

The present study found an important role for collagen content, but not collagen cross-linking, in the pulsatile right ventricular (RV) afterload, which is correlated with RV hypertrophy. These results uncover a new collagen-mediated mechanical mechanism of RV dysfunction in early pulmonary hypertension progression. Furthermore, our results suggest that measures and metrics of pulsatile hemodynamics such as pulse pressure and pulse wave velocity are potentially important to cardiovascular mortality in patients with pulmonary hypertension.
Keywords: collagen, pulmonary vascular impedance, pulmonary artery remodeling, hypoxic pulmonary hypertension
Abstract
Hypoxic pulmonary hypertension (HPH) is associated with pulmonary artery (PA) remodeling and right ventricular (RV) overload. We have previously uncovered collagen-mediated mechanisms of proximal PA stiffening in early HPH by manipulating collagen degradation and cross-linking using a transgenic mouse strain and a potent collagen cross-link inhibitor, β-aminopropionitrile (BAPN). However, the roles of collagen in distal PA remodeling, overall RV afterload, and RV hypertrophy in HPH remain unknown. Here, we used the same experimental strategy to investigate the effect of pulmonary vascular collagen content and cross-linking on steady and pulsatile RV afterload and on RV hypertrophy in early HPH. Collagenase-resistant mice (Col1a1R/R) and their littermate controls (Col1a1+/+) were exposed to normobaric hypoxia for 10 days with or without BAPN treatment. In vivo pulmonary vascular impedance, a comprehensive measure of RV afterload, was measured via simultaneous RV catheterization and echocardiography. Morphology and collagen accumulation were examined using histological techniques and ELISA in lungs and RVs. In both mouse strains, BAPN did not limit increases in pulmonary arterial pressure or pulmonary vascular resistance, indicating a negligible effect of either collagen content or cross-linking on steady RV afterload. However, BAPN prevented the increase in pulse pressure and RV hypertrophy in Col1a1+/+ mice and these effects were absent in Col1a1R/R mice, suggesting a role for PA collagen content, not cross-linking, in the pulsatile RV afterload. Moreover, we found a significant correlation between pulse pressure and RV hypertrophy, indicating an important role for pulsatile RV afterload in RV overload in early HPH.
NEW & NOTEWORTHY The present study found an important role for collagen content, but not collagen cross-linking, in the pulsatile right ventricular (RV) afterload, which is correlated with RV hypertrophy. These results uncover a new collagen-mediated mechanical mechanism of RV dysfunction in early pulmonary hypertension progression. Furthermore, our results suggest that measures and metrics of pulsatile hemodynamics such as pulse pressure and pulse wave velocity are potentially important to cardiovascular mortality in patients with pulmonary hypertension.
hypoxic pulmonary hypertension (HPH) occurs in individuals who live at high altitudes and in patients with lung diseases such as chronic obstructive pulmonary disease (1), cystic fibrosis (7), and sleep apnea (8), and contributes significantly to morbidity and mortality. The devastating consequence of persistently high pulmonary pressure is right ventricular (RV) overload, and ultimately, RV failure. HPH often begins with narrowing of the distal, small pulmonary arteries (PAs) and soon causes biological, mechanical, and hemodynamic changes throughout the pulmonary vasculature (28), which are important contributors to RV overload that can progress into RV failure (37).
Collagen, a major component of the extracellular matrix, markedly accumulates in the PAs in chronic HPH and is a key factor in large, proximal PA stiffening (4, 12, 13, 19, 21, 33, 38, 39). Collagen accumulation is also found in distal PAs (6, 11, 20, 27), although its roles in distal PA mechanics and consequent RV afterload changes remain unclear. We have previously examined the differential contributions of collagen content and cross-linking to extralobar PA mechanical properties using the combination of a transgenic mouse strain (Col1a1), which has a defect in type I collagen degradation, and an antifibrotic drug (β-aminopropionitrile, BAPN), which prevents new cross-link formation (38, 39) (Fig. 1). We found that with 10 days of hypoxia exposure, BAPN treatment limited the PA collagen cross-linking and stiffening in both mouse strains, but only in the wild-type mice did BAPN limit the PA collagen content increase. Therefore, we concluded that collagen cross-linking, not collagen content, is responsible for proximal PA stiffening in early HPH (39). However, how collagen content and cross-linking affect distal PA narrowing, overall RV afterload, and RV hypertrophy in this experimental model of HPH is unknown. We hypothesized that PA collagen cross-linking, not content, contributes to increases in steady and pulsatile RV afterload in early HPH.
