

Significance
αB-crystallin (CRYAB) is a protein involved in the protection of cells from stress and cell death, and is very highly produced in brain lesions in multiple sclerosis (MS). Astrocytes are the main non-neuronal cell type in the central nervous system. Although they are involved in many processes in health and injury, their precise role in MS is poorly understood. Using a mouse model of MS, as well as brain tissue from MS patients, we found that CRYAB enables astrocytes to become activated and that these activated astrocytes play a pathogenic role in MS, worsening injury. Furthermore, we found that a specific stress-induced modification of CRYAB supports its role in astrocyte activation and affects the intracellular signaling pathways it regulates.
Keywords: astrogliosis, demyelination, cuprizone, proteomics, phosphorylation
Abstract
The small heat shock protein αB-crystallin (CRYAB) has been implicated in multiple sclerosis (MS) pathogenesis. Earlier studies have indicated that CRYAB inhibits inflammation and attenuates clinical disease when administered in the experimental autoimmune encephalomyelitis model of MS. In this study, we evaluated the role of CRYAB in primary demyelinating events. Using the cuprizone model of demyelination, a noninflammatory model that allows the analysis of glial responses in MS, we show that endogenous CRYAB expression is associated with increased severity of demyelination. Moreover, we demonstrate a strong correlation between the expression of CRYAB and the extent of reactive astrogliosis in demyelinating areas and in in vitro assays. In addition, we reveal that CRYAB is differentially phosphorylated in astrocytes in active demyelinating MS lesions, as well as in cuprizone-induced lesions, and that this phosphorylation is required for the reactive astrocyte response associated with demyelination. Furthermore, taking a proteomics approach to identify proteins that are bound by the phosphorylated forms of CRYAB in primary cultured astrocytes, we show that there is clear differential binding of protein targets due to the specific phosphorylation of CRYAB. Subsequent Ingenuity Pathway Analysis of these targets reveals implications for intracellular pathways and biological processes that could be affected by these modifications. Together, these findings demonstrate that astrocytes play a pivotal role in demyelination, making them a potential target for therapeutic intervention, and that phosphorylation of CRYAB is a key factor supporting the pathogenic response of astrocytes to oligodendrocyte injury.
Multiple sclerosis (MS) is an autoimmune demyelinating disease of the central nervous system (CNS) and is the major cause of neurological disability among young adults in Western countries. Its pathology is characterized by infiltration of inflammatory cells, localized destruction of myelin, oligodendrocyte cell death, and axonal degeneration (1, 2). Although the role of infiltrating lymphocytes and macrophages has been well described in the (late) active stages of demyelination, the exact mechanism of oligodendrocyte pathology and early demyelinating events are not well understood.
In addition to infiltrating macrophages and lymphocytes, astrocytes have been implicated in the pathogenesis of MS (3). The most abundant cell type in the brain, astrocytes perform an array of physiological functions, including maintenance of CNS homeostasis, regulation of blood–brain barrier integrity, modulation of blood flow, and support of synaptic transmission (4). In addition, upon activation by danger signals, astrocytes actively participate in the CNS innate immune response and are the main source of inflammatory mediators, including several complement components, cytokines, and chemokines (5). Astrocyte (dys)function has been implicated in many neurodegenerative and neuroinflammatory diseases (4, 6). Although it is well established that chronic stages of MS lesions are characterized by the abundant presence of fibrous astrocytes (7), the role of astrocytes in early demyelinating events remains conjectural.
αB-crystallin (CRYAB) is a member of the small heat shock protein family and is constitutively expressed in the vertebrate lens, striated muscle tissue, and the CNS (8). In addition, expression of the protein can be induced by a variety of stress stimuli (9, 10), and altered expression of CRYAB has been implicated in many neurological diseases (11). CRYAB is the most abundantly enriched gene transcript in active demyelinating MS lesions and is present in oligodendrocytes and astrocytes at different stages of MS lesions (12–16). Expression of CRYAB in astrocytes has been shown to protect these cells from apoptosis, by binding to mediators of cell death and inhibiting caspase activity (17–19). Phosphorylation is an important posttranslational modification of CRYAB, affecting both the localization and function of the protein (19–23). CRYAB can be phosphorylated at three serine residues, Ser-19, Ser-45, and Ser-59, and although the former does not contribute to cytoprotection, Ser-59 and Ser-45 phosphorylation are required for the protective function of CRYAB, with a dominant role being reported for Ser-59 (19, 20, 22, 23).
Here, we reveal a role for CRYAB in astrocyte cell function. We demonstrate that CRYAB enhances the local astrocyte response to oligodendrocyte injury induced by the toxin cuprizone. Moreover, we show that this reactive astrogliosis response is a key aspect in cuprizone-induced demyelination and that interfering with this response by abolishing CRYAB expression significantly reduces CNS pathology. In addition, we show that preferential phosphorylation of CRYAB at Ser-59 is present in astrocytes in actively demyelinating MS tissue and cuprizone-induced lesions and that this phosphorylation, mediated through activation of the protein kinase p38 pathway, supports reactive astrogliosis in vitro. Finally, applying a proteomics approach, we demonstrate that CRYAB displays differential binding to protein targets upon phosphorylation, which has implications for its role in various biological processes. Together, these findings indicate that reactive astrogliosis may play a prominent role in primary demyelination and that phosphorylation of CRYAB at Ser-59 supports this role. Consequently, specific targeting of the reactive astrocyte pool might prove efficacious at specific stages of demyelination.
Results
CRYAB Expression Correlates with Severity of Cuprizone-Induced Demyelination.
The cuprizone model of demyelination is based on the oligodendrocyte-toxic properties of the copper chelator bis-(cyclohexanone)-oxaldihydrazone (cuprizone). Chronic exposure to cuprizone results in demyelination in preferential areas in the rodent brain (24, 25). To evaluate the impact of endogenous CRYAB on primary demyelination in vivo, we applied the cuprizone model of demyelination to Cryab-null (Cryab−/−) mice, which are maintained on a 129S6 background, a strain in which cuprizone-induced pathology has not been described before. Luxol fast blue (LFB) staining of myelin revealed that treatment with cuprizone leads to distinct demyelinating lesions in the brains of wild-type (WT) 129S6 mice, which were particularly substantial in the cerebellum (Fig. 1 A and C). Moreover, presence of CRYAB impacted the severity of demyelination greatly, resulting in smaller lesions upon cuprizone treatment in Cryab−/− mice (Fig. 1 B and D). Quantification of lesion size revealed that, after 4.5–8 wk of cuprizone treatment, lesion size was significantly decreased in Cryab−/− mice, compared with WT mice (Fig. 1E, 71.55, 55.63, and 49.95% reduction at weeks 4.5, 6, and 8, respectively). Immunofluorescent staining for myelin basic protein (MBP) confirmed the effect of CRYAB expression on the severity of demyelination (Fig. S1 A–D).
Fig. 1.

CRYAB expression determines severity of cuprizone-induced demyelinating pathology. (A–D) LFB staining of myelin in cerebellar tissue of control mice (A and B) or mice treated with cuprizone for 8 wk (C and D) shows that cuprizone-induced demyelination is more severe in WT mice (A and C) than in Cryab−/− mice (B and D). (E) Quantification of cerebellar lesion size, as percentage of white matter area, in WT (closed symbols) and Cryab−/− mice (open symbols) after 4.5, 6, and 8 wk of cuprizone diet, reveals that at all time points lesions in WT mice are significantly larger than in Cryab−/− mice. Shown are data points representing individuals animals, mean ± SEM, and P values determined by unpaired t test (n = 4; *P ≤ 0.05, **P < 0.01). (F) Motor skill performance, as measured by a rotarod assay and depicted as percentage change from baseline performance, reveals that WT mice (blue boxes) are more susceptible to cuprizone-induced motor skill deficits than Cryab−/− mice (red boxes). Data are shown as box-and-whisker plots, with whiskers depicting minimal and maximal values. P values were determined by unpaired t test (n = 8–16; †P < 0.1, *P < 0.05).
Fig. S1.

CRYAB expression determines severity of cuprizone-induced symptoms, but treatment with exogenous CRYAB does not affect severity of cuprizone-induced demyelinating pathology. (A–D) Immunofluorescence staining for myelin basic protein (MBP) in cerebellar tissue of mice treated with cuprizone for 6 (A and B) or 8 wk (C and D) shows that cuprizone-induced demyelination is more severe in WT mice (A and C) than in Cryab−/− mice (B and D). Myelin damage is visible in WT mice after 6 wk of cuprizone treatment (as increased availability of MBP antigen; A), whereas myelin loss is prominent after 8 wk (C). (E and F) Assessing body weight as a measure of clinical outcome of cuprizone-induced pathology shows that WT mice (F) are more susceptible to cuprizone-induced symptoms than in Cryab−/− mice (G). Data shown are mean ± SEM and P values determined by unpaired t test (n = 4–8; †P < 0.1, *P < 0.05, **P < 0.01). (G and H) LFB staining of myelin in corpus callosum of WT C57/Bl6 mice treated with vehicle (A) or CRYAB (B; 10 µg, i.p., every other day), showing extensive demyelination and inflammation in the caudal region of the corpus callosum. (I and J) Motor skill performance (C) and body weight (D) as outcome measures of cuprizone-induced pathology, reveal that clinical outcome is not different between mice treated with vehicle (red) or CRYAB (blue). Data shown are mean ± SEM (n = 4).
It has been reported before that cuprizone-induced toxicity leads to motor skill deficiencies (26). We therefore assessed how CRYAB affects motor skill performance by rotarod testing during cuprizone treatment. Correlating with demyelination severity, WT mice displayed significantly more severe motor skill impairment after 8 wk of cuprizone treatment than Cryab−/− mice (Fig. 1F; 16.6% reduction in motor skill performance). The increased severity of cuprizone-induced pathology in the presence of CRYAB was also evident from a delayed gain of body weight, which was much more pronounced in WT mice than in Cryab−/− mice (Fig. S1 E and F).
We have previously shown that CRYAB, in addition to its intracellular function, can also act when delivered exogenously, by binding misfolded and unfolded proteins in the blood as a molecular chaperone (27). To investigate whether the effect of CRYAB is due to intracellular expression in CNS-resident cells, or an extracellular effect of circulating CRYAB, we assessed whether administration of exogenous CRYAB affected cuprizone-induced demyelination. Treatment of mice with CRYAB did not affect severity of demyelination, motor skill impairment, or gain of body weight (Fig. S1 G–J), indicating that the observed effect of endogenous CRYAB is likely due to its intracellular expression by CNS-resident cells.
CRYAB Expression Correlates with the Extent of Cuprizone-Induced Reactive Astrogliosis.
Microglial activation and, to a lesser extent, reactive astrogliosis have been described as possible secondary effector mechanisms of cuprizone-induced demyelination (25). We therefore assessed how the glial response to cuprizone-induced pathology is affected by CRYAB. Cuprizone treatment resulted in a profound microglial and astrocyte response in demyelinating areas, characterized by macrophage-like morphology of ionized calcium binding adaptor molecule 1 (Iba1)-positive microglia and the presence of large hypertrophic glial fibrillary acid protein (GFAP)-expressing reactive astrocytes, respectively, which was more pronounced in WT lesions than Cryab−/− lesions (Fig. 2 A–D). Assessment of reactive astrocyte and microglia cell numbers revealed that CRYAB affected the response of both cell types to the cuprizone-induced insult (Fig. 2 E and F), whereas cell numbers in healthy control tissue (animals on normal diet) were not affected. Reactive astrocyte cell numbers were affected to a greater extent (56.0 ± 7.36% reduction in Cryab−/−) than microglia cell numbers (32.6 ± 14.7% reduction in Cryab−/−). This difference in the extent of the decrease in cellular response between astrocytes and microglia was also reflected by the ratio of reactive astrocyte cell numbers inside lesions over numbers in unaffected areas, which was reduced in Cryab−/− animals to a significantly greater extent than the ratio of microglia numbers inside lesions over unaffected areas (Fig. S2). Together, these data indicate that reactive astrogliosis is the most pronounced determinant of the diminishing effect of CRYAB expression on the severity of cuprizone-induced demyelination.
Fig. 2.

