

Abstract
Plasma corticosteroid-binding globulin (CBG) plays a critical role in regulating glucocorticoid bioavailability and is an acute phase “negative” protein during inflammation. In an adjuvant-induced arthritis model, plasma CBG levels decrease in rats that develop severe inflammation, and we have now determined when and how these reductions in CBG occur. After administering complete Freund's adjuvant or saline intra-dermally at the tail base, blood samples were taken periodically for 16 days. In adjuvant-treated rats, decreases in plasma CBG levels matched the severity of inflammation, and decreases were observed 4 days before any clinical signs of inflammation. Decreases in CBG levels coincided with an ∼5kDa reduction in its apparent size, consistent with proteolytic cleavage, and cleaved CBG lacked steroid-binding activity. At the termination of the experimental period, hepatic Cbg mRNA levels were decreased in rats with severe inflammation. While plasma TNF-α increased in all adjuvant-treated rats, increases in Il-4, IL-6, IL-10, IL-13, and IFN-γ were only observed in rats with cleaved CBG. Rats with cleaved CBG also exhibited increased spleen weights, and strong negative correlations were observed between CBG, IL-6 and spleen weights, respectively. However, there were no differences in hepatic Cbg mRNA levels in relation to the apparent proteolysis of CBG, suggesting that CBG cleavage occurs prior to changes in hepatic Cbg expression. Our results indicate that the levels and integrity of plasma CBG are biomarkers of the onset and severity of inflammation. Dynamic changes in the levels and function of CBG likely modulate the tissue availability of corticosterone during inflammation.
Keywords: corticosterone, cytokines, spleen weight, proteolysis, serine protease inhibitor
Introduction
Produced primarily by the liver, corticosteroid-binding globulin (CBG) is a plasma glycoprotein that binds ∼ 90% of circulating glucocorticoids, and regulates their bioavailability in target tissues (Lin, et al. 2010). Plasma CBG shares structural similarities with clade A serine protease inhibitor (SerpinA) family members (Lin, et al. 2010). However, CBG is not a protease inhibitor, but rather serves as a reservoir for glucocorticoids that are released when its reactive center loop (RCL) is cleaved by proteases, including neutrophil elastase (Hammond, et al. 1990), chymotrypsin (Lewis & Elder. 2014) or the bacterial proteinase, LasB (Simard, et al. 2014).
During inflammation, CBG acts as an acute phase “negative” protein, and reductions in its plasma levels can be attributed to both proteolytic cleavage (Hammond, et al. 1990) and down-regulation of its production by the liver (Bernier, et al. 1998, Emptoz-Bonneton, et al. 1997, Smith & Hammond. 1992). In humans, proteolysis of the RCL of CBG by neutrophil elastase appears to be an early event during inflammatory reactions, rendering CBG non-functional and promoting the localised release of CBG-bound glucocorticoids at sites of inflammation (Hammond. 1990, Perogamvros, et al. 2012). Enhanced exposure of tissues to anti-inflammatory glucocorticoids represses cytokine production and activity (Brattsand & Linden. 1996), thereby limiting cytokine-mediated tissue damage (Simon. 2003). At the same time, plasma glucocorticoid concentrations increase acutely as a result of hypothalamic-pituitary-adrenal axis activation in response to stress (Sapolsky, et al. 2000), and likely act in synergy with elevations in inflammatory cytokines, such as interleukin-6 (IL-6), to further reduce plasma CBG production, further amplifying free glucocorticoid exposures (Bartalena, et al. 1993, Bernier, et al. 1998, Emptoz-Bonneton, et al. 1997). During recovery from inflammation, normalization of CBG levels likely plays a role in determining when, and to what extent, glucocorticoids act to restore the normal homeostatic balance.
Several studies in CBG deficient animals support the idea that CBG plays a vital role during the inflammatory process. For instance, a genetic study revealed that C57BL/6 mice are more sensitive to an acute challenge with TNF-α than DBA/2 mice, and this trait was mapped to the Cbg (SerpinA6) locus (Libert, et al. 1999). Recently, we have found that Harlan Sprague Dawley (SD) rats are deficient in plasma CBG (50% lower levels), when compared to Charles River SD rats (Bodnar, et al. 2015). Harlan SD rats are also more sensitive to inflammatory challenges than Charles River SD rats in an adjuvant-induced arthritis model (Bodnar, et al. 2015) or after treatment with lipopolysaccharide (Turnbull & Rivier. 1999). A key role for CBG in these differential responses is further supported by studies in Cbg-/- mice in which survival rates after an acute inflammatory challenge are compromised (Petersen, et al. 2006).
Low plasma cortisol levels in patients have led to the identification of several individuals with CBG deficiencies (Gagliardi, et al. 2010). Genome-wide sequencing of human populations has also identified numerous other single nucleotide polymorphisms that cause decreased CBG production or defects in steroid binding, some of which are enriched in specific ethnic groups (Simard, et al. 2015). Although patients with CBG deficiencies have been reported to suffer from a variety of symptoms including chronic pain, fatigue, depression, hypotension and excess body weight (Gagliardi, et al. 2010), it remains to be determined how well they cope with severe, acute inflammation. To explore this, we have determined when changes in plasma CBG levels occur during the course of acute inflammation in rats and how this relates to the severity of the inflammatory response.
Materials and Methods
Animals and treatments
Female Sprague Dawley rats (n=24, 51-52 days old) were received from Charles River Laboratories International, Inc. (St. Constant, Canada), and maintained as previously described (Bodnar, et al. 2015). Following an acclimatization period of 5 days, pre-treatment (baseline) blood samples were taken from the tail vein, under light isofluorane anesthesia, between 11 am and 1 pm for the preparation of serum, which was stored at -80°C until analyzed. Three days later, rats were anesthetized with isoflurane, and injected with 0.6 mg complete Freund's adjuvant (n=20) prepared as described previously (Zhang, et al. 2012) or with physiological saline (control; n=4). Rats were then housed with CareFRESH® (Healthy Pet, Ferndale, USA) bedding to minimize discomfort, and monitored for signs of pain, discomfort or infection, and for general signs of health including activity, coat quality, and ability to rear.
Post-treatment, rats were split into two groups (n=12 per group; 2 controls and 10 adjuvant-treated rats). Blood samples (∼50 μL) from the two groups were alternatively collected between 11 am and 1 pm under light isoflurane anesthesia from the tail vein over a 16 day experimental period. Serum was prepared and stored at -80°C until assayed. At the time of blood sampling, rats were weighed and clinical signs of inflammation were scored and recorded. To calculate clinical scores, each of the four paws was given a score of 0-4, where 0 = no signs of inflammation, 1 = single focus of redness or swelling, 2 = two or more foci of redness or swelling, 3 = confluent but not global swelling, 4 = severe global swelling (Zhang, et al. 2012). Rats achieving an overall clinical score ≥ 8 (out of a possible 16) at any point during the study were classified as developing severe inflammation, whereas rats with a clinical score < 8 were classified as developing mild-moderate inflammation. One animal with severe inflammation was sacrificed on experimental day 14 for humane reasons, while all other animals were sacrificed on experimental day 16. On the day of termination (8-10 am), rats were removed from the colony room and decapitated (< 1 min). Trunk blood was collected in tubes containing EDTA, and plasma was stored at -80°C until analyzed. In addition, livers and spleens were removed, flash frozen and stored at -80°C.
The animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and approved by the University of British Columbia Animal Care Committee.
Plasma CBG analysis
Plasma corticosterone-binding capacity of CBG was measured using an established ligand-saturation assay (Hammond & Lahteenmaki. 1983). Briefly, samples were diluted (1:1000-1:3000) in phosphate buffered saline and stripped of endogenous steroids by incubation with dextran-coated charcoal (DCC) for 30 min at room temperature followed by centrifugation. Samples were then incubated with ∼10 nmol l-1 [3H]-corticosterone (PerkinElmer Life Sciences, Waltham, USA) in the absence or presence of excess corticosterone to monitor non-specific binding. After separation of free [3H]-corticosterone by adsorption with DCC for 10 min and centrifugation at 0°C, CBG-bound [3H]-corticosterone in the supernatants was determined in a scintillation spectrophotometer.
To assess CBG integrity, diluted (1:200) plasma samples were subjected to SDS-PAGE and transferred to PVDF membranes using a Trans-Blot turbo transfer system (BioRad, Mississauga, Canada). Blots were blocked with 5% milk-PBST for 2 hrs at room temperature and incubated overnight at 4°C with polyclonal rabbit anti-mouse CBG antiserum diluted 1:4,000 in the 5% milk-PBST (Scrocchi, et al. 1993), followed by a horseradish peroxidase-labeled goat anti-rabbit IgG antibody (1:10,000; Sigma-Aldrich, Oakville, Canada) for 1 hr at room temperature. Immunoreactive CBG was then detected with ECL reagent using an ImageQuant LAS4000 (GE Health Care, Mississauga, Canada). Total protein concentrations in plasma samples were measured using a Micro BCA™ Protein Assay Kit (Thermo Fisher Scientific Inc., Mississauga, Canada).
Separation and characterization of functional and non-functional CBG in plasma samples
To characterize CBG in plasma samples from rats with severe inflammation, plasma from two rats were pooled, treated with DCC to remove endogenous steroids, and then applied to an 11β-hydroxy-andros-4-en-3-oxo-17β-carboxylic acid (HACA)-Sepharose affinity column (Seralini, et al. 1989). After washing the column with 100 mmol l-1 Tris-NaCl, steroid-bound CBG was eluted with 1 μmol l-1 cortisol in the same buffer. The flow-through, wash and eluent fractions were analyzed by corticosterone-binding capacity assay and western blotting, as described above.
CBG mRNA measurements
Total RNA was extracted from liver using Trizol reagent (Thermo Fisher Scientific Inc., Mississauga, Canada), followed by purification using an RNeasy kit (Qiagen, Valencia, USA), as per the manufacturer's instructions. To obtain cDNA, 1 ug of RNA was reversed transcribed using High-Capacity cDNA Reverse Transcription Kits (Thermo Fisher Scientific Inc., Mississauga, Canada). Real time quantitative PCR (qRT-PCR) was completed using a pre validated rat Serpina6 (Cbg) PrimeTime Std qPCR Assay (Integrated DNA Technologies (IDT), Coralville, USA: Assay ID# Rn.PT.58.02619945) and 5 ng of cDNA per reaction. The qRT-PCR reactions were run in duplicate on the Applied Biosystems 7500 Fast Real-Time PCR system (Thermo Fisher Scientific Inc., Mississauga, Canada). Hepatic Cbg mRNA levels were normalized to those of rat Gapdh (IDT: Assay ID# Rn.PT.58.35727291) using the delta-delta Ct method.
Multiplex cytokine immunoassays
Plasma cytokines (IL-4, IL-5, IL-6, IL-10, IL-13, IFN-ɣ, and TNF-α) were measured in plasma samples collected at termination using the Proinflammatory Panel 2 (rat) V-PLEX kit from Meso Scale Discovery (MSD, Rockville, USA; catalog #: K15059D-1). The cytokine assays were performed according to the established MSD protocol. The assay plate was read using a MESO QuickPlex SQ 120 and data were analyzed using the MSD Discovery Workbench software v. 4.0. Lower limits of detection for cytokines (pg/mL) are as follows: IL-4 (0.16), IL-5 (6.89), IL-6 (7.18), IL-10 (6.18), IL-13 (0.45), TNF-α (1.04), IFN-ɣ (1.48).
Statistical analyses
Data (expressed as mean ± SD or SEM, as indicated) were analyzed using t-tests or analysis of variance (ANOVA), with repeated measures as required, followed by Fisher or Tukey's post hoc tests to examine significant main effects. Differences were considered significant at p ≤ 0.05, and trends (0.06 > P > 0.05) were examined, as appropriate. T-test, ANOVA and post hoc p values are shown in results; ANOVA F statistics are included in the figure legends.
Results
Body weights and clinical scores
The body weights of all animals increased over the course of the experiment (i.e., 242.0 ± 8.7 g to 285.0 ± 10.4 g), but there were no differences in body weight between groups. Five of the 20 adjuvant-treated animals (25%) developed inflammation, as indicated by their clinical scores (Figure 1 in parenthesis), which generally increased over time. The mean time of inflammation onset (i.e. the first day of a clinical score > 0) was 12.8 ± 0.8 days.
Figure 1.