Fig. 1.
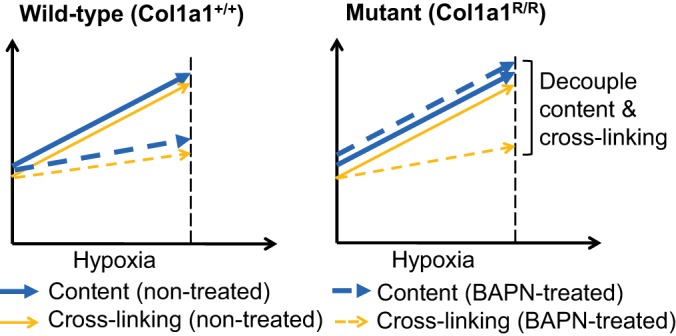
Schematic of the experimental design to decouple pulmonary artery collagen content and cross-linking using a Col1a1 mouse strain and β-aminopropionitrile (BAPN) treatment.
RV afterload can be comprehensively quantified by the pulmonary vascular impedance (PVZ), which is obtained from pressure-flow relationships ex vivo or in vivo (9, 18, 34, 37). The steady component of impedance that represents the opposition to steady flow (i.e., the resistance) is Z0; the impedance that represents the opposition to pulsatile flow (i.e., the characteristic impedance) is ZC; and the degree of pulse wave reflections can be represented by the index of wave reflection, Pb/Pf, or pulse wave velocity (PWV) (16). How these steady and pulsatile hemodynamic components contribute to RV afterload is not fully understood.
In the present study, using identically treated animal groups as we did previously in extralobar PA studies (38, 39), we investigated the changes in PVZ with decoupled PA collagen content and cross-linking (Fig. 1) using techniques established by our group (27, 31, 36). We found that BAPN did not prevent hypoxia-induced increases in steady RV afterload in either mouse strain, suggesting that collagen content and cross-linking played negligible roles in distal PA narrowing. However, BAPN limited the pulsatile RV afterload increase only in wild-type mice, suggesting that PA collagen content has an important effect on the pulsatile hemodynamic component. Furthermore, the correlation between pulsatile RV afterload and RV hypertrophy indicates that pulsatile RV afterload is a key factor in RV overload in early HPH.
MATERIALS AND METHODS
Animal handling.
All procedures were approved by the University of Wisconsin–Madison Institutional Animal Care and Use Committee. Breeding pairs of Col1a1tmJae mice on a B6/129 background were obtained from The Jackson Laboratory (Bar Harbor, ME). Genotyping by PCR was performed as described previously. Col1a1R/R mice and littermate controls (Col1a1+/+), both 16–18 wk old, were randomized into three groups: normoxia, 10 days of hypoxia with or without BAPN treatment (400 mg·kg−1·day−1, ip injection; Sigma-Aldrich). Both female and male mice were included in each group. During hypoxia, animals were maintained in a normobaric hypoxic chamber with a controlled O2 concentration of 10% with 4 l/min airflow to maintain the CO2 level at <600 parts per million. The chambers were opened for less than 30 min at a time for regular animal care or BAPN injections. Details that describe animal housing and handling were reported previously (19).
C57BL6 male mice (age, 12 wk) obtained from The Jackson Laboratory were housed in normal room air for up to 8 wk with and without BAPN drinking water at the same dose (400 mg·kg−1·day−1) as a control for the effects of BAPN alone.
In vivo hemodynamic measurements.
