [DOI] [PMC free article] [PubMed] [Google Scholar]