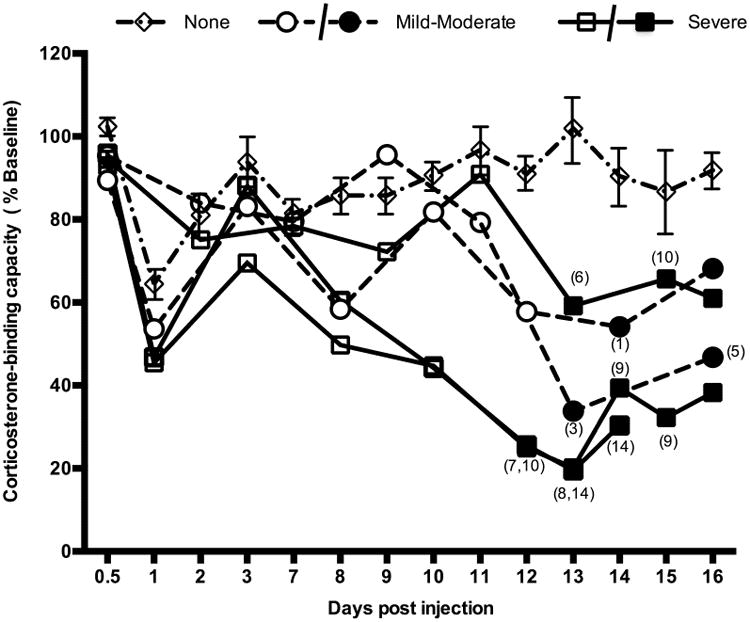
Time course of plasma CBG levels during inflammatory responses. Blood samples were collected from each animal prior to treatment (baseline values) and after Complete Freund's adjuvant injection, as indicated. A reduction in the corticosterone-binding capacity of serum CBG was observed 24 hrs post injection in all adjuvant-treated animals. Following this, rats that developed inflammation had large decreases in CBG levels, whereas the mean CBG levels in animals without clinical signs of inflammation (none, diamonds) were unchanged. Rats with mild-moderate inflammation (circles) displayed reduced CBG levels 12-13 days post-injection. Two of three rats with severe inflammation (squares) had reduced CBG levels as early as day 8, several days before clinical signs of inflammation were evident. Solid symbols denote the presence of clinical symptoms with the corresponding clinical score given in parentheses, while open symbols denote the absence of clinical signs of inflammation. Data for rats (n=15) without inflammation are presented as mean ± SEM for comparison.
Time-course of changes in plasma CBG levels
Adjuvant-treated rats had a 30-50% reduction in plasma CBG levels 24 hrs post injection, irrespective of whether or not they developed inflammation, and this decrease resolved by day 3 post-injection (Figure 1). This was not seen in the saline-treated control rats. When compared to baseline values, 40-80% decreases in CBG-corticosterone-binding capacity were subsequently observed over the experimental period in rats that developed inflammation (Figure 1), but mean plasma CBG levels were unchanged in saline-treated rats (not shown) or in adjuvant-treated rats that did not develop inflammation (Figure 1).
Overall, the magnitude of decline in plasma CBG levels matched the severity of inflammation, with animals that developed severe inflammation showing the largest decreases in CBG levels (Figure 1). In two severely inflamed rats, 50% reductions in plasma CBG were seen as early as day 8 post-injection, which was 4 days before any clinical signs of inflammation were evident. In these animals, CBG levels reached as low as 20% of baseline by 12-13 days post injection. The two rats with mild-moderate inflammation also had significant reductions (35% and 55% of baseline values at 13-14 days post-injection) in CBG levels. Total protein levels in plasma samples did not differ in rats over the course of the study in relation to the severity of inflammation (data not shown).
Plasma CBG and hepatic Cbg mRNA levels in relation to clinical score
Plasma CBG levels at termination declined in relation to clinical score (main effect of inflammation severity, P = 0.0001; Figure 2A). Rats with severe inflammation had significantly lower CBG levels when compared to saline-treated controls or adjuvant-treated rats that did not develop inflammation (P < 0.001). Significant differences were also seen in Cbg mRNA levels in livers collected on the day of termination (main effect of inflammation severity, P < 0.02; Figure 2B). When compared to saline-treated rats or adjuvant-treated rats that did not develop inflammation, post hoc tests revealed that animals with severe inflammation had lower Cbg mRNA levels (P < 0.05).
Figure 2.
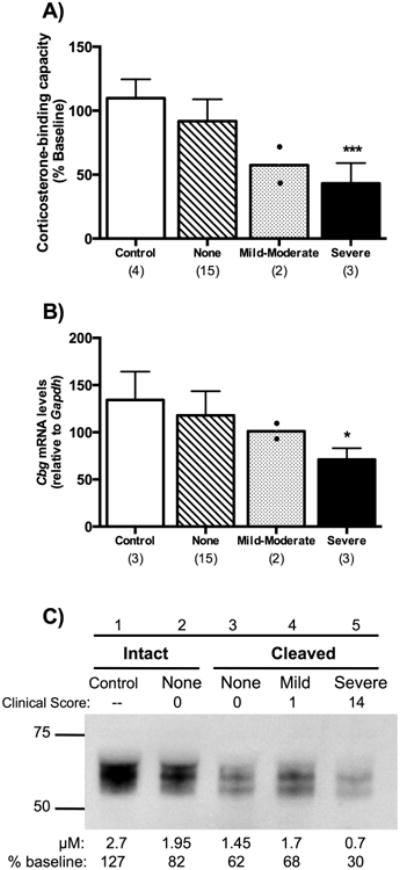
Plasma CBG (A) and liver Cbg mRNA (B) levels, and evidence of CBG proteolysis (C) in relation to clinical score (None, Mild-Moderate, or Severe). In A and B, the numbers of animals in each group are shown in parentheses, and rats (n=2) with mild-moderate inflammation were not included in statistical analyses. Rats with severe inflammation were compared to both saline-treated control animals and adjuvant-treated animals without clinincal signs of inflammation (None). (A) Plasma CBG levels decreased in relation to clinical score with a significant decrease in rats with severe inflammation [main effect of inflammation severity, F (2,19) = 14.85, P = 0.0001]. (B) Liver Cbg mRNA levels were significantly lower in animals with severe inflammation [main effect of inflammation severity, F(2,17) = 5.59, P < 0.02]. Data in A and B are presented as mean ± SD. Tukey's Post hoc: *P < 0.05; ***P < 0.001, (C) Representative western blot illustrating the proteolysis of plasma CBG, as indicated by an ∼5 kDa size reduction (lanes 3-5), in concert with reductions in CBG corticosterone-binding capacity (μM) when compared as a percentage of pretreatment values (shown below). Note that CBG proteolysis was also observed in some animals prior to clinical signs of inflammation (None, lane 3).
Western blotting was used to assess the integrity of CBG in plasma samples taken at termination. An ∼5 kDa reduction in the apparent molecular size of CBG was evident in rats with mild-moderate and severe inflammation (Figure 2C, lanes 4 and 5), consistent with cleavage of its RCL (Gardill, et al. 2012). This was also observed in some adjuvant-treated rats without clinical signs of inflammation (Figure 2C, lane 3), suggesting the presence of an underlying inflammatory state in those animals. As expected, this evidence of CBG proteolysis coincided with decreased plasma CBG levels as determined in the corticosterone-binding capacity assay (Figure 2C). Therefore, on the day of termination, decreases in the corticosterone-binding capacity of plasma CBG in animals with severe inflammation are associated with decreased Cbg mRNA levels, as well as evidence of proteolytic cleavage of CBG.