Astrocyte cell number is the primary determinant of cuprizone-induced pathology. (A–D) Immunofluorescent staining for GFAP (red) and Iba1 (blue) shows a dramatic astrocyte and microglial response in cerebellar lesions induced by 8 wk of cuprizone diet in WT mice (A and B), which is reduced in Cryab−/− mice (C and D). Shown are composite images of tile scans performed with a 40× objective. (B and D) Magnified images of the boxed areas in A and C, respectively. DAPI counterstain of nuclei is shown in white. (E and F) Quantification of reactive astrocyte and microglia cell numbers, by counting nuclei associated with GFAP or Iba1 staining, respectively, in control tissue, outside and inside cuprizone-induced lesions reveals that the number of reactive astrocytes (E) is significantly higher in WT lesions than in Cryab−/− lesions and more severely affected than the number of microglia (F). Shown are data points representing individual animals, mean ± SEM, and P values determined by Mann–Whitney test (n = 4; *P < 0.05).
Fig. S2.

The local astrocyte response to cuprizone-induced pathology is affected by CRYAB expression. Quantification of the local response of astrocytes (Left) and microglia (Right) as ratio of number of cells inside cuprizone-induced lesions, compared with number of cells outside lesions, reveals that the response of astrocytes is significantly higher in WT mice than in Cryab−/− mice, whereas the ratio of microglia is not affected by CRYAB expression. Shown are data points representing individual animals, mean ± SEM, and P values determined by Mann–Whitney test (n = 4) and two-way ANOVA (cell type effect).
CRYAB Enables Reactive Astrogliosis in Vitro.
Considering the protective role that has been described for CRYAB in astrocyte biology, we next hypothesized that CRYAB, as a consequence of this cytoprotective function, enables the reactive astrocyte response. To confirm this, we performed an in vitro reactive astrogliosis assay after down-regulation of CRYAB expression by siRNA transfection in primary cultured astrocytes. We found that knockdown of CRYAB expression resulted in a less efficient response to an in vitro scratch insult (Fig. 3 A and B, and Fig. S3). Both the area that was repopulated after the insult as well as the number of astrocytes repopulating the scratched area were significantly reduced in astrocytes with reduced CRYAB expression (Fig. 3C and Fig. S3), which indicates that CRYAB expression facilitates reactive astrogliosis in vitro.
Fig. 3.

Expression of CRYAB affects reactive astrogliosis. (A–C) Representative pictures of an in vitro scratch assay with primary astrocyte cultures transfected with either control (A) or CRYAB (B) siRNA smartpools, revealing that loss of CRYAB in astrocytes results in a significantly reduced response to a scratch insult, quantified as area of the scratch occupied by invading cells (C). Data are representative of three independent experiments, values depict mean + SEM of triplicate culture wells and P values determined by unpaired t test (*P < 0.05). (D) IL-6 secretion by primary astrocyte cultures transfected with either control (light gray bars) or CRYAB siRNAs (dark gray bars), measured by ELISA, shows that loss of CRYAB results in a reduced cytokine response upon stress induced by stimulation with LPS. Shown is a representative of four experiments, values depict mean + SEM of duplicate measurements and P values determined by two-way ANOVA (*P < 0.05, ***P < 0.001). (E and F) Proliferating astrocytes, identified as cells colabeled with GFAP (red) and nuclear staining of Ki67 (blue), are present in demyelinating areas in cuprizone-treated WT mice (E, arrows) but are not present in similar areas in Cryab−/− mice (F). DAPI counterstain of nuclei is shown in white.
Fig. S3.

Expression of CRYAB affects reactive astrogliosis in vitro. (A) Confirmation of siRNA-mediated knockdown of CRYAB expression. Western blot for CRYAB (Top) and GAPDH as a control (Lower) on lysates of primary cultured astrocytes after transfection of CRYAB siRNAs (100 nM; smartpool containing three siRNAs) reveals that CRYAB protein expression is markedly down-regulated 24–72 h after transfection. (B) Representative quantification of an in vitro scratch assay with primary astrocyte cultures transfected with either control or CRYAB siRNA smartpools, revealing that loss of CRYAB in astrocytes results in a significantly reduced response to a scratch insult, quantified as number of cells invading the scratched area. Shown is a representative of three independent experiments; values depict mean + SEM of triplicate culture wells and P values determined by unpaired t test (*P < 0.05).
In addition, considering that proinflammatory cytokines, such as interleukin-6 (IL-6), are harmful when present in high concentrations in the brain parenchyma, and astrocytes are the main source of IL-6 in the CNS (5), we evaluated whether IL-6 production by astrocytes was affected by CRYAB expression. Down-regulation of CRYAB expression resulted in significantly reduced secretion of IL-6 upon stress induced by LPS stimulation (Fig. 3D), indicating that secretion of cytokines might be one of the effector mechanisms contributing to the pathogenic reactive astrocyte response observed in the presence of CRYAB.
CRYAB Expression Correlates with Reactive Astrogliosis in Vivo.
In addition to in vitro analysis of reactive astrogliosis, we analyzed whether CRYAB expression affects reactive astrogliosis in vivo. To investigate this, we assessed signs of active proliferation of astrocytes, the main hallmark of reactive astrogliosis, by immunostaining for the proliferation marker Ki67. Under homeostatic conditions, the astrocyte pool in the CNS is stable. However, in cuprizone-induced demyelinating lesions, we found many Ki67-immunopositive, GFAP-expressing, astrocytes, indicating an active proliferative response in response to the cuprizone insult (Fig. 3E). Many Ki67-positive astrocytes were present in WT lesions, whereas they were not detectable in Cryab−/− animals (Fig. 3F), indicating that the in vivo reactive astrocyte response is impaired in the absence of CRYAB.
CRYAB Is Differentially Phosphorylated in MS Lesions.
Considering the importance of phosphorylation for the cytoprotective function of CRYAB, we determined the phosphorylation status of CRYAB in active demyelinating MS lesions and healthy control brain samples. For this, we analyzed data from a phosphoproteomic screen of posttranslationally modified proteins in phosphopeptide-enriched brain lysates obtained from active MS lesion tissue and healthy control tissue (28). Mass-spectrometric analysis of these phosphopeptide-enriched lysates yielded three phosphopeptides derived from CRYAB that were present in both MS and healthy control tissue. All three peptides mapped to the same region in the protein, containing Ser-59. Two peptides (peptides 1 and 2) contained phosphorylated Ser-59, whereas the third peptide contained unphosphorylated Ser-59. The peptides that included P-Ser-59 were present at medium (peptide 2, 30 spectral counts) to high (peptide 1, 40 spectral counts) levels in control brain tissue and were increased 2.5- to 3.5-fold, respectively, in active demyelinating MS tissue (peptide 2, 74 spectral counts; peptide 1, 140 spectral counts; Table 1) (29). In contrast, the peptide containing unphosphorylated Ser-59 was only present at low levels in both control and MS tissue.
Table 1.
Phosphorylation of serine-59 is markedly up-regulated in MS lesions
| No. | Sequence* | Modifications | Spectral counts† | |
| Con | MS | |||
| 1 | 57-APsWFDTGLSEmR-70 | S59 (P), M69 (O) | 40 | 140 |
| 2 | 57-APsWFDTGLSEMR-70 | S59 (P) | 30 | 74 |
| 3 | 57-APSWFDtGLSEmR-70 | T63 (P), M69 (O) | 1 | 2 |
Depicted are three peptides, derived from CRYAB, that were identified in a mass spectrometry screen for posttranslationally modified proteins present in brain tissue from healthy controls or active demyelinating areas from MS brain tissue.
Residues that were modified are shown in the peptide sequences (bold lowercase), as well as their modification (P, phosphorylation; O, oxidation) and location in the CRYAB sequence (UniProt ID P02511).
Spectral counts for each peptide in control (Con) and MS tissue depict low (0–5 counts), medium (6–33 counts), and high (>33 counts) levels, according to Huttlin et al. (29).
We then evaluated the expression and phosphorylation status of CRYAB in active MS lesions and surrounding normal-appearing white-matter (NAWM) tissue of MS patients by immunohistochemistry. We found that native CRYAB is present in cells with astrocyte and oligodendrocyte morphology in NAWM and that this expression is noticeably increased in cells with reactive astrocyte morphology in areas with active demyelination (Fig. 4 A and B). Immunolabeling with specific antibodies detecting CRYAB phosphorylated either at Ser-59 or Ser-45 revealed that phosphorylation of Ser-59 is dramatically increased in astrocytes in active demyelinating MS lesions, whereas it is not present in NAWM (Fig. 4 C and D). In contrast, phosphorylation of Ser-45 is constitutively present in astrocytes in NAWM and was not altered in areas with active demyelination (Fig. 4 E and F). Together, this indicates that CRYAB is preferentially phosphorylated at Ser-59 in reactive astrocytes inside active demyelinating MS lesions.
Fig. 4.