In vivo pressure and flow measurements were performed as previously reported (27, 31). Mice were anesthetized with an injection of urethane solution (2 mg/g body wt ip), intubated, and placed supine on a ventilator (Harvard Apparatus, Holliston, MA) using a tidal volume of ~225 ml and respiratory rate of ~200 breaths/min. A central midline skin incision was made, and the chest was carefully removed to expose the right ventricle. To confirm the absence of systemic hypertension, the right carotid artery was cannulated with a 1.2-Fr catheter-tip pressure transducer (Millar Instruments, Houston, TX) and advanced into the ascending aorta. Hydroxyethyl-starch was used to restore vascular volume due to blood loss.
Subsequently, the apex of the right ventricle was localized and a 1.0-Fr pressure-tip catheter (Scisense, London, ON, Canada) was introduced using a 20-gauge needle. After instrumentation was established and pressure was stabilized, the catheter was advanced to the main pulmonary artery (MPA) for pressure measurement. The pressure tracing was recorded at 5 kHz on a hemodynamic analysis workstation (Cardiovascular Engineering, Norwood, MA). The blood flow velocity and MPA inner diameter (MPA ID) measurements were performed via ultrasound (Visualsonics, Toronto, ON, Canada) with a 40-MHz probe during catheterization and recorded with the same hemodynamic analysis workstation. The probe was positioned to image the MPA just distal to the pulmonary valve in a right parasternal long-axis orientation in the same location as the catheter. The probe was angled until the maximal velocity signal was obtained. Measurement at this point allowed for better detection of the MPA ID. Measurement of the MPA ID was taken from leading edge to leading edge during end systole from three different cardiac cycles; we report the average of those three. Pulmonary arterial flow velocity was calculated by spectral analysis of the digitized broadband Doppler audio signal (typically represented as an image on ultrasound machines), which is an output of the hemodynamic analysis workstation. The spectral envelope was traced to provide a signal-averaged blood flow velocity waveform. We then used MPA ID to convert the instantaneous peak centerline flow velocity signal to instantaneous volume flow rate (Q), assuming a circular cross-section and a blunt velocity profile. Both the flow rate and pressure waveforms were signal-averaged using the ECG as a fiducial point, and then processed and analyzed using custom software (Cardiovascular Engineering). Twenty consecutive cardiac cycles free of extra systolic beats were selected and averaged.
Pressure and flow waveforms were obtained with mice ventilated with room air. After all hemodynamic measurements were completed, a sample of blood was extracted to measure hematocrit. RV hypertrophy was quantified by 1) the tissue weight ratio of the right ventricle to the left ventricle and septum (LV+S) (i.e., the Fulton index) and 2) the weight ratio of right ventricle to body weight (BW).
In vivo hemodynamic analysis – time domain analysis.
PVZ was calculated using wave intensity analysis as previously described (14). Total pulmonary vascular resistance (or Z0) was calculated as mean PA pressure divided by mean flow rate (i.e., cardiac output, CO), where CO was calculated as the time integral of Q over the cardiac cycle. Characteristic impedance (ZC) was calculated from the ratio of the change in pressure to the change in flow in early ejection. That is,
where dP and dQ are taken before when Q reaches 95% of its maximum value, assuming the system is free from wave reflections.
Next, total pulmonary arterial compliance was calculated by the following two methods: 1) C = stroke volume/pulse pressure (SV/PP), and 2) Ca = the time constant from an exponential fit of the diastolic part of the pressure waveform (29). To contrast this global compliance change with the local compliance change measured previously on extralobar PAs isolated and tested ex vivo (39), we calculated volumetric compliance of proximal PAs by ΔV/ΔP, which is the volume change assuming a unit length [1 mm; so ΔV = ΔA × 1 mm and A = π/4 (OD)2] for a comparable pressure change (i.e., ΔP = ~systolic PA pressure – ~diastolic PA pressure) for each group.
To examine the dynamic hemodynamic component involved during the arterial remodeling, we calculated PP as the pressure difference between systolic pressure and diastolic pressure, and PWV from ZC as
(41) assuming the density of blood, ρ = 1,060 kg/m3, and cross-sectional area, A = π/4 (MPA ID)2.
The in vivo pressure and flow measurement and analysis methods described above for mice have been well established within our group of researchers (27, 31).
PVZ was also calculated by the traditional frequency domain method, which also has been established by our group (34, 36). Briefly, PVZ was calculated as the ratio of the Fourier transformed pressure to flow at each harmonic (18):
Z0 was obtained as the magnitude of PVZ at 0 Hz; ZC was obtained by averaging Z from the 4th to 10th harmonics (22).