Plasma CBG and hepatic Cbg mRNA levels in relation to CBG proteolysis
Plasma samples taken at termination were classified into two groups depending on the integrity of CBG as assessed by western blotting: i.e., samples in which CBG proteolysis was either evident (cleaved) or not (intact), as illustrated in Figure 2C. Reductions in CBG levels were found in rats with evidence of cleaved CBG (Figure 3A), when compared to rats with intact CBG (P < 0.0001). When classifying samples in this way, no differences were found in corresponding liver Cbg mRNA levels (Figure 3B), suggesting that CBG proteolysis precedes changes in liver Cbg mRNA levels.
Figure 3.
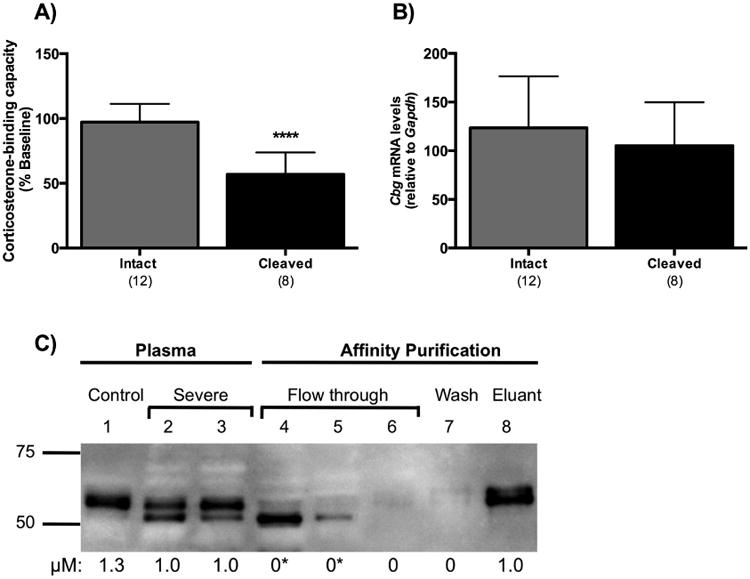
Proteolysis of CBG is associated with reduced plasma CBG levels without changes in liver Cbg mRNA levels, and evidence that cleaved CBG in plasma lacks steroid-binding activity. (A) Rats with cleaved CBG had significantly lower plasma CBG values than rats with intact CBG. (B) Liver Cbg mRNA levels were similar irrespective of CBG proteolysis status. In A and B, samples were grouped for analysis based on CBG integrity, as assessed by western blotting (see Figure 2C), and classified as being either intact or cleaved. The numbers of animals in each group are shown in parentheses, and data are presented as mean ± SD. **** P < 0.0001. (C) Plasma from rats with severe inflammation (lanes 2 and 3) were pooled and purified by steroid affinity chromatography. Cleaved CBG did not bind the steroid-affinity matrix and eluted in the flow through (lanes 4 and 5). Intact CBG bound to the steroid-affinity matrix and was eluted with buffer containing excess corticosterone (lane 8). CBG-corticosterone binding capacity values (μM) are shown under each lane. There was no detectable CBG steroid-binding activity (*) in the flow through fractions that contained cleaved CBG (lanes 4 and 5). Intact CBG (lane 8) exhibited full steroid-binding activity. An intact control (saline) sample is shown (lane 1) for comparison.
Next, we investigated whether cleaved CBG in plasma samples retained high-affinity corticosterone-binding activity. Plasma samples from rats with severe inflammation (Figure 3C, lanes 2 and 3) were pooled and CBG was captured by HACA-Sepharose-affinity column chromatography. Cleaved CBG, as indicated by an ∼5 kDa size reduction, failed to bind the affinity column and eluted in the flow through fractions (Figure 3C, lanes 4 and 5). Moreover, the flow through fractions containing the cleaved CBG had no detectable CBG-corticosterone binding activity. There was no immunoreactive CBG or corticosterone-binding activity in the wash fraction (Figure 3C, lane 7). Importantly, the CBG eluting from the affinity column using excess (200 μM) corticosterone (Figure 3C, lane 8) appeared to be intact and retained full corticosterone-binding activity.
Plasma cytokine biomarkers of inflammation and spleen weight
Pro- and anti-inflammatory cytokine levels were measured in plasma samples collected at termination (Figure 4 A-G). Measurements in samples from saline-treated control animals and adjuvant-treated animals were classified according to whether CBG was intact or cleaved (Figure 2C). No significant differences were found in plasma levels of IL-5 (Figure 4A). Trends for a main effect of CBG proteolysis were found for the anti-inflammatory cytokines IL-4 (Figure 4B, P = 0.051) and IL-10 (Figure 4C, P = 0.056), as well as the pro-inflammatory cytokine IFN-ɣ (Figure 4D, P = 0.057). Planned pairwise comparisons revealed significantly elevated IL-4, IL-10 and IFN-ɣ levels only in rats with cleaved CBG, when compared to saline-treated controls (P < 0.05).
Figure 4.
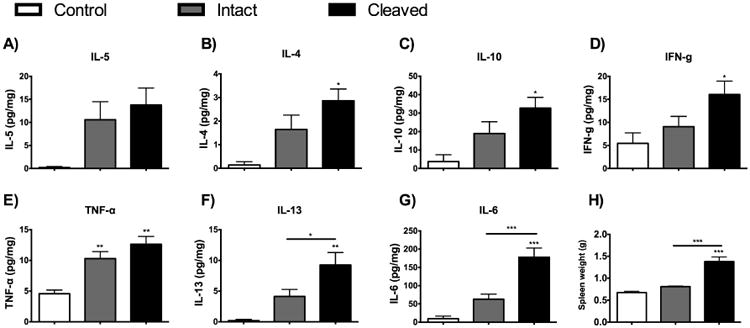
Associations between CBG proteolysis, plasma cytokine levels (A-G) and spleen weights (H). As in Figure 3, results from adjuvant-treated rats were grouped based on CBG proteolysis status (i.e., cleaved versus intact CBG). When compared to control saline-treated) rats, all plasma cytokines, apart from IL-5 (A), were significantly increased in rats with evidence of CBG proteolysis, as was spleen weight (H). TNF-α (E, main effect of CBG proteolysis, F(2,23) = 7.30, p < 0.004) showed a treatment effect with significantly increased levels in adjuvant-treated animals (intact and cleaved CBG) when compared to controls. Plasma levels of IL-13 (F, main effect of CBG proteolysis, F(2,23) = 6.36, p < 0.007), IL-6 (G, main effect of CBG proteolysis, F(2,23) = 16.50, p < 0.001) and spleen weight (H, main effect of CBG proteolysis, F(2,23) = 30.47, p < 0.001) were significantly increased in samples with cleaved CBG versus intact CBG. Cytokine levels were Blom transformed for statistical analysis; untransformed data (pg/ml) are presented as mean ± SEM. Control, n=4; Intact, n=12; Cleaved, n=8. Fischer's Post hoc: *P < 0.05; **P < 0.01; ***P < 0.001.