CRYAB in astrocytes in MS lesions is preferentially phosphorylated at serine-59. (A and B) Immunohistochemical analysis of CRYAB in NAWM (A) and areas with active demyelination (B) demonstrates that CRYAB is expressed by astrocytes (arrows) and oligodendrocytes (arrowheads) in NAWM (A), and that expression of CRYAB is increased in astrocytes with reactive morphology in active demyelinating areas (B). (C–F) Analysis of CRYAB, phosphorylated at Ser-59 (C and D) and Ser-45 (E and F), in NAWM (C and E) and active demyelinating areas in MS tissue (D and F), reveals that phosphorylation of Ser-59 is not present in NAWM (C) and is dramatically increased in cells with reactive astrocyte morphology in active demyelinating lesions (D), whereas phosphorylation of Ser-45 is present in both NAWM (E) and active demyelinating areas (F). Nuclei were counterstained with hematoxylin. Insets depict magnified images of positive-staining astrocytes.
P38 and ERK Are Activated and CRYAB Is Preferentially Phosphorylated at Ser-59 in Reactive Astrocytes in Cuprizone Lesions.
We additionally investigated the phosphorylation status of CRYAB in cuprizone-induced lesions. Similar to our findings in MS lesions, immunolabeling with phosphospecific antibodies revealed that phosphorylation of Ser-59 is highly abundant in reactive astrocytes in areas with active demyelination (Fig. 5A). The localization of P-Ser-59-CRYAB in these reactive astrocytes is exclusively cytoplasmic. In contrast, we found that P-Ser-45-CRYAB is present mainly in axonal structures within lesions, and in nuclei of cells that were identified as oligodendrocytes (Fig. 5B and Fig. S4). No presence of P-Ser-45-CRYAB, either cytoplasmic or nuclear, could be found in astrocytes.
Fig. 5.

Phosphorylation of serine-59, and the MAP kinases p38 and ERK, is present in reactive astrocytes in cuprizone lesions. (A–D) Immunofluorescent staining for CRYAB, phosphorylated at Ser-59 (A, red), or Ser-45 (B, red), and phosphorylated p38 (C, red) or ERK (D, red) and GFAP (C and D, blue) of a cuprizone-induced cerebellar lesion (8-wk treatment). Shown are composite images of tile scans performed with a 40× objective. DAPI counterstain of nuclei is shown in white. (A and B) Phospho-Ser-59 is abundant in the cytoplasm of cells with reactive astrocyte morphology, characterized by large translucent nuclei and hypertrophic cell bodies (A, Inset, arrows), whereas phospho-Ser-45 is present in nuclei of cells identified as mature oligodendrocytes (B, Inset, arrows; Fig. S7) and axonal structures (B, Inset, arrowheads). (C and D) Phosphorylated p38 is abundant in the nucleus of many cells, including a subset of astrocytes (C, Inset, arrows), inside the lesion, whereas phosphorylated ERK is present in the cytoplasm and the nucleus of a subset of reactive astrocytes (D, Inset, arrows, arrowheads: ERK-negative astrocytes). DAPI counterstain of nuclei is shown in white.
Fig. S4.

Nuclear localization of P-Ser-45-CRYAB is restricted to oligodendrocytes. (A) Immunofluorescent staining for P-Ser-59-CRYAB (blue) and the mature oligodendrocyte marker CC1 (red) of a cerebellar lesion induced by cuprizone treatment for 8 wk, shows a cytoplasmic staining pattern of P-Ser-59-CRYAB in reactive astrocytes that is not associated with CC1 expression. (B) Immunostaining for P-Ser-45-CRYAB (blue) and CC1 (red) of the same region shows that nuclear localization of P-Ser-45-CRYAB is present in a subset of CC1-positive oligodendrocytes (arrows). DAPI counterstain of nuclei is shown in white.
CRYAB is preferentially phosphorylated through the activation of different mitogen-activated protein kinase (MAPK) pathways in response to specific stress stimuli: p38 preferentially phosphorylates Ser-59, whereas activation of the extracellular-regulated kinase (ERK) pathway leads to preferential phosphorylation of Ser-45 (19, 21, 22, 30, 31). We therefore assessed the activation of these pathways inside cuprizone-induced lesions. Immunostaining for the phosphorylated forms of p38 and ERK revealed that both pathways are activated in cells inside demyelinating areas. Particularly intense staining for phosphorylated p38 was present in many nuclei inside lesions, including those of a subset of GFAP-positive astrocytes (Fig. 5C). Likewise, staining for phosphorylated ERK was present, both cytoplasmic and nuclear, in a subset of GFAP-positive astrocytes (Fig. 5D).
Phosphorylation of Ser-59, Mediated by p38, Is Required for Reactive Astrogliosis in Vitro.
To further investigate the role of CRYAB phosphorylation in astrocyte reactivity, we assessed the phosphorylation of Ser-59 and Ser-45, and of p38 and ERK, upon stress in primary cultured astrocytes. Phosphorylation of Ser-59, localized primarily in the cytoplasm, was markedly increased upon oxidative stress and stress induced by LPS treatment (Fig. S5 A–C), and this increase was sustained for up to 24 h (Fig. 6 A and B). In contrast, constitutive phosphorylation of Ser-45, exclusively present in the nucleus of culture astrocytes, was not altered by either stress stimulus (Fig. S5 D–F, and Fig. 6 C and D). In parallel, phosphorylation of p38 was induced upon stress, whereas phosphorylation of ERK was not affected by these stimuli (Fig. S5 G–L).
Fig. S5.

Stress stimuli increase phosphorylation of CRYAB serine-59 and p38, but not CRYAB serine-45 or ERK, in cultured astrocytes. (A–L) Immunocytochemical staining for phospho-Ser-59 (A–C, red), phospho-Ser-45 (D–F, red), phospho-p38 (G–I, red), or phospho-ERK (J–L, red), and unphosphorylated CRYAB (A–L, blue) of primary astrocytes either untreated (A, D, G, and J), treated with H2O2 (B, E, H, and K; 500 μM, 1 h), or treated with LPS (C, F, I, and L; 1,000 ng/mL, 1 h) reveals that phosphorylation of CRYAB Ser-59 and p38 is enhanced by stress stimuli, whereas CRYAB Ser-45 and ERK phosphorylation is not affected.
Fig. 6.

Stress-induced serine-59 phosphorylation is necessary for CRYAB-dependent IL-6 production by astrocytes. (A–D) Immunocytochemical staining for phospho-Ser-59 (A and B, red) or phospho-Ser-45 (C and D, red) and unphosphorylated CRYAB (A–D, blue) of primary astrocytes either untreated (A and C) or treated with LPS (B and D; 1,000 ng/mL, 24 h) reveals that phosphorylation of Ser-59, localized in the cytoplasm, is enhanced upon stress, whereas Ser-45 phosphorylation, localized in the nucleus, is not affected. (E) ELISA analysis of IL-6 secretion by primary astrocyte cultures treated with inhibitors of p38 (SB203580; 10 μM) and ERK (PD98059; 100 μM), shows that inhibition of p38 and, to a lesser extent, ERK reduces constitutive and stress-induced secretion of IL-6 (1,000 ng/mL LPS; 24 h), depicted as percentage of unstimulated control values. Values depict mean + SEM of four independent experiments and P values determined by unpaired t test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
To determine how p38 and ERK-mediated phosphorylation of CRYAB affects its function in the reactive astrocyte response, we analyzed production of IL-6 by primary astrocytes after treatment with specific inhibitors of p38 (SB203580) and ERK (PD98059), which results in reduced phosphorylation of Ser-59 and Ser-45, respectively (Fig. S6). Inhibition of p38 activity resulted in a dramatic reduction of both constitutive and LPS-induced IL-6 production, indicating that Ser-59 phosphorylation is required for reactive astrogliosis in vitro (Fig. 6E). Inhibition of ERK also reduced IL-6 production, however to a lesser extent than p38 inhibition, and inhibiting both pathways resulted in an additional reduction of IL-6 production, suggesting that phosphorylation of Ser-45 has an additional role in vitro astrocyte activation.
Fig. S6.

Inhibition of p38 and ERK results in decreased Ser-59 and Ser-45 phosphorylation, respectively. (A–P) Immunocytochemical staining for phospho-Ser-59 (A–H, red) or phospho-Ser-45 (I–P, red) and unphosphorylated CRYAB (A–P, blue) of primary astrocytes shows that treatment with a p38 inhibitor (SB203580; 10 μM; 16-h pretreatment) results in a reduction of Ser-59 phosphorylation that is enhanced by LPS stimulation (1,000 ng/mL; 24 h), whereas treatment with an ERK inhibitor (PD98059; 100 μM; 16-h pretreatment) results in a reduction of Ser-45 phosphorylation.
Phosphorylated CRYAB Binds Differential Sets of Proteins in Astrocytes.
Considering the nature of CRYAB to bind other proteins as a molecular chaperone (27), we next aimed to identify protein targets that are bound by different phosphorylation states of CRYAB in astrocytes, to obtain clues as to which other cellular functions might be affected by the phosphorylation of CRYAB. To this end, we coimmunoprecipitated CRYAB-bound proteins in lysates of primary astrocyte cultures using the same phosphorylation-specific antibodies used for immunohistochemistry and identified these binding targets by mass spectrometry. This yielded a list of 240 protein targets, 84 of which are bound by native CRYAB and 131, 96, and 71 bound by CRYAB phosphorylated at Ser-19, Ser-45, and Ser-59, respectively (Fig. 7 and Dataset S1). Unsupervised clustering of all targets bound by (phosphorylated) CRYAB revealed that there are distinct sets of proteins that are bound by the different phosphorylated states of CRYAB (Fig. S7). We then determined which known biological pathways were enriched for these differential targets, using the Web-based gene set analysis toolkit WebGestalt (32) and the WikiPathways platform of curated biological pathways. For this analysis, all bound target proteins for each phosphorylated form of CRYAB were assessed, without taking the extent of enrichment into account. This analysis revealed that there are distinct pathways that are enriched for either native, P-Ser-59, and P-Ser-45 CRYAB targets (Fig. 7 and Table S1). Pathways enriched for native CRYAB targets greatly overlapped with those for P-Ser-19 CRYAB. They include many cytoplasmic ribosomal proteins and are mainly involved in mRNA processing (Fig. 7 and Table S1, Native CRYAB and P-Ser19 CRYAB). Conversely, pathways enriched for P-Ser-59 CRYAB binding targets are involved in energy metabolism, such as glycolysis and gluconeogenesis, and the tricarboxylic acid (TCA) cycle, as well as stress/immune-activated pathways such as the T-cell receptor and FAS signaling pathways (Fig. 7 and Table S1, P-Ser59 CRYAB), whereas P-Ser-45 CRYAB binding targets are enriched in pathways involved in DNA replication and cell cycle control, as well as proteasome degradation (Fig. 7 and Table S1, P-Ser45 CRYAB).
Fig. 7.