Ex vivo pulmonary vascular resistance.
Because we observed different CO between the BAPN-treated and nontreated hypoxia groups in wild-type mice and because the in vivo measurement of Z0 (or PVR) depends on CO, we added additional groups of Col1a1+/+ mice (control, hypoxia, and hypoxia+BAPN) and performed ex vivo PVR measurements with a controlled, constant flow rate (Q) to confirm the changes in PVR after hypoxia exposure with and without BAPN treatment. The methodology of ex vivo lung isolation, ventilation, and perfusion is well established in our group and has been described previously (34, 35).
In vivo RV pressure-volume measurement.
To examine the effect of BAPN treatment alone on the right ventricle, we measured RV function as described previously (40). Briefly, the mice were anesthetized with urethane solution (1,000–1,200 mg/kg body wt ip), intubated, and placed on a ventilator (Harvard Apparatus) using a tidal volume of ~225 µl and respiratory rate of ~125 breaths/min. A ventral midline skin incision was made and the chest wall and lungs were carefully retracted to expose the right ventricle. The left carotid artery was cannulated with a 1.2-Fr catheter pressure transducer (Scisense, London, ON) and advanced into the ascending aorta to measure systemic blood pressure. Subsequently, a 1.4-Fr admittance pressure-volume catheter (Scisense, Ithaca, NY) was inserted from the apex of the RV for pressure-volume (PV) recordings. After instrumentation was established and initial RV PV measurements were obtained, the inferior vena cava was isolated and briefly occluded to obtain alterations in venous return for determination of end-systolic and end-diastolic pressure relations. This vena cava occlusion was performed at least three times.
The magnitude and phase of the electrical admittance as well as the RV pressure were continuously recorded at 1,000 Hz and analyzed on commercially available software (Notocord Systems, Croissy Sur Seine, France) as described previously (30).
Histological examination.
Right ventricle, aorta, and left lungs were harvested and saved for histological examination by fixing in 10% formalin for ≥48 h and preserving in 70% ethanol. The tissues were then embedded, sectioned, and stained with hematoxylin and eosin for aorta and cardiomyocyte morphology measurement and with PicroSirius Red to identify collagen in right ventricle and lungs. Sections were imaged on an inverted microscope (TE-2000; Nikon, Melville, NY) and captured using a Spot camera and software for image capture and analysis (Meta Vue; Optical Analysis Systems, Nashua, NH). The cardiomyocyte (aorta) size was measured by the cell area (outside diameter, OD) in cross-section. For right ventricle tissues, 5–10 regions were selected for each sample and then the average myocyte cross-section area was calculated. The RV fibrosis score was calculated as the (collagen area/total tissue area) × 100 using color thresh-holding under brightfield light. In lungs, using polarized light, the area positive for collagen isoforms type I and type III was identified by color thresh-holding. Specifically, areas of green (G), orange (O), and yellow (Y) in the cross-section of each vessel ring were measured as area percentage of the total image. Then, percentages of type I and type III collagen were calculated by the combined area of O+Y and the area of G, respectively (39).
We also measured the degree of distal PA narrowing in PicroSirius Red-stained sections by quantifying the ratio of vessel wall thickness (WT) to OD (2 × WT/OD × 100%) for each sample. This method has been used as an assessment of PA muscularization previously (32). Only transversally cut PAs were included in the measurement.
All histological analyses were performed by individual observers who were blinded to the condition/group of the histology slides during the quantification.
Type I collagen concentration.
After the in vivo hemodynamic measurements, right lungs were snap-frozen in liquid nitrogen and saved for type I collagen ELISA assay. Briefly, mouse lung tissues are solubilized by the pretreatment with 0.05 M acetic acid and digested with pepsin and elastase overnight two or three times. Supernatants were then collected for the type I ELISA assay. Details of ELISA assay are provided by the company (Mouse Type I Collagen Detection Kit; Chondrex, Redmond, WA). The results are presented as the concentration of collagen (µg/ml) normalized by wet tissue weight (µg·ml−1·mg−1).