A significant main effect of CBG proteolysis was found for TNF-α (P < 0.004), IL-13 (P = 0.007) and IL-6 (P < 0.001), respectively. A treatment effect was found for the pro-inflammatory cytokine TNF-α, with post hoc tests revealing that adjuvant treated animals (both intact and cleaved CBG) had higher cytokine levels compared to control animals (Figure 4E, P < 0.01). A different pattern was observed for the anti-inflammatory cytokine IL-13 for which post hoc tests indicated that plasma levels were significantly increased in rats with cleaved CBG, when compared to saline-treated controls (P < 0.001) or rats with intact CBG (P < 0.05), respectively (Figure 4F). In terms of changes in plasma cytokine levels, the largest increases were seen for IL- 6 (Figure 4G), with post-hoc tests indicating significant increases in rats with cleaved CBG, as compared to the saline-treated controls or rats with intact CBG (P < 0.001). Similar to IL-6, spleen weight (Figure 4H) was very significantly associated with CBG proteolysis (main effect of CBG proteolysis, P < 0.001). Post-hoc tests also indicated that rats with cleaved CBG had significantly elevated spleen weights, when compared to the saline-treated controls or adjuvant treated rats with intact CBG (P < 0.001). Furthermore, there were strong negative correlations between plasma CBG and IL-6 levels (Figure 5A, r2=0.71) and between plasma CBG levels and spleen weights (Figure 5B, r2=0.73) but not between plasma CBG and IL-13 levels (r2= 0.22).
Figure 5.
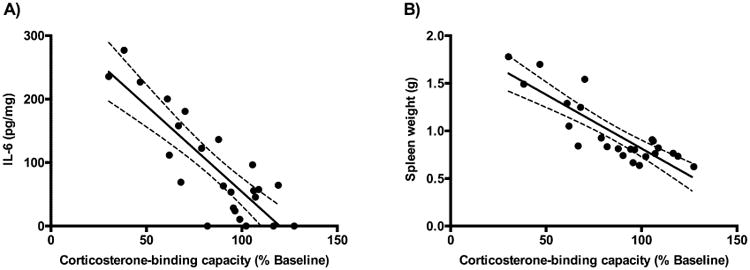
Strong negative correlations between plasma CBG and IL-6 levels and CBG and spleen weights. Data were analyzed using linear regression, with plasma CBG levels expressed as a % of pretreatment values. (A) Plasma CBG and IL-6 levels (r2 =0.71) and (B) plasma CBG and spleen weights (r2 =0.73), were negatively correlated. The best-fit linear regression line (solid) and 95% confidence interval (dotted) are also shown.
Discussion
Changes in plasma CBG levels occur during inflammation in humans, with very low levels reported during sepsis (Ho, et al. 2006), septic shock (Bendel, et al. 2008, Ho, et al. 2006, Pugeat, et al. 1989), burn injuries (Bernier, et al. 1998) and after open heart surgery (Tinnikov, et al. 1996). In addition, decreased CBG levels have been reported after thermal injuries to mice and rats (D'Elia, et al. 2005, Garrel, et al. 1993) and in pigs treated with lipopolysaccharide (Carroll, et al. 2003). These latter studies model acute, all-or-none, inflammation, while the adjuvant induced inflammation model we have used allows those rats that developed inflammation to be compared with those that did not. In doing so, it was possible to monitor temporal changes in the plasma levels of CBG and inflammatory markers, as inflammation developed at different rates and degrees of severity.
In adjuvant-treated rats, the corticosterone-binding capacity of plasma CBG decreased 30-50% at 24 hrs post-injection, irrespective of whether or not rats eventually developed inflammation. Based on previous reports (Billiau & Matthys. 2001), we suspect that an initial inflammatory response to the complete Freund's adjuvant is responsible for decreases in plasma CBG levels because this did not occur in saline-treated controls. However, this initial response was short-lived and CBG levels in adjuvant-treated animals returned to baseline by day 3 post-injection. Over the subsequent experimental period we found consistent decreases in plasma CBG levels of 40-80% in those animals that developed inflammation, and the magnitude of this decline aligned with the severity of inflammation, as indexed by clinical scores. Notably, marked (50%) decreases in plasma CBG levels occurred 4 days before any clinical symptoms of inflammation were evident. Such dynamic changes in CBG levels prior to or during inflammation are expected to modulate the availability of corticosterone to its target cells, thereby affecting the inflammatory reaction as well as the healing process. When considered together, these results suggest that CBG may be a useful biomarker of inflammation onset and severity.
As noted previously (Bodnar, et al. 2015), significant decreases in plasma CBG levels occurred in adjuvant-treated rats that developed severe inflammation, and we have now defined the mechanisms responsible for this. At day 14 or 16 post-adjuvant injection (termination), rats with severe inflammation had reduced plasma CBG levels and this was associated with an ∼5 kDa reduction in CBG molecular size by western blotting. Changes in the carbohydrate composition of plasma proteins have been reported to occur during acute inflammation, including decreases in core fucosylation (Rombouts, et al. 2016). However, small changes in the composition of the six N-linked oligosaccharides associated with rat CBG will not result in detectable differences in molecular size by western blotting. It is also known that the complete loss of N-glycosylation of human CBG at Asn238 causes a loss of steroid-binding activity (Avvakumov, et al. 1993), however there is no evidence that compositional changes in the N-linked glycosylation of CBG, in any species, adversely effects its steroid-binding activity. Differences in N-linked oligosaccharide composition will not account for the substantial reduction in the molecular size of CBG, or the complete loss of steroid-binding activity associated with immune-reactive CBG that does not interact with the steroid-affinity chromatography matrix, and we therefore conclude that these observations are the result of proteolytic cleavage.
It has been reported that cleaved CBG can be detected in human blood samples using ELISAs with highly specific monoclonal antibodies that discriminated between CBG with an intact versus cleaved RCL (Lewis & Elder. 2011), but direct evidence that RCL cleavage of CBG actually occurred in these samples is lacking. A size reduction of CBG consistent with RCL cleavage has never been observed in human blood samples, and it has been postulated that human CBG is rapidly removed from the blood circulation after RCL cleavage (Mast, et al. 1991). In rats, this does not seem to be the case and it appears that cleaved CBG is cleared more slowly from the circulation. However, the site of RCL cleavage and the protease responsible remain to be identified.