Overview of protein binding by (phosphorylated) CRYAB. Venn diagram depicting the number of identified proteins bound by the different phosphorylated states of CRYAB. Also indicated are the biological pathways that are associated with the target proteins bound by native and/or phospho-Ser-19 CRYAB, phospho-Ser-45 CRYAB, and phospho-Ser-59 CRYAB.
Fig. S7.

Clustering of (phosphorylated) CRYAB-bound protein targets reveal distinct sets of proteins differentially bound by phosphorylated states of CRYAB. Coimmunoprecipitated protein targets of CRYAB were identified my mass spectrometry and clustered using the unsupervised multidimensional clustering method AutoSOME (54).
Table S1.
Biological pathways enriched for binding targets of (phosphorylated) CRYAB, yielded by WebGestalt analysis, using the Wikipathways database of biological pathways
| Rank | Pathway name | No. of targets/reference proteins | Adjusted P value* |
| Native CRYAB | |||
| 1 | Cytoplasmic Ribosomal Proteins | 24/78 | 4.32E-42 |
| 2 | mRNA Processing | 13/456 | 5.80E-09 |
| 3 | Prostaglandin Synthesis and Regulation | 3/31 | 0.0005 |
| 4 | Regulation of Actin Cytoskeleton | 4/160 | 0.0049 |
| 5 | Urea Cycle and Metabolism of Amino Groups | 2/22 | 0.0057 |
| 6 | Oxidative Phosphorylation | 2/60 | 0.0335 |
| P-Ser59 CRYAB | |||
| 1 | Cytoplasmic Ribosomal Proteins | 8/78 | 2.54E-10 |
| 2 | Glycolysis and Gluconeogenesis | 6/50 | 1.84E-08 |
| 3 | mRNA Processing | 11/456 | 6.70E-08 |
| 4 | TCA Cycle | 4/32 | 4.16E-06 |
| 5 | Electron Transport Chain | 5/101 | 1.47E-05 |
| 6 | Amino Acid Metabolism | 4/112 | 0.0004 |
| 7 | Striated Muscle Contraction | 3/44 | 0.0004 |
| 8 | T Cell Receptor Signaling Pathway | 4/143 | 0.0007 |
| 9 | Oxidative Phosphorylation | 3/60 | 0.0007 |
| 10 | Regulation of Actin Cytoskeleton | 4/160 | 0.0009 |
| 11 | Circadian Exercise | 2/49 | 0.0084 |
| 12 | FAS Pathway and Stress Induction of HSP Regulation | 2/50 | 0.0084 |
| 13 | MAPK Signaling Pathway | 3/165 | 0.0089 |
| P-Ser45 CRYAB | |||
| 1 | Cytoplasmic Ribosomal Proteins | 8/78 | 5.15E-09 |
| 2 | Glycolysis and Gluconeogenesis | 6/50 | 1.98E-07 |
| 3 | DNA Replication | 5/41 | 2.03E-06 |
| 4 | Proteasome Degradation | 5/59 | 9.69E-06 |
| 5 | Cell Cycle | 5/95 | 8.15E-05 |
| 6 | TNF-α NF-κB Signaling Pathway | 6/213 | 0.0003 |
| 7 | G1 to S Cell Cycle Control | 4/65 | 0.0003 |
| 8 | Myometrial Relaxation and Contraction Pathways | 5/158 | 0.0006 |
| 9 | Striated Muscle Contraction | 3/44 | 0.0011 |
| 10 | mRNA Processing | 7/456 | 0.002 |
| 11 | PluriNetWork | 5/289 | 0.0064 |
| 12 | Circadian Exercise | 2/49 | 0.0221 |
| 13 | Wnt Signaling Pathway NetPath | 3/141 | 0.0221 |
| 14 | Calcium Regulation in the Cardiac Cell | 3/152 | 0.0252 |
| 15 | Oxidative Phosphorylation | 2/60 | 0.0282 |
| 16 | IL-5 Signaling Pathway | 2/80 | 0.0453 |
| P-Ser19 CRYAB | |||
| 1 | Cytoplasmic Ribosomal Proteins | 31/78 | 4.19E-52 |
| 2 | mRNA Processing | 23/456 | 9.76E-17 |
| 3 | Electron Transport Chain | 5/101 | 0.0005 |
| 4 | Oxidative Phosphorylation | 3/60 | 0.0104 |
Listed are pathways that are enriched for binding targets of (i) native, (ii) phospho-Ser-59, (iii) phospho-Ser-45, and (iv) phospho-Ser-19 CRYAB. Most highly enriched pathways (compared with other CRYAB phosphorylation states) are depicted in bold, and pathways uniquely enriched for targets of a specific phosphorylation state are depicted in italics.
P value determined by hypergeometric testing, after Benjamini–Hochberg adjustment for multiple testing.
Next, using Ingenuity Pathway Analysis (IPA), we assessed how phosphorylation of CRYAB differentially affects its binding in a quantitative way and the possible implications of these changes for signaling networks and biological functions. For this analysis, we used quantitative protein values (spectral counts) of native CRYAB-bound targets and values of phosphorylated CRYAB-bound targets relative to native CRYAB binding values (phosphorylated CRYAB spectral counts minus native CRYAB spectral counts) and determined the canonical pathways and biological functions that were predicted to be affected by CRYAB binding and phosphorylation.
Table 2 provides an overview of the main pathways (more detailed description in Table S2) and functions (more detailed description in Table S3) predicted to be affected by protein binding to native, phospho-Ser-59, and phospho-Ser-45 CRYAB. In line with the preliminary pathway analysis, pathways and functions predicted to be affected by Ser-19 phosphorylation do not differ substantially from those associated with native CRYAB binding (Tables S2, P-Ser19–induced changes, and S3, P-Ser19–induced changes). Protein targets bound by native CRYAB are mainly associated with differential Rho signaling (reduced RhoGDI signaling, increased RhoA signaling), inhibition of cell death and cell morphological changes, and an increase in EIF2 signaling, cytoskeleton signaling and reorganization, cell differentiation and proliferation, and integrin signaling (Table 2 and Tables S2, Native CRYAB binding targets, and S3, Native CRYAB binding targets). Protein targets that are differentially affected by Ser-59 phosphorylation are associated with reduced EIF2 and eNOS signaling, cytoskeletal organization and proliferation, and an increase in integrin signaling, cell death, and ATP metabolism (Table 2 and Tables S2, P-Ser59–induced changes, and S3, P-Ser59–induced changes). Protein targets affected by Ser-45 phosphorylation are associated with reduced HIPPO signaling, cytoskeletal rearrangement and proliferation, and an increase in 14-3-3, PKA, and PI3K/AKT signaling, necrosis, protein metabolism, and cell cycle progression (Table 2 and Tables S2, P-Ser45–induced changes, and S3, P-Ser45–induced changes). These data suggest that many cellular functions might be affected in astrocytes, depending on the phosphorylation state of CRYAB.
Table 2.
Summary of canonical pathways and biological functions predicted to be differentially affected by binding to native and phosphorylated CRYAB, identified by IPA
| Protein state | Canonical pathways affected | Biological functions affected |
| Native CRYAB | ⇓ RhoGDI signaling | ⇓ Cell death and survival, cell morphology |
| ⇑ EIF2 signaling, RhoA signaling, actin cytoskeleton signaling, integrin signaling | ⇑ Cellular assembly and organization, cell proliferation and differentiation, cell movement | |
| P-Ser-59 CRYAB | ⇓ EIF2 signaling, eNOS signaling | ⇓ Cellular assembly and organization, cell growth and proliferation |
| ⇑ Integrin signaling | ⇑ Cell death and survival, cell morphology, ATP metabolism | |
| P-Ser-45 CRYAB | ⇓ EIF2 signaling, HIPPO signaling | ⇓ Cellular assembly and organization, cell growth and proliferation |
| ⇑ 14-3-3-mediated signaling, protein kinase A signaling, PI3K/AKT signaling | ⇑ Cell death and survival, cell morphology, protein synthesis, cell cycle control |
⇓ depicts pathways and functions enriched for CRYAB binding targets, expression of which is associated with pathway inhibition or reduction of function. ⇑ depicts pathways and functions enriched for CRYAB binding targets, expression of which is associated with pathway activation or increase in function.
Table S2.
Canonical pathways predicted to be differentially affected by phosphorylation of CRYAB, identified by IPA analysis
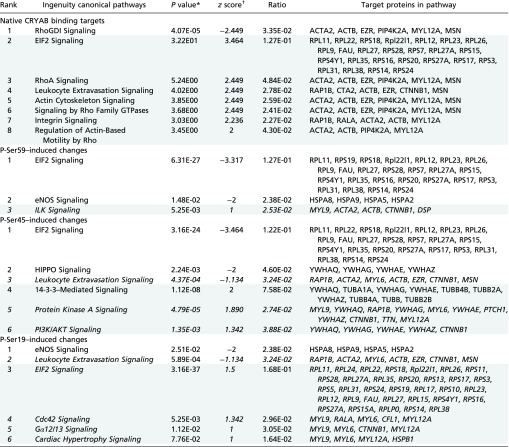 |
Listed are pathways that are predicted to be affected, based on enrichment for binding targets of (i) native CRYAB, or changes in enrichment/binding by phosphorylation of (ii) Ser-59, (iii) Ser-45, and (iv) Ser-19. Targets associated with pathway inhibition are listed first, and targets associated with pathway activation are listed second and are shaded.
P value calculated using right-tailed Fisher exact test and Benjamini–Hochberg correction for multiple testing.
The IPA z score measures the correlation between expected gene/protein expression values and actual protein enrichment values, weighted by relationship, relationship bias, data bias, a positive score correlating to an activated pathway, and a negative score correlating to an inhibited pathway. A z score >2 or less than −2 was considered significant. z scores between 1 and −1 are depicted in italics.
Table S3.
Biological functions predicted to be differentially affected by phosphorylation of CRYAB
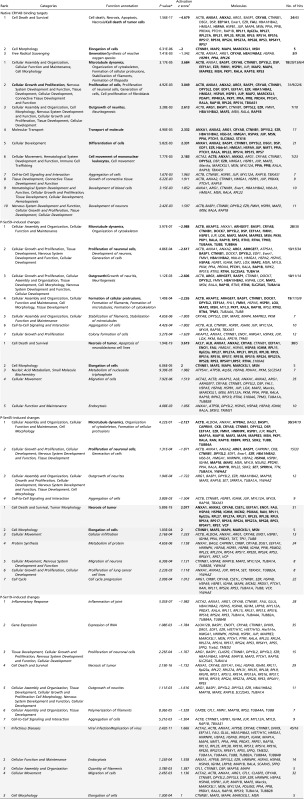 |
Listed are functions predicted to be affected based on enrichment for binding targets of (i) native CRYAB, or changes in enrichment/binding by phosphorylation of (ii) Ser-59, (iii) Ser-45, and (iv) Ser-19. Targets associated with reduced functions are listed first, targets associated with increased functions second and are shaded. Shown are all results with a z score less than −1.5 or >1.5, unless this cutoff yielded less than five results, in which case results with a z score less than −1 or >1 are shown. Overlapping functional annotations are grouped per category. Functions and corresponding molecules with the highest activation z score within categories containing overlapping functions are depicted in bold. Functions and corresponding molecules with an activation z score between −2 and −1, or 1 and 2, are italicized.
P value calculated using right-tailed Fisher exact test and Benjamini–Hochberg correction for multiple testing.
The IPA z score measures the correlation between expected gene/protein expression values and actual protein enrichment values, weighted by relationship, relationship bias, data bias, a positive score correlating to an increased process, and a negative score correlating to a decreased process.
Discussion