Statistical analyses.
All results are presented as means ± SE. For each mouse genotype, comparisons between exposure groups were performed by using one-way ANOVA or a Student's t-test. The Tukey’s honestly significant difference method was used to control the type I error for the pairwise comparisons between exposure groups. Data analysis was conducted using R software version 2.5.1 (R Foundation for Statistical Computing) or Microsoft Excel.
The nonparametric Spearman rank correlation test with a permutation analysis (permutation size M = 10,000) was used to compute the Spearman’s correlation coefficient (rs) and P values for the correlation between the averages of PP and PWV for both strains combined using SAS version 9.2. To assess linearity, a simple linear correlation coefficient (R2) was determined for the averages of PP or PWV for both strains combined using Microsoft Excel. Finally, a Pearson correlation test was performed between PP and RV hypertrophy index for all experimental animals using Microsoft Excel.
All P values were two-sided and P < 0.05 was considered statistically significant (in figures, *vs. normoxia and †vs. hypoxia).
RESULTS
HPH progression and RV hypertrophy.
In both wild-type (Col1a1+/+) and mutant (Col1a1R/R) mice, chronic hypoxia led to an increase in the mean pulmonary arterial pressure (mPAP) as expected; the elevated pressure persisted with BAPN treatment (P < 0.05, Fig. 2, left). The same trend was found for PA systolic and diastolic pressures of both genotypes (P < 0.05, Table 1).
Fig. 2.

Development of hypoxic pulmonary hypertension (HPH) evidenced by mean pulmonary arterial pressure (mPAP) (left) and Fulton index (right) in experimental groups. Results are presented as means ± SE. Col1a1+/+ mice, n = 8 for normoxia and hypoxia+BAPN groups, n = 6 for the hypoxia group; Col1a1R/R mice, n = 8 for normoxia, hypoxia, and hypoxia+BAPN groups. *P < 0.05 vs. normoxia, †P < 0.05 vs. hypoxia.
Table 1.
Biological, morphometric, and hemodynamic changes in six experimental groups
| Strain | Experimental Group | BW | Hct | dPAP | sPAP | MPA ID | RV/BW | CO | C | Ca |
|---|---|---|---|---|---|---|---|---|---|---|
| Col1a1+/+ | Normoxia | 26 ± 3 | 44 ± 2 | 8 ± 1 | 20 ± 1 | 1.3 ± 0.3 | 0.11 ± 0.01 | 11 ± 1 | 1.8 ± 0.2 | 1.9 ± 0.3 |
| Hypoxia | 26 ± 3 | 65 ± 2* | 14 ± 2* | 30 ± 2* | 1.4 ± 0.3* | 0.14 ± 0.01* | 13 ± 1 | 1.4 ± 0.1 | 1.7 ± 0.2 | |
| Hypoxia +BAPN | 23 ± 1 | 62 ± 1* | 17 ± 1* | 29 ± 1* | 1.4 ± 0.2 | 0.11 ± 0.01 | 9 ± 1† | 1.4 ± 0.2 | 1.4 ± 0.2 | |
| Col1a1R/R | Normoxia | 23 ± 1 | 50 ± 1 | 9 ± 1 | 17 ± 1 | 1.4 ± 0.3 | 0.11 ± 0.01 | 9 ± 1 | 2.2 ± 0.2 | 2.5 ± 0.9 |
| Hypoxia | 23 ± 2 | 66 ± 2* | 15 ± 2* | 28 ± 2* | 1.4 ± 0.3 | 0.15 ± 0.01* | 10 ± 1 | 1.5 ± 0.1* | 1.6 ± 0.2* | |
| Hypoxia +BAPN | 24 ± 2 | 64 ± 2* | 14 ± 1* | 26 ± 2* | 1.4 ± 0.2 | 0.13 ± 0.01 | 10 ± 1 | 1.5 ± 0.2* | 1.5 ± 0.2* |
Values are means ± SE. BW, body weight (g); Hct, hematocrit (%); dPAP, diastolic pulmonary arterial pressure (mmHg); sPAP, systolic pulmonary arterial pressure (mmHg); MPA ID, main pulmonary artery inner diameter (mm); RV/BW, right ventricle to body weight ratio (%); CO, cardiac output (ml/min). Total pulmonary arterial compliance (µl/mmHg) was measured by stroke volume/pulse pressure (C) or by exponential fit of the diastolic part of the pressure curve (Ca). Col1a1+/+ mice, n = 8 for the normoxia and hypoxia+BAPN groups, and n = 6 for the hypoxia group. Col1a1R/R mice, n = 8 for the normoxia, hypoxia, and hypoxia+BAPN groups.