The steroid-binding activity of CBG is undetectable in rat plasma samples in which CBG appears to be have undergone proteolysis, and this is in line with a marked loss in cortisol-binding affinity observed when the RCL of human CBG is cleaved by neutrophil elastase (Hammond, et al. 1990), chymotrypsin (Simard, et al. 2015) or the bacterial proteinase, LasB (Simard, et al. 2014). However, our observations of in vivo rat CBG proteolysis under pathophysiological conditions contrasts with a previous report that E-coli produced rat CBG, mutated to allow for cleavage by human neutrophil elastase, only undergoes a 2-fold reduction in binding affinity (Gardill, et al. 2012). We attribute this discrepancy to the fact that E.coli expressed CBG is not glycosylated and has about a 10-fold lower affinity for corticosterone when compared to native CBG in rat blood samples (Gardill, et al. 2012). It is known that the N-glycosylation of human CBG is critically important for the formation of a high-affinity binding site (Avvakumov, et al. 1993, Avvakumov & Hammond. 1994), and the RCLs of human and rat CBGs both contain N-glycosylation sites, but in different locations (Hammond, et al. 1991). It is possible that N-glycosylation within the RCL of rat CBG influences how, and in what location, it is cleaved by a protease that allows for RCL insertion and the subsequent protein conformational rearrangement that disrupts the high affinity steroid-binding properties of CBG (Lin, et al. 2010). Unexpectedly, plasma CBG proteolysis was also evident in rats that developed mild-moderate inflammation, as well as in a subset of rats that did not display clinical signs of inflammation. However, significant reductions in plasma CBG were evident in rats where CBG proteolysis appears to have occurred, despite the fact that this did not coincide with a reduction in liver Cbg mRNA levels, which suggests that plasma CBG proteolysis occurs prior to changes in liver Cbg mRNA levels.
Plasma CBG production by the liver is down regulated by glucocorticoids (Smith & Hammond. 1992) and cytokines (Emptoz-Bonneton, et al. 1997). Although plasma corticosterone levels were not measured in this study, they were increased in our previous study using the same experimental protocol, with the highest levels found in animals with severe inflammation (Bodnar, et al. 2015). In this model, increased levels of IL-6, as seen here, and corticosterone, as seen previously (Bodnar, et al. 2015), are likely contributors to the significant decreases in liver Cbg mRNA levels that we have now observed in rats with severe inflammation. This is not entirely surprising given the presence of a cis-regulatory element for IL-6 in the rat Cbg promoter (Underhill & Hammond. 1995), and the established role of IL-6 in the acute phase response during inflammation (Fonseca, et al. 2009). This is also in line with inverse relationships between IL-6 and plasma CBG levels in humans (Bernier, et al. 1998, Tsigos, et al. 1998), and studies in human hepatoblastoma-derived (HepG2) cells where an IL-6 induced reduction in CBG production (Emptoz-Bonneton, et al. 1997) was associated with decreased Cbg mRNA stability (Bartalena, et al. 1993). Moreover, the strong negative correlation we observed between plasma CBG and IL-6 levels, further supports the proposition that IL-6 inhibits CBG production during inflammation. Together, our results suggest that the mechanisms responsible for decreases in plasma CBG levels during inflammation are multi-factorial and occur in a sequential manner. First, the RCL of CBG undergoes proteolysis rendering it essentially non-functional as a steroid binding protein, thereby amplifying free plasma corticosterone levels. Further decreases to plasma CBG levels are then caused by decreases in liver Cbg mRNA levels, mediated by increased IL-6 and corticosterone levels.
Activated immune cells produce a wide variety of cytokines and cytokine levels increase in the circulation during inflammation (Choy & Panayi. 2001, Ramadori & Armbrust. 2001). We have now found that animals that showed evidence of CBG proteolysis also had increased cytokine (IFN-γ, TFNα, IL-4, IL-6, IL-10 and IL-13) levels, in association with large increases in spleen weight, which is a recognized marker of inflammation. These changes in plasma cytokine levels are expected to act together with increases in corticosterone levels and bioavailability to repress the production of cytokines in an attempt to alleviate cytokine-mediated tissue damage in rats with cleaved CBG. Increases in the plasma levels of positive acute phase plasma proteins, including other SERPINA family members, such as alpha-1 antitrypsin (SERPINA1) that inhibits the activities of neutrophil elastase in humans, may have occurred in animals that developed inflammation. Although this may afford some protection to CBG proteolysis, there is little information about the roles of alpha-1 antitrypsin and other related SERPINAs, or their target proteases, in rats in relation to inflammation.
The fact that both plasma IL-6 and IL-13 levels are markedly elevated in animals with cleaved CBG, while only IL-6 levels were inversely correlated with plasma CBG levels, suggests that these cytokines function in different ways. In contrast to IL-6, the lack of any relationship between plasma CBG and IL-13 levels suggests that IL-13 does not influence CBG production or proteolysis. Moreover, the coincidence of increased cytokine levels and spleen weights in rats with cleaved CBG indicates an underlying inflammatory response, even in the absence of clinical symptoms. Although some rats with cleaved CBG did not display overt signs of inflammation over the experimental period, it is possible that these may have developed if the study had been extended. Nevertheless, these data indicate that evidence of CBG proteolysis in plasma samples is a potential pre-symptomatic biomarker of inflammation.
In conclusion, our time course study of CBG changes during inflammation demonstrates that significant decreases in the corticosterone-binding capacity of plasma CBG occur in rats that developed inflammation, with the magnitude of the decline matching the clinical severity. Notably, large decreases in plasma CBG levels occur prior to clinical signs of inflammation. At termination, significant decreases in the corticosterone-binding capacity of CBG appear to coincide with CBG proteolysis that causes a loss of its steroid-binding activity. Significant increases in pro- and anti-inflammatory plasma cytokine levels, as well as increased spleen weights, were all associated with evidence of CBG proteolysis. These novel findings suggest that CBG proteolysis is a marker of active inflammation and, perhaps even more importantly, is a prognostic indicator of inflammation onset. In addition, the fact that rats with evidence of plasma CBG proteolysis had similar liver Cbg mRNA levels to rats with intact CBG, suggests that cleavage of plasma CBG occurs before any reductions in hepatic CBG production occur. Overall, our data suggest that changes in CBG, particularly CBG proteolysis, are an early, pre symptomatic, marker of inflammation and a useful biomarker of inflammation onset and severity.
Acknowledgments
The authors wish to thank Dr. Timothy Kieffer for generously providing access to the QuickPlex, and Travis Webber for his assistance. We also thank Wayne Yu and Linda Ellis for their assistance with animal procedures.
Funding: This work was supported by: the Canadian Institutes of Health Research (Operating Grant MOP-111102) to G.L.H.; NIH/NIAAA (RO1 AA022460 and R37 AA007789) and NeuroDevNet (Canadian Networks of Centres of Excellence) to JW; and the Natural Sciences and Engineering Research Council of Canada (CGS-D award) to TSB. GLH holds a Tier 1 Canada Research Chair in Reproductive Health.