In this study, we aimed to further uncover the role of CRYAB in demyelination in MS. Using the cuprizone model, we were able to evaluate the function of CRYAB in the glial response to a primary oligodendrocyte insult and in subsequent demyelination. We find that, in the context of cuprizone-induced damage to oligodendrocytes, CRYAB expression supports a reactive astrogliosis response that contributes to demyelination. Concordantly, using a combination of phosphoproteomics and immunohistochemistry, we show that astrocytes in active demyelinating MS lesions display a similar phenotype to that of astrocytes in cuprizone-induced lesions, defined by enhanced and preferential phosphorylation of CRYAB at Ser-59. Finally, we demonstrate that this phosphorylation, mediated by the p38 MAPK pathway, is required for the reactive astrocyte response and has implications for the activation of a range of biological pathways. Together, this suggests that, in astrocytes, phosphorylation of CRYAB serves to support their pathogenic role in the early events underlying demyelination in MS.
Historically, the role of astrocytes in MS has been thought to be limited to the chronic stage of lesion development, where they populate the hypocellular areas that remain after active demyelination has expanded and died out, resulting in a glial scar. Our findings, however, point toward an active role for reactive astrocytes in early demyelinating events and/or perpetuating the cascade of myelin destruction. This hypothesis is supported by the report that astrocyte-specific inhibition of NF-κB activation leads to a reduction in the astrocyte response and subsequently reduced cuprizone-induced demyelination (33), as well as the notion that ablation of reactive astrocytes during cuprizone treatment dramatically diminishes demyelination, due to reduced recruitment of microglia, which remove myelin debris (34). In addition, recent reports using the experimental autoimmune encephalomyelitis (EAE) model of MS, demonstrate an instrumental role for astrocytes in regulating neuroinflammation (35, 36). Therefore, the pathogenic role of astrocytes we describe here is not unprecedented.
We propose that, during the course of reactive astrogliosis in the context of demyelination, stress-induced activation of, among others, p38 and ERK MAPK pathways leads to the phosphorylation of CRYAB, altering its intracellular localization and function and ultimately leading to a stronger reactive response. We further argue that phosphorylation of Ser-59 has a dominant role in this reactive response, considering the observation that Ser-59 phosphorylation is increased upon stress in vitro, whereas Ser-45 phosphorylation is not affected. In addition, phosphorylation of Ser-59 is highly abundant in reactive astrocytes in MS and cuprizone-induced lesions, whereas Ser-45 phosphorylation is unaffected in MS lesions and not present in astrocytes in cuprizone-induced lesions. Moreover, inhibition of p38, resulting in reduced Ser-59 phosphorylation, has a greater effect on in vitro reactivity than inhibition of ERK, which directs Ser-45 phosphorylation.
Some discrepancies in our findings warrant further discussion, though. We show that, in MS brain tissue, as in cuprizone-induced lesions, phosphorylation of Ser-59 is present exclusively in reactive astrocytes in areas with active demyelination, whereas in vitro, phosphorylated Ser-59 is constitutively present at low levels and is enhanced by stress stimuli. In contrast, phosphorylation of Ser-45 is present in astrocytes in both NAWM and active MS lesions and in cultured primary astrocytes, whereas it is not found in astrocytes in cuprizone-induced lesions. Interestingly, localization of phospho-Ser-45 in astrocytes seems to be species specific. We find cytoplasmic localization in human astrocytes in vivo and in human astroglioma-derived cells (own observations), whereas phospho-Ser-45 is confined to the nucleus in murine cultured astrocytes. In contrast, phospho-Ser-59 is exclusively localized in the cytoplasm in both human and murine astrocytes. An additional discrepancy lies in the observed activation of the MAPK pathways responsible for CRYAB phosphorylation. In vitro, the level of phosphorylation of both Ser-59 and Ser-45 correlates with the activation of p38 and ERK, respectively. In vivo, however, we observe that p38 activation is present in a subset of reactive astrocytes, whereas many reactive astrocytes contain phosphorylated Ser-59. Likewise, activated ERK is present in a subset of reactive astrocytes, although phosphorylated Ser-45 is not present in astrocytes in either cuprizone-affected or unaffected tissue. These incongruities might be explained by the highly versatile system of differential phosphorylation upon specific types of stress that regulates the activity, localization, and function of CRYAB (30), and that might be affected by species and context-driven differences. These variations could also explain the discrepancy of the current data with our previous findings in the EAE model of neuroinflammation, where administration of recombinant CRYAB attenuates disease and reduces proinflammatory cytokine production by T cells (17). We observed there that expression of ERK and p38 is increased in Cryab−/− astrocytes, compared with WT astrocytes, correlating with increased IL-6 production, as well as cell death, of these cells. However, phosphorylation of ERK is induced in WT cells 72 h after stimulation with TNF, as well as phosphorylation of CRYAB Ser-59, but not Ser-45, whereas phospho-ERK levels are slightly lower in Cryab−/− astrocytes and phospho-p38 was not detected in either type of cells. This suggested that, in the setting of this stimulus and within this time frame, other signaling pathways, as well as possible feedback and/or compensatory mechanisms, might contribute to the susceptibility of astrocytes to stress and their production of cytokines. More insight in the kinetics of MAPK signaling, as well as of potential, currently unknown, phosphatases involved in the dephosphorylation, of CRYAB could also further illuminate the regulation of CRYAB in astrocytes.
In addition, as pointed out before, in EAE the binding of exogenously administered CRYAB to proinflammatory factors in the blood underlies its therapeutic effect (27). The same might provide the basis for the beneficial outcome that has been shown in a recent clinical trial in MS patients after i.v. administration of CRYAB. In this trial, repeated administration of CRYAB resulted in decreased lesion activity on MRI, which the investigators suggested was due to an alteration of peripheral immune cell responses (37). In contrast to this, we propose that the effect of CRYAB we describe here is due to its intracellular expression and impact on signaling pathways in astrocytes, rather than an effect on peripheral immunologic mechanisms. This contention is supported by the fact that administration of exogenous CRYAB has no effect on cuprizone-induced pathology.
The versatility in functions of (phosphorylated) CRYAB is also highlighted by our analysis of the proteins it binds to, which shows clear differences in binding targets and associated functions based on the state of phosphorylation. However, our analyses merely reveal associations with previously described functional interactions of the protein binding targets bound by (phosphorylated) CRYAB, not necessarily how this binding affects downstream pathways and functions. If binding by (phosphorylated) CRYAB hinders the normal function of these targets, though, our results predict that native CRYAB might suppress the integration of stress signals to regulate mRNA translation, associated with EIF2 signaling (38), whereas signals inducing Ser-59 or Ser-45 phosphorylation could lift this inhibition (Table 2).
Phosphorylation of Ser-59 might promote the antiapoptotic function of eNOS and its described role in neurovascular coupling by astrocytes (39, 40), as well as enable proliferation and cytoskeletal rearrangement to support cellular movement, whereas it might inhibit integrin-linked kinase signaling, cell death, and ATP metabolism (Table 2 and Tables S2, P-Ser59–induced changes, and S3, P-Ser59–induced changes). In contrast, phosphorylation of Ser-45 could support HIPPO signaling, which is involved in the regulation of proliferation and a key component of which has been shown to inhibit reactive astrogliosis (41), whereas it might inhibit cell cycle and metabolism control by affecting 14-3-3, PKA, and PI3K/AKT signaling (Table 2 and Tables S2, P-Ser45–induced changes, and S3, P-Ser45–induced changes).
Together, these observations strengthen our hypothesis that Ser-59 phosphorylation has a dominant role in supporting reactive astrogliosis. However, further in-depth analyses of the pathways implicated here in the regulation of astrocyte functioning are warranted.
Many studies show a correlation between astrocyte activation and CRYAB levels in reactive astrocytes in injury or disease, such as intracerebral hemorrhage, stroke, and Parkinson’s disease (42–44). In most models, however, CRYAB is reported to have a protective role. In spinal cord contusion injury (SCI), for example, treatment with CRYAB improves recovery (45). Interestingly, this beneficial effect of CRYAB treatment is accompanied by enhanced GFAP immunoreactivity around the lesion and enhanced levels of several cytokines and chemokines, possibly representing—in the context of SCI—a beneficial acute reactive astrocyte response, which subsides at the chronic stage. As important initiators of the inflammatory response after SCI, astrocytes might be the source of (some of) these inflammatory mediators, which is consistent with our finding that CRYAB supports the production of IL-6 by reactive astrocytes. In addition, a series of studies recently reported a central role for CRYAB in the dopamine D2 receptor (DRD2)-mediated suppression of the inflammatory astrocyte response (42, 43, 46). They demonstrate how CRYAB prevents NF-κB and STAT3 signaling, resulting in reduced neuroinflammation. This discordance with our findings could reflect differences in the activation state induced by different kinds of stress and/or regional differences in the astrocyte response, given that dopamine signals through Gi, which might have diverse downstream effects, depending on the area of the brain and the signals received (47). Furthermore, these studies did not address how CRYAB phosphorylation is affected by altered DRD2 expression or signaling. Interestingly, however, activation of several kinases, including p38 and ERK, was increased in the striatum of DRD2−/− mice, indicating that phosphorylation of CRYAB might be affected in these mice (46).