P < 0.05 vs. normoxia;
P < 0.05 vs. hypoxia.
Next, we examined the degree of RV hypertrophy, which is typically assessed by the Fulton index and RV/BW in these groups (Fig. 2, right, and Table 1). Despite comparable pressure increases in Col1a1+/+ and Col1a1R/R mice after BAPN treatment, RV hypertrophy was limited in Col1a1+/+ mice but remained significant (P < 0.05) in Col1a1R/R mice.
No differences were observed in body weight or heart rate (data not shown) between experimental groups. Hematocrit was significantly increased with hypoxia exposure, and BAPN treatment did not affect the increase in hematocrit (P < 0.05, Table 1).
Pulmonary hemodynamic changes.
With hypoxia, total PVR (or Z0) tended to increase (P = 0.09) in Col1a1+/+ mice, and it increased significantly (P < 0.05) in Col1a1R/R mice. Treatment did not prevent the increase in Z0 in either mouse strain because Z0 was significantly higher than in control mice in both strains (P < 0.05). Moreover, BAPN led to a further increase in Col1a1+/+ mice (P < 0.05). These results suggest a persistently increased RV steady afterload that was not attenuated by either reduced collagen content or reduced collagen cross-linking. Neither hypoxia nor BAPN treatment changed ZC in either genotype (Fig. 3). Z0 and ZC derived from the frequency domain method were consistent with those derived from the time domain method (data not shown).
Fig. 3.

Changes in total pulmonary vascular resistance (Z0) and characteristic impedance (Zc) in experimental groups. Results are presented as means ± SE. Col1a1+/+ mice, n = 8 in the normoxia and hypoxia+BAPN groups, n = 6 in the hypoxia group. Col1a1R/R mice, n = 8 for the normoxia, hypoxia, and hypoxia+BAPN groups. *P < 0.05 vs. normoxia, †P < 0.05 vs. hypoxia.
CO was not changed by hypoxia exposure or BAPN treatment in either mouse strain; however, CO was significantly lower in the BAPN-treated Col1a1+/+ group compared with the hypoxia-alone Col1a1+/+ group (P < 0.05, Table 1). Because the in vivo measurement of Z0 (or PVR) depends on CO, we measured the PVR ex vivo at a constant flow rate in additional groups (control, hypoxia, and hypoxia+BAPN) to examine the effect of BAPN on PVR in wild-type mice. We found that hypoxia significantly increased PVR (hypoxia, 2.42 ± 0.08 vs. control, 2.06 ± 0.11 mmHg min/ml, P < 0.05) and BAPN treatment did not affect this increase (hypoxia+BAPN, 2.43 ± 0.11 mmHg min/ml, P = 0.94 vs. hypoxia) at the flow rate Q = 3 ml/min. These data confirmed a lack of effect of BAPN on the PVR. Moreover, for all lungs, PVR decreased as Q increased in an inverse power law relationship, as expected due to recruitment and vasodilation (34). Extrapolating from these ex vivo data obtained at flow rates of Q = 1 to 5 ml/min (data not shown), we predict that a drop in CO from ~13 ml/min to ~9 ml/min, as found in our in vivo measurements, would lead to a 40% increase in PVR, which is consistent with the in vivo difference (Fig. 3). These data therefore confirm that the steady RV afterload increase was not prevented by either reduced collagen accumulation or reduced collagen cross-linking.