Footnotes
Declaration of Interest: The authors have declare that they have no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Author contributions: LAH and TSB carried out the experiments and analyzed the data. LAH and GLH wrote the manuscript. All authors designed the study, had final approval of the submitted version of the manuscript and have read and agreed with the manuscript written.
References
- Avvakumov GV, Hammond GL. Glycosylation of human corticosteroid-binding globulin. differential processing and significance of carbohydrate chains at individual sites. Biochemistry. 1994;33:5759–5765. doi: 10.1021/bi00185a012. [DOI] [PubMed] [Google Scholar]
- Avvakumov GV, Warmels-Rodenhiser S, Hammond GL. Glycosylation of human corticosteroid-binding globulin at aspargine 238 is necessary for steroid binding. The Journal of biological chemistry. 1993;268:862–866. [PubMed] [Google Scholar]
- Bartalena L, Hammond GL, Farsetti A, Flink IL, Robbins J. Interleukin-6 inhibits corticosteroid-binding globulin synthesis by human hepatoblastoma-derived (hep G2) cells. Endocrinology. 1993;133:291–296. doi: 10.1210/endo.133.1.8391424. [DOI] [PubMed] [Google Scholar]
- Bendel S, Karlsson S, Pettila V, Loisa P, Varpula M, Ruokonen E Finnsepsis Study Group. Free cortisol in sepsis and septic shock. Anesthesia and Analgesia. 2008;106:1813–1819. doi: 10.1213/ane.0b013e318172fdba. [DOI] [PubMed] [Google Scholar]
- Bernier J, Jobin N, Emptoz-Bonneton A, Pugeat MM, Garrel DR. Decreased corticosteroid-binding globulin in burn patients: Relationship with interleukin-6 and fat in nutritional support. Critical care medicine. 1998;26:452–460. doi: 10.1097/00003246-199803000-00014. [DOI] [PubMed] [Google Scholar]
- Billiau A, Matthys P. Modes of action of freund's adjuvants in experimental models of autoimmune diseases. Journal of leukocyte biology. 2001;70:849–860. [PubMed] [Google Scholar]
- Bodnar TS, Hill LA, Taves MD, Yu W, Soma KK, Hammond GL, Weinberg J. Colony-specific differences in endocrine and immune responses to an inflammatory challenge in female sprague dawley rats. Endocrinology. 2015;156:4604–4617. doi: 10.1210/en.2015-1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brattsand R, Linden M. Cytokine modulation by glucocorticoids: Mechanisms and actions in cellular studies. Alimentary Pharmacology & Therapeutics. 1996;10 Suppl 2:81–90. doi: 10.1046/j.1365-2036.1996.22164025.x. discussion 91-2. [DOI] [PubMed] [Google Scholar]
- Carroll JA, Gaines AM, Spencer JD, Allee GL, Kattesh HG, Roberts MP, Zannelli ME. Effect of menhaden fish oil supplementation and lipopolysaccharide exposure on nursery pigs. I. effects on the immune axis when fed diets containing spray-dried plasma. Domestic animal endocrinology. 2003;24:341–351. doi: 10.1016/s0739-7240(03)00017-1. [DOI] [PubMed] [Google Scholar]
- Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. The New England journal of medicine. 2001;344:907–916. doi: 10.1056/NEJM200103223441207. [DOI] [PubMed] [Google Scholar]
- D'Elia M, Patenaude J, Hamelin C, Garrel DR, Bernier J. Corticosterone binding globulin regulation and thymus changes after thermal injury in mice. American journal of physiology Endocrinology and metabolism. 2005;288:E852–60. doi: 10.1152/ajpendo.00407.2004. [DOI] [PubMed] [Google Scholar]
- Emptoz-Bonneton A, Crave JC, LeJeune H, Brebant C, Pugeat M. Corticosteroid-binding globulin synthesis regulation by cytokines and glucocorticoids in human hepatoblastoma-derived (HepG2) cells. The Journal of clinical endocrinology and metabolism. 1997;82:3758–3762. doi: 10.1210/jcem.82.11.4362. [DOI] [PubMed] [Google Scholar]
- Fonseca JE, Santos MJ, Canhao H, Choy E. Interleukin-6 as a key player in systemic inflammation and joint destruction. Autoimmunity reviews. 2009;8:538–542. doi: 10.1016/j.autrev.2009.01.012. [DOI] [PubMed] [Google Scholar]
- Gagliardi L, Ho JT, Torpy DJ. Corticosteroid-binding globulin: The clinical significance of altered levels and heritable mutations. Molecular and cellular endocrinology. 2010;316:24–34. doi: 10.1016/j.mce.2009.07.015. [DOI] [PubMed] [Google Scholar]
- Gardill BR, Vogl MR, Lin HY, Hammond GL, Muller YA. Corticosteroid-binding globulin: Structure-function implications from species differences. PloS one. 2012;7:e52759. doi: 10.1371/journal.pone.0052759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrel DR, Zhang L, Zhao XF, Hammond GL. Effect of burn injury on corticosteroid-binding globulin levels in plasma and wound fluid. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society. 1993;1:10–14. doi: 10.1046/j.1524-475X.1993.10105.x. [DOI] [PubMed] [Google Scholar]
- Hammond GL. Molecular properties of corticosteroid binding globulin and the sex-steroid binding proteins. Endocrine reviews. 1990;11:65–79. doi: 10.1210/edrv-11-1-65. [DOI] [PubMed] [Google Scholar]
- Hammond GL, Lahteenmaki PL. A versatile method for the determination of serum cortisol binding globulin and sex hormone binding globulin binding capacities. Clinica chimica acta; international journal of clinical chemistry. 1983;132:101–110. doi: 10.1016/0009-8981(83)90237-1. [DOI] [PubMed] [Google Scholar]
- Hammond GL, Smith CL, Underhill DA. Molecular studies of corticosteroid binding globulin structure, biosynthesis and function. The Journal of steroid biochemistry and molecular biology. 1991;40:755–762. doi: 10.1016/0960-0760(91)90300-t. [DOI] [PubMed] [Google Scholar]
- Hammond GL, Smith CL, Paterson NA, Sibbald WJ. A role for corticosteroid-binding globulin in delivery of cortisol to activated neutrophils. The Journal of clinical endocrinology and metabolism. 1990;71:34–39. doi: 10.1210/jcem-71-1-34. [DOI] [PubMed] [Google Scholar]
- Ho JT, Al-Musalhi H, Chapman MJ, Quach T, Thomas PD, Bagley CJ, Lewis JG, Torpy DJ. Septic shock and sepsis: A comparison of total and free plasma cortisol levels. The Journal of clinical endocrinology and metabolism. 2006;91:105–114. doi: 10.1210/jc.2005-0265. [DOI] [PubMed] [Google Scholar]