Phosphorylation of CRYAB has been associated with several neurodegenerative diseases, such as Alexander’s disease, Alzheimer's, and Down syndrome (48, 49). Here, we provide an evaluation, however, of the phosphorylation status of CRYAB in MS lesions and its function in demyelination. Exploring this additional level of complexity in the regulation of key proteins implicated in MS pathogenesis, by investigating the posttranslational modifications that might lead to altered function of these proteins, might ultimately lead to new targets for therapeutic intervention. Moreover, in doing so, we have uncovered a potential pivotal role for astrocyte activation in the pathogenesis of MS. Considering this role of astrocytes in the process of active demyelination, these findings open up alternative avenues in the development of treatments for MS. It is becoming clear that there is a fine balance of astrocyte activation stages that result in either beneficial or detrimental functions during demyelinating events. A better understanding of the extracellular and intracellular factors that determine the reactive astrocyte phenotype is needed to target this delicate balance. Phosphorylation of CRYAB, as one of the main regulators of reactive astrogliosis, could prove to be one of those defining factors controlling the tipping point of the pathogenic astrocyte response.
Materials and Methods
Mice.
All animal procedures were approved by the Stanford University Administrative Panel on Laboratory Animal Care. Cryab−/− mice were first generated on a 129S4/SvJae background and maintained on a 129S6/SvEvTac × 129S4/SvJae background (50). At the ages used for this study, Cryab−/− mice are fertile and viable with normal lens transparency and without any apparent prenatal defects. Colonies of Cryab−/− mice and WT controls were maintained and bred in our animal colony according to regulations approved by the Stanford University Institutional Animal Care and Use Committee.
Cuprizone Feeding and Rotarod Testing.
Eight- to 10-wk-old 129S6 WT and Cryab−/− were fed a diet containing 0.2% cuprizone for 4.5–8 wk to induce demyelination. Motor skill was tested by rotarod assay twice a week from week 0.5 before start of the cuprizone diet. Rotor speed increased from 5 to 45 rpm over 300 s, and animals were tested over four trials with a minimum of 5 min rest between trials. Motor skill was scored on a combined 0- to 14-point scale based on lag time to first fall or flip (0–5 points), total time spent on the rotarod (0–5 points), and number of flips made (0–4 points), and the average score from the best three trials was calculated.
Tissue Preparation and Histology.
Mice were deeply anesthetized with ketamine/xylazine (100 and 7 mg/kg, respectively), and then transcardially perfused [cold saline followed by cold 4% (wt/vol) paraformaldehyde in 0.1 M phosphate buffer], and brains were collected for immunohistochemistry. All brains were postfixed overnight in 4% (wt/vol) paraformaldehyde at 4 °C, after which brains were cut in half sagittally. One-half was then cryoprotected in 30% (wt/vol) sucrose and stored at 4 °C until they were embedded in Optimal Cutting Temperature compound (Tissue-Tek) and frozen at −80 °C. The other half was transferred to 10% (vol/vol) formalin in PBS and embedded in paraffin. Paraffin sections (5 μm) were used for analysis of demyelination and remyelination by LFB staining. Light-microscopic pictures of LFB-stained tissue (50×) were analyzed using ImageJ (version 1.44; National Institutes of Health).
Cryosections [20 μm, stored at − 20 °C in cryoprotectant consisting of 30% (vol/vol) ethylene glycol, 30% (vol/vol) glycerin in 0.1 M phosphate buffer] were stained with free-floating immunohistochemistry. Briefly, sections were incubated in Tris-buffered saline (TBS) with 0.3% Triton X-100 and 1% normal donkey serum at 4 °C overnight containing the following primary antibodies: goat anti-GFAP (1:1,000; Abcam; ab53554), mouse anti-Iba1 (1:1,000; Wako; 019-19741), rat anti-MBP (1:500; Millipore; MAB386), rabbit anti-phospho-Ser-59-CRYAB (1:500; Abcam; ab5577), rabbit anti-phospho-Ser-45-CRYAB (1:500; Abcam; ab5598), mouse anti-CC1 (1:500; Calbiochem; OP80), rabbit anti-phopho-p38 (1:500; Cell Signaling Technology; 4511), and rabbit anti-phopho-Erk1/2 (1:500; Cell Signaling Technology; 4370). Species-specific fluorescent secondary antibodies from donkey conjugated with FITC, Cy3, or Cy5 were obtained from Jackson Laboratories and all used at 1:500. Fluorescent signals were detected using a confocal laser-scanning microscope (Zeiss; LSM 700). All analyses were performed using multichannel configuration with a 40× objective and electronic zoom of 1. Number of astrocytes and microglia in comparable regions inside and outside the lesions and in control tissue, were quantified by counting nuclei associated with GFAP or Iba1 staining, respectively, using ImageJ.
Primary Astrocyte Cultures and Stimulations.
Primary astrocyte cultures were derived from 7-d-old WT mouse pups and enriched using a modified immunopanning method described before (51). Briefly, brain tissue was dissected and enzymatically dissociated to a single-cell suspension. Microglia were first depleted by two panning steps on plates coated with BSL-1 (Vector Laboratories). Oligodendrocyte progenitor cells were then depleted on an anti-PDGFRα (BD Pharmingen; 558774)-coated plate, and the remaining astrocyte-enriched cell suspension was plated in uncoated T75 flasks in DMEM (HyClone) containing 10% (vol/vol) FBS, 4.5 g/L glucose, 4 mM l-glutamine, 1 mM sodium pyruvate, 100 U/mL penicillin, and 0.1 mg/mL streptomycin. Astrocytes were passaged at least once before being used in the described assays. Serum concentration in the media was lowered in steps [10, 5, 1% (vol/vol)] to serum-free conditions before start of the stimulation experiments. Cells were stimulated for 24–48 h with increasing doses of LPS (Sigma; doses indicated in figures) and with 1,000 ng/mL LPS following 16-h pretreatment with specific inhibitors for p38 (10 μM; SB203580; Calbiochem) and ERK (100 μM; PD98059; Calbiochem) in serum-free media.
siRNA Transfection.
Primary WT astrocytes were transfected with control and CRYAB ON-TARGETplus SMARTpool siRNAs (mouse CRYAB, 12955, and nontargeting control pool; Dharmacon) using the Amaxa Nucleofector system and a Basic Glial Cell Nucleofector kit (Lonza). Serum concentration in the media was lowered in steps [10, 5, 1% (vol/vol)] to serum-free conditions before start of the stimulation experiments. Cells were plated on poly-d-lysine (PDL) (BD)-coated 12-well plates for in vitro scratch assays or uncoated 6-well plates and stimulated with LPS for 48 h for analysis of IL-6 secretion by ELISA.
In Vitro Scratch Assay.
Transfected cells, grown in PDL-coated 12-well plates until confluency, were scratched with a 20- to 200-μL pipet tip and left to proliferate for 48 h. Pictures were taken with light microscopy immediately and 48 h after the scratch insult. Sets of pictures were then analyzed using ImageJ.
ELISA.
Untransfected and transfected cells were stimulated for 24–48 h with increasing doses of LPS (Sigma; doses indicated in figures) and with 1,000 ng/mL LPS following 16-h pretreatment with specific inhibitors for p38 (10 μM; SB203580; Calbiochem) and ERK (100 μM; PD98059; Calbiochem) in serum-free media. Culture supernatants were harvested 48 h after stimulation, and IL-6 was measured in the supernatants using a commercial ELISA kit (OptEIA mouse IL-6; BD).
Identification of CRYAB Phosphopeptides in Human Brain Tissue.
Frozen MS brain tissue samples were histologically classified as described before in refs. 28 and 52. Active MS brain lesions (pooled from three MS samples) and control (pooled from two age-, sex-, and brain region-matched) samples (each with a wet weight of ∼1 g) were pulverized on dry ice in a Dounce homogenizer in lysis buffer (53). High-speed supernatants from the tissue extracts were subjected to reduction, alkylation, protease digestion with trypsin, and phosphoprotein enrichment on IMAC columns followed by nanoscale liquid chromatography coupled to tandem mass spectrometry (nano-LC-MS/MS) on an LTQ velos-Orbitrap (Thermo Fisher Scientific) (29). Mass spectra were searched against a human IPI protein database (version 3.75) concatenated with a reversed database by the SEQUEST algorithm (Proteome Discoverer software, version 1.2; Thermo Scientific), and mass addition for oxidation (15.995 Da) on methionine and phosphorylation (79.966 Da) on Ser, Thr, and Tyr was tolerated for posttranslational modification. A complete list with mass spectra is archived in Peptide database PASS00275–phosphoproteome of human multiple sclerosis brain lesion (https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/PASS_View?identifier=PASS00275).
MS Brain Tissue and Immunohistochemistry.
Brain tissue, taken from patients with clinically diagnosed and neuropathologically confirmed MS, was obtained at rapid autopsy and immediately frozen in liquid nitrogen (in collaboration with The Netherlands Brain Bank, Amsterdam, The Netherlands; coordinator, Inge Huitinga). White-matter MS tissue samples were selected on the basis of postmortem MRI. All patients, or their next of kin, had given informed consent for autopsy and use of their brain tissue for research purposes. The use of tissue and access to medical records for research purposes was approved by the Ethical Committee of the VU University Medical Center (Amsterdam, The Netherlands).
For immunohistochemical staining, 5-μm cryosections were air-dried and fixed in acetone for 10 min. Sections were incubated overnight at 4 °C with primary antibodies for native CRYAB (Enzo; ADI-SPA-223), phospho-Ser-59-CRYAB or phospho-Ser-45-CRYAB, all diluted in PBS containing 0.1% BSA (Boehringer-Mannheim), after which slides were incubated with EnVision Kit mouse/rabbit-labeled horseradish peroxidase (DAKO) for 30 min at room temperature. Peroxidase activity was demonstrated with 0.5 mg/mL 3,30-diaminobenzidine tetrachloride (DAB) (Sigma) in PBS containing 0.02% hydrogen peroxide. Between incubation steps, sections were thoroughly washed with PBS. After DAB development, sections were counterstained with hematoxylin for 1 min and dehydrated with ethanol followed by xylenes and mounted with Entellan (Merck).
Immunocytochemistry.
Cells were cultured on glass coverslips and stimulated with 1,000 ng/mL LPS alone or following 16-h pretreatment with specific inhibitors for p38 (10 μM; SB203580; Calbiochem) and ERK (100 μM; PD98059; Calbiochem) in serum-free media. After stimulation, cells were fixed in 4% (wt/vol) paraformaldehyde in PBS for 10 min and then washed with PBS and kept in PBS until immunocytochemistry. Fixed cells were stained with the following primary antibodies: mouse anti-CRYAB (Abcam; ab13496), rabbit anti-phospho-Ser-59-CRYAB, and rabbit anti-phospho-Ser-45-CRYAB. Species-specific fluorescent secondary antibodies from donkey conjugated with Al488, Cy3, or Cy5 were obtained from Jackson Laboratories and all used at 1:500. Fluorescent signals were detected using a confocal laser-scanning microscope (Zeiss; LSM 700). All analyses were performed using multichannel configuration with a 40× objective and electronic zoom of 1.
Coimmunoprecipitation and Protein Identification by Mass Spectrometry.
Astrocyte cultures were grown up in bulk in T175 flasks, and lysates were made and coimmunoprecipitated with the following antibodies: rabbit anti-CRYAB, phospho-Ser-59-, phospho-Ser-45-, phospho-Ser-19-CRYAB (Thermo Fisher; PA1-20795, PA1-012, PA1-011, PA1-010, respectively), above using a Pierce Co-IP kit, according to manufacturer’s directions. An isotype control was used to identify background binding. The resulting eluates were subjected to reduction, alkylation, and protease digestion with trypsin followed by nano-LC-MS/MS on an LTQ velos-Orbitrap. Proteins were identified using Scaffold4 software, with peptide and protein thresholds of 95.0% minimum and 1 peptide minimum, and peptide and protein false-discovery rates of 0.9 and 0.7%, respectively. Quantitative values, corrected for background binding (as determined by binding to the isotype control), using total spectra outputs, were used for subsequent analyses.
AutoSOME Clustering of Protein Binding Targets.
AutoSOME (autosome.stanford.edu) (54) clustering was performed with all protein targets and background-corrected quantitative values (240 entries) over 100 ensemble runs, using a minimum spanning tree approach with unit variance normalization, a Euclidean distance matrix, and a P-value cutoff of 0.05.
WebGestalt Identification of Enriched Protein Pathways.
WebGestalt, a Web-based enrichment tool for the functional annotation and pathway analysis of sets of genes or proteins, was performed, and enrichment analysis of GO terms and pathways in the sets of proteins bound by (phosphorylated) CRYAB that were identified by mass spectrometry, using the Wikipathways database of biological pathways (www.webgestalt.org). A hypergeometric test was performed with Mus musculus as organism, and using identified entrezgene number as ID types and the entrezgene_protein-coding reference set, followed by Benjamini–Hochberg adjustment for multiple testing. An adjusted P value of 0.05 was considered significant and pathways containing a minimum of two identified proteins were included.
IPA of Predicted Differentially Affected Pathways and Functions.
IPA (Qiagen; www.ingenuity.com/) was used to identify curated canonical pathways and biological pathways and disease annotations that were enriched for binding targets of native CRYAB, as well as changes to binding patterns induced by phosphorylation of Cryab, by subtracting quantitative protein values (spectral counts) bound by native CRYAB from those bound by phosphorylated CRYAB. Analysis was run using the Ingenuity Knowledge Base reference set containing genes and endogenous chemicals, considering direct and indirect relationships. Confidence was set at experimentally observed in data curated from all human, mouse, and rat tissue and cell line sources.
Statistical Analysis.
Statistical analysis was performed using GraphPad Prism software, version 5.0a. A Kolmogorov–Smirnov test was used to verify normality of samples. In samples with Gaussian distribution, an unpaired t test was used to determine significant differences between groups, or a two-way ANOVA was used to identify effects of multiple parameters. In samples without Gaussian distribution, a nonparametric Mann–Whitney test was used to determine significant differences. Values of P < 0.01, < 0.05, and < 0.1 are indicated in the figures.
SI Materials and Methods
In Vivo CRYAB Treatment.
C57BL/6J mice were fed cuprizone-containing chow for 6 wk and treated with 10 µg of recombinant human CRYAB in 200 µL of PBS or PBS alone intraperitoneally every other day. Mice were weighed, and motor skill was tested by rotarod assay twice a week from week 0.5 before start of the cuprizone diet.
Western Blotting.
Cells were detached using trypsin (GIBCO), washed with ice-cold PBS, and lysed in radioimmunoprecipitation assay (RIPA) lysis buffer containing 1× Halt protease and phosphatase inhibitor mixture (Pierce). Fifteen micrograms of each sample were subjected to SDS/PAGE electrophoresis. Proteins were transferred to PVDF membranes and immunoblotted with antibodies against CRYAB and GAPDH. Bound antibodies were visualized by enhanced chemiluminescence.
Cultured Astrocyte Stimulations and Immunocytochemistry.
Cells were cultured on coverslips and stimulated with 500 μM H2O2 or 1 μM LPS for 1 h, or for 24 h with LPS alone or following 16-h pretreatment with specific inhibitors for p38 (10 μM; SB203580; Calbiochem) and ERK (100 μM; PD98059; Calbiochem) in serum-free media. After stimulation, cells were fixed in 4% (wt/vol) paraformaldehyde in PBS for 10 min and then washed with PBS and kept in PBS until immunocytochemistry. Fixed cells were stained with the following primary antibodies: mouse anti-CRYAB, rabbit anti-phospho-Ser-59-CRYAB, rabbit anti-phospho-Ser-45-CRYAB, rabbit anti-phopho-p38, and rabbit anti-phopho-Erk1/2. Species-specific fluorescent secondary antibodies from donkey conjugated with Al488, Cy3, or Cy5 were obtained from Jackson Laboratories and all were used at 1:500. Fluorescent signals were detected using a confocal laser-scanning microscope (Zeiss; LSM 700). All analyses were performed using multichannel configuration with a 40× objective and electronic zoom of 1.
Supplementary Material
Acknowledgments
We thank M. Rivera, S. van der Pol, J. Vister, R. Becker, N. Epstein, and X. Mignot for their technical assistance, and B. Ajami and J. J. Bajramovic for critically reading the manuscript. This research was funded by grants from the Human Frontier Science Program (LT000830/2009-L) and from the California Institute for Regenerative Medicine (postdoctoral scholarship; Grant TG2- 01159) (to H.F.K.), and the Endriz Fund and the National Multiple Sclerosis Society (Grant RG 4240B12/2) (to L.S.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1621314114/-/DCSupplemental.
References
- 1.Compston A, Coles A. Multiple sclerosis. Lancet. 2002;359(9313):1221–1231. doi: 10.1016/S0140-6736(02)08220-X. [DOI] [PubMed] [Google Scholar]
- 2.Frohman EM, Racke MK, Raine CS. Multiple sclerosis—the plaque and its pathogenesis. N Engl J Med. 2006;354(9):942–955. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- 3.Trapp BD, Bö L, Mörk S, Chang A. Pathogenesis of tissue injury in MS lesions. J Neuroimmunol. 1999;98(1):49–56. doi: 10.1016/s0165-5728(99)00081-8. [DOI] [PubMed] [Google Scholar]
- 4.Sofroniew MV, Vinters HV. Astrocytes: Biology and pathology. Acta Neuropathol. 2010;119(1):7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36(2):180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- 6.Verkhratsky A, et al. Neurological diseases as primary gliopathies: A reassessment of neurocentrism. ASN Neuro. 2012;4(3):131–149. doi: 10.1042/AN20120010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Valk P, De Groot CJ. Staging of multiple sclerosis (MS) lesions: Pathology of the time frame of MS. Neuropathol Appl Neurobiol. 2000;26(1):2–10. doi: 10.1046/j.1365-2990.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- 8.Dubin RA, Wawrousek EF, Piatigorsky J. Expression of the murine αB-crystallin gene is not restricted to the lens. Mol Cell Biol. 1989;9(3):1083–1091. doi: 10.1128/mcb.9.3.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Head MW, Corbin E, Goldman JE. Coordinate and independent regulation of αB-crystallin and hsp27 expression in response to physiological stress. J Cell Physiol. 1994;159(1):41–50. doi: 10.1002/jcp.1041590107. [DOI] [PubMed] [Google Scholar]
- 10.Bajramović JJ, et al. Differential expression of stress proteins in human adult astrocytes in response to cytokines. J Neuroimmunol. 2000;106(1-2):14–22. doi: 10.1016/s0165-5728(99)00260-x. [DOI] [PubMed] [Google Scholar]