- Lewis JG, Elder PA. The reactive centre loop of corticosteroid-binding globulin (CBG) is a protease target for cortisol release. Molecular and cellular endocrinology. 2014;384:96–101. doi: 10.1016/j.mce.2014.01.005. [DOI] [PubMed] [Google Scholar]
- Lewis JG, Elder PA. Corticosteroid-binding globulin reactive centre loop antibodies recognise only the intact natured protein: Elastase cleaved and uncleaved CBG may coexist in circulation. The Journal of steroid biochemistry and molecular biology. 2011;127:289–294. doi: 10.1016/j.jsbmb.2011.08.006. [DOI] [PubMed] [Google Scholar]
- Libert C, Wielockx B, Hammond GL, Brouckaert P, Fiers W, Elliott RW. Identification of a locus on distal mouse chromosome 12 that controls resistance to tumor necrosis factor-induced lethal shock. Genomics. 1999;55:284–289. doi: 10.1006/geno.1998.5677. [DOI] [PubMed] [Google Scholar]
- Lin HY, Muller YA, Hammond GL. Molecular and structural basis of steroid hormone binding and release from corticosteroid-binding globulin. Molecular and cellular endocrinology. 2010;316:3–12. doi: 10.1016/j.mce.2009.06.015. [DOI] [PubMed] [Google Scholar]
- Mast AE, Enghild JJ, Pizzo SV, Salvesen G. Analysis of the plasma elimination kinetics and conformational stabilities of native, proteinase-complexed, and reactive site cleaved serpins: Comparison of alpha 1-proteinase inhibitor, alpha 1-antichymotrypsin, antithrombin III, alpha 2-antiplasmin, angiotensinogen, and ovalbumin. Biochemistry. 1991;30:1723–1730. doi: 10.1021/bi00220a039. [DOI] [PubMed] [Google Scholar]
- Perogamvros I, Ray DW, Trainer PJ. Regulation of cortisol bioavailability--effects on hormone measurement and action. Nature reviews Endocrinology. 2012;8:717–727. doi: 10.1038/nrendo.2012.134. [DOI] [PubMed] [Google Scholar]
- Petersen HH, Andreassen TK, Breiderhoff T, Brasen JH, Schulz H, Gross V, Grone HJ, Nykjaer A, Willnow TE. Hyporesponsiveness to glucocorticoids in mice genetically deficient for the corticosteroid binding globulin. Molecular and cellular biology. 2006;26:7236–7245. doi: 10.1128/MCB.00400-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugeat M, Bonneton A, Perrot D, Rocle-Nicolas B, Lejeune H, Grenot C, Dechaud H, Brebant C, Motin J, Cuilleron CY. Decreased immunoreactivity and binding activity of corticosteroid-binding globulin in serum in septic shock. Clinical chemistry. 1989;35:1675–1679. [PubMed] [Google Scholar]
- Ramadori G, Armbrust T. Cytokines in the liver. European journal of gastroenterology & hepatology. 2001;13:777–784. doi: 10.1097/00042737-200107000-00004. [DOI] [PubMed] [Google Scholar]
- Rombouts Y, Jonasdottis HS, Hipgrave Ederveen AL, Reidine KR, Jansen BC, Freysdottir J, Hardardottir I, Ioan-Facsinay A, Giera M, Wuhrer M. Acute phase inflammation is characterized by rapid changes in plasma/peritoneal fluid N-glycosylation in mice. Glycoconjugate Journal. 2016 doi: 10.1007/s10719-015-9648-9. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? integrating permissive, suppressive, stimulatory, and preparative actions. Endocrine reviews. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- Scrocchi LA, Hearn SA, Han VK, Hammond GL. Corticosteroid-binding globulin biosynthesis in the mouse liver and kidney during postnatal development. Endocrinology. 1993;132:910–916. doi: 10.1210/endo.132.2.8425503. [DOI] [PubMed] [Google Scholar]
- Seralini GE, Underhill CM, Smith CL, Nguyen VT, Hammond GL. Biological half-life and transfer of maternal corticosteroid-binding globulin to amniotic fluid in the rabbit. Endocrinology. 1989;125:1321–1325. doi: 10.1210/endo-125-3-1321. [DOI] [PubMed] [Google Scholar]
- Simard M, Hill LA, Lewis JG, Hammond GL. Naturally occurring mutations of human corticosteroid-binding globulin. The Journal of clinical endocrinology and metabolism. 2015;100:E129–39. doi: 10.1210/jc.2014-3130. [DOI] [PubMed] [Google Scholar]
- Simard M, Hill LA, Underhill CM, Keller BO, Villanueva I, Hancock RE, Hammond GL. Pseudomonas aeruginosa elastase disrupts the cortisol-binding activity of corticosteroid-binding globulin. Endocrinology. 2014;155:2900–2908. doi: 10.1210/en.2014-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon HU. Neutrophil apoptosis pathways and their modifications in inflammation. Immunological reviews. 2003;193:101–110. doi: 10.1034/j.1600-065x.2003.00038.x. [DOI] [PubMed] [Google Scholar]
- Smith CL, Hammond GL. Hormonal regulation of corticosteroid-binding globulin biosynthesis in the male rat. Endocrinology. 1992;130:2245–2251. doi: 10.1210/endo.130.4.1547738. [DOI] [PubMed] [Google Scholar]
- Tinnikov AA, Legan MV, Sheveluk NA, Cvetovskaya GA, Naumenko SE, Sidelnikov SG. Corticosteroid and immune responses to cardiac surgery. Steroids. 1996;61:411–415. doi: 10.1016/0039-128x(96)00060-8. [DOI] [PubMed] [Google Scholar]
- Tsigos C, Kyrou I, Chrousos GP, Papanicolaou DA. Prolonged suppression of corticosteroid-binding globulin by recombinant human interleukin-6 in man. The Journal of clinical endocrinology and metabolism. 1998;83:3379. doi: 10.1210/jcem.83.9.5100-5. [DOI] [PubMed] [Google Scholar]
- Turnbull AV, Rivier CL. Sprague-dawley rats obtained from different vendors exhibit distinct adrenocorticotropin responses to inflammatory stimuli. Neuroendocrinology. 1999;70:186–195. doi: 10.1159/000054475. [DOI] [PubMed] [Google Scholar]
- Underhill DA, Hammond GL. Cis-regulatory elements within the proximal promoter of the rat gene encoding corticosteroid-binding globulin. Gene. 1995;162:205–211. doi: 10.1016/0378-1119(95)00337-6. [DOI] [PubMed] [Google Scholar]
- Zhang X, Lan N, Bach P, Nordstokke D, Yu W, Ellis L, Meadows GG, Weinberg J. Prenatal alcohol exposure alters the course and severity of adjuvant-induced arthritis in female rats. Brain, behavior, and immunity. 2012;26:439–450. doi: 10.1016/j.bbi.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]