- 11.Brownell SE, Becker RA, Steinman L. The protective and therapeutic function of small heat shock proteins in neurological diseases. Front Immunol. 2012;3(May):74. doi: 10.3389/fimmu.2012.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chabas D, et al. The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science. 2001;294(5547):1731–1735. doi: 10.1126/science.1062960. [DOI] [PubMed] [Google Scholar]
- 13.van Noort JM, et al. The small heat-shock protein αB-crystallin as candidate autoantigen in multiple sclerosis. Nature. 1995;375(6534):798–801. doi: 10.1038/375798a0. [DOI] [PubMed] [Google Scholar]
- 14.Bajramović JJ, Lassmann H, van Noort JM. Expression of αB-crystallin in glia cells during lesional development in multiple sclerosis. J Neuroimmunol. 1997;78(1-2):143–151. doi: 10.1016/s0165-5728(97)00092-1. [DOI] [PubMed] [Google Scholar]
- 15.van Noort JM, et al. αB-crystallin is a target for adaptive immune responses and a trigger of innate responses in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol. 2010;69(7):694–703. doi: 10.1097/NEN.0b013e3181e4939c. [DOI] [PubMed] [Google Scholar]
- 16.Bsibsi M, et al. α-B-crystallin induces an immune-regulatory and antiviral microglial response in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol. 2013;72(10):970–979. doi: 10.1097/NEN.0b013e3182a776bf. [DOI] [PubMed] [Google Scholar]
- 17.Ousman SS, et al. Protective and therapeutic role for αB-crystallin in autoimmune demyelination. Nature. 2007;448(7152):474–479. doi: 10.1038/nature05935. [DOI] [PubMed] [Google Scholar]
- 18.Shin J-H, et al. αB-crystallin suppresses oxidative stress-induced astrocyte apoptosis by inhibiting caspase-3 activation. Neurosci Res. 2009;64(4):355–361. doi: 10.1016/j.neures.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 19.Li R, Reiser G. Phosphorylation of Ser45 and Ser59 of αB-crystallin and p38/extracellular regulated kinase activity determine αB-crystallin-mediated protection of rat brain astrocytes from C2-ceramide- and staurosporine-induced cell death. J Neurochem. 2011;118(3):354–364. doi: 10.1111/j.1471-4159.2011.07317.x. [DOI] [PubMed] [Google Scholar]
- 20.Morrison LE, Hoover HE, Thuerauf DJ, Glembotski CC. Mimicking phosphorylation of αB-crystallin on serine-59 is necessary and sufficient to provide maximal protection of cardiac myocytes from apoptosis. Circ Res. 2003;92(2):203–211. doi: 10.1161/01.res.0000052989.83995.a5. [DOI] [PubMed] [Google Scholar]
- 21.Launay N, Goudeau B, Kato K, Vicart P, Lilienbaum A. Cell signaling pathways to αB-crystallin following stresses of the cytoskeleton. Exp Cell Res. 2006;312(18):3570–3584. doi: 10.1016/j.yexcr.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 22.Aggeli I-KS, Beis I, Gaitanaki C. Oxidative stress and calpain inhibition induce α B-crystallin phosphorylation via p38-MAPK and calcium signalling pathways in H9c2 cells. Cell Signal. 2008;20(7):1292–1302. doi: 10.1016/j.cellsig.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 23.Chis R, et al. α-Crystallin B prevents apoptosis after H2O2 exposure in mouse neonatal cardiomyocytes. Am J Physiol Heart Circ Physiol. 2012;303(8):H967–H978. doi: 10.1152/ajpheart.00040.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blakemore WF. Demyelination of the superior cerebellar peduncle in the mouse induced by cuprizone. J Neurol Sci. 1973;20(1):63–72. doi: 10.1016/0022-510x(73)90118-4. [DOI] [PubMed] [Google Scholar]
- 25.Skripuletz T, Gudi V, Hackstette D, Stangel M. De- and remyelination in the CNS white and grey matter induced by cuprizone: The old, the new, and the unexpected. Histol Histopathol. 2011;26(12):1585–1597. doi: 10.14670/HH-26.1585. [DOI] [PubMed] [Google Scholar]
- 26.Liebetanz D, Merkler D. Effects of commissural de- and remyelination on motor skill behaviour in the cuprizone mouse model of multiple sclerosis. Exp Neurol. 2006;202(1):217–224. doi: 10.1016/j.expneurol.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 27.Rothbard JB, et al. Therapeutic effects of systemic administration of chaperone αB-crystallin associated with binding proinflammatory plasma proteins. J Biol Chem. 2012;287(13):9708–9721. doi: 10.1074/jbc.M111.337691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garris CS, et al. Defective sphingosine 1-phosphate receptor 1 (S1P1) phosphorylation exacerbates TH17-mediated autoimmune neuroinflammation. Nat Immunol. 2013;14(11):1166–1172. doi: 10.1038/ni.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huttlin EL, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143(7):1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito H, Okamoto K, Nakayama H, Isobe T, Kato K. Phosphorylation of αB-crystallin in response to various types of stress. J Biol Chem. 1997;272(47):29934–29941. doi: 10.1074/jbc.272.47.29934. [DOI] [PubMed] [Google Scholar]
- 31.Kato K, et al. Phosphorylation of αB-crystallin in mitotic cells and identification of enzymatic activities responsible for phosphorylation. J Biol Chem. 1998;273(43):28346–28354. doi: 10.1074/jbc.273.43.28346. [DOI] [PubMed] [Google Scholar]
- 32.Zhang B, Kirov S, Snoddy J. WebGestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33(Web Server issue) SUPPL. 2:W741–W748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raasch J, et al. IkappaB kinase 2 determines oligodendrocyte loss by non-cell-autonomous activation of NF-kappaB in the central nervous system. Brain. 2011;134(Pt 4):1184–1198. doi: 10.1093/brain/awq359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skripuletz T, et al. Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination. Brain. 2013;136(Pt 1):147–167. doi: 10.1093/brain/aws262. [DOI] [PubMed] [Google Scholar]
- 35.Mayo L, et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat Med. 2014;20(10):1147–1156. doi: 10.1038/nm.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mayo L, et al. IL-10-dependent Tr1 cells attenuate astrocyte activation and ameliorate chronic central nervous system inflammation. Brain. 2016;139(Pt 7):1939–1957. doi: 10.1093/brain/aww113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Noort JM, Bsibsi M, Nacken PJ, Verbeek R, Venneker EHG. Therapeutic intervention in multiple sclerosis with α B-crystallin: A randomized controlled phase IIa trial. PLoS One. 2015;10(11):e0143366. doi: 10.1371/journal.pone.0143366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wek RC, Jiang H-Y, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34(Pt 1):7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- 39.Sessa WC. eNOS at a glance. J Cell Sci. 2004;117(Pt 12):2427–2429. doi: 10.1242/jcs.01165. [DOI] [PubMed] [Google Scholar]
- 40.Muñoz MF, Puebla M, Figueroa XF. Control of the neurovascular coupling by nitric oxide-dependent regulation of astrocytic Ca2+ signaling. Front Cell Neurosci. 2015;9(March):59. doi: 10.3389/fncel.2015.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang Z, et al. YAP is a critical inducer of SOCS3, preventing reactive astrogliosis. Cereb Cortex. 2016;26(5):2299–2310. doi: 10.1093/cercor/bhv292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y, et al. Activation of dopamine D2 receptor suppresses neuroinflammation through αB-crystalline by inhibition of NF-κB nuclear translocation in experimental ICH mice model. Stroke. 2015;46(9):2637–2646. doi: 10.1161/STROKEAHA.115.009792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiu J, et al. Sinomenine activates astrocytic dopamine D2 receptors and alleviates neuroinflammatory injury via the CRYAB/STAT3 pathway after ischemic stroke in mice. J Neuroinflammation. 2016;13(1):263. doi: 10.1186/s12974-016-0739-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Y, et al. Upregulation of αB-crystallin expression in the substantia nigra of patients with Parkinson’s disease. Neurobiol Aging. 2015;36(4):1686–1691. doi: 10.1016/j.neurobiolaging.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 45.Klopstein A, et al. Beneficial effects of αB-crystallin in spinal cord contusion injury. J Neurosci. 2012;32(42):14478–14488. doi: 10.1523/JNEUROSCI.0923-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shao W, et al. Suppression of neuroinflammation by astrocytic dopamine D2 receptors via αB-crystallin. Nature. 2013;494(7435):90–94. doi: 10.1038/nature11748. [DOI] [PubMed] [Google Scholar]
- 47.Davila D, Thibault K, Fiacco TA, Agulhon C. Recent molecular approaches to understanding astrocyte function in vivo. Front Cell Neurosci. 2013;7:272. doi: 10.3389/fncel.2013.00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kato K, et al. Ser-59 is the major phosphorylation site in αB-crystallin accumulated in the brains of patients with Alexander’s disease. J Neurochem. 2001;76(3):730–736. doi: 10.1046/j.1471-4159.2001.00038.x. [DOI] [PubMed] [Google Scholar]
- 49.Palminiello S, et al. Upregulation of phosphorylated αB-crystallin in the brain of children and young adults with Down syndrome. Brain Res. 2009;1268:162–173. doi: 10.1016/j.brainres.2009.02.058. [DOI] [PubMed] [Google Scholar]
- 50.Brady JP, et al. αB-crystallin in lens development and muscle integrity: A gene knockout approach. Invest Ophthalmol Vis Sci. 2001;42(12):2924–2934. [PubMed] [Google Scholar]
- 51.Foo LC, et al. Development of a method for the purification and culture of rodent astrocytes. Neuron. 2011;71(5):799–811. doi: 10.1016/j.neuron.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Han MH, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451(7182):1076–1081. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- 53.Villén J, Gygi SP. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat Protoc. 2008;3(10):1630–1638. doi: 10.1038/nprot.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Newman AM, Cooper JB. AutoSOME: A clustering method for identifying gene expression modules without prior knowledge of cluster number. BMC Bioinformatics. 2010;11(1):117. doi: 10.1186/1471-2105-11-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.