To quantify the dynamic hemodynamic component of the pulsatile flow, we examined PP and PVW. We found that hypoxia led to an increase in PP in both mouse strains with BAPN treatment, but BAPN had different effects in the different genotypes: PP was maintained at control levels in Col1a1+/+ mice and was elevated in Col1a1R/R mice (P < 0.05, Fig. 4A). The same trends in changes were also observed in PWV, although these changes did not reach statistical significance (Fig. 4B). Therefore, BAPN treatment prevented the increase in the dynamic hemodynamic component in Col1a1+/+ mice but not Col1a1R/R mice. Because similar changes occurred in PA collagen content (Fig. 1) (38), our data suggest that pulmonary vascular collagen content, not cross-linking, is a key contributor to pulsatile RV afterload in early HPH.
Figure 4.

Changes in pulse pressure (PP) (A) and pulse wave velocity (PWV) (B) in experimental groups. C: Pearson correlation between PP and RV hypertrophy index (RV/BW) in all experimental groups (r = 0.32, P < 0.05). D: linear correlation between PP and PWV in all experimental groups. Results are presented as means ± SE. Col1a1+/+ mice, n = 8 for the normoxia (open squares) and hypoxia+BAPN (closed squares) groups, and n = 6 for the hypoxia (gray squares) group. Col1a1R/R mice, n = 8 for the normoxia (open triangles), hypoxia (gray triangles), and hypoxia+BAPN (closed triangles) groups. *P < 0.05 vs. normoxia, †P < 0.05 vs. hypoxia.
For all groups, the PP was moderately and significantly correlated to the RV hypertrophy index, RV/BW (r = 0.32, P < 0.05, Fig. 4C). A similar trend was found between PP and the Fulton index (r = 0.25, P = 0.09). A strong and significant correlation was found between PP and PWV (R2 = 0.87, ρ = 0.89, P < 0.001, Fig. 4D).
Because arterial stiffness contributes to the pulsatile RV afterload, we next examined arterial compliance for the whole pulmonary vasculature (C and Ca) as well as the large proximal arteries. For both mouse strains, we found that hypoxia decreased total pulmonary arterial compliance and BAPN did not affect the decrease (P < 0.05, Table 1 and Fig. 5). This result indicates persistent stiffening of the entire pulmonary vasculature despite BAPN treatment. Interestingly, in contrast, BAPN had different effects on proximal PA compliance between the mouse strains: the proximal PA compliance decrease was not affected by BAPN in Col1a1+/+ mice, but it was prevented in Col1a1R/R mice (Fig. 5). These results show that with BAPN treatment, proximal and distal PA stiffening was concomitant in Col1a1+/+ mice but not in Col1a1R/R mice.
Fig. 5.

Changes in total pulmonary arterial compliance (CLUNG calculated as SV/PP; dark lines and diamonds) and proximal, left pulmonary artery (PA) compliance (CLPA calculated as ΔV/ΔP; gray lines and open squares) in all experimental groups in Col1a1+/+ mice (A) and Col1a1R/R (B) mice. Values are presented as means ± SE. In CLUNG calculations, for Col1a1+/+ mice, n = 8 for the normoxia and hypoxia+BAPN groups, and n = 6 for the hypoxia group; for Col1a1R/R mice, n = 8 for the normoxia, hypoxia, and hypoxia+BAPN groups. In CLPA calculations, for Col1a1+/+ mice, n = 5 for the normoxia group, and n = 6 for the hypoxia and hypoxia+BAPN groups; for Col1a1R/R mice, n = 5 for the normoxia group and n = 6 for the hypoxia and hypoxia+BAPN groups. *P < 0.05 vs. normoxia, †P < 0.05 vs. hypoxia.
Small pulmonary artery geometry and structure.
To gain more insight into collagen-mediated mechanisms of remodeling in the distal vasculature, we further examined the percentage of collagen by area fraction in arterioles within the lung. The examined PAs had an OD range of approximately 30–100 µm, and the average OD was similar (range, 60–70 µm) for all groups. In these vessels, we did not observe significant changes in collagen percentage with hypoxia or BAPN treatment (Table 2). Furthermore, the percentage of type I (or III) collagen was not altered (Table 2). The type I collagen concentration normalized by lung tissue weight was further confirmed by an ELISA assay. The type I collagen concentration was higher at baseline in Col1a1R/R mice, but it did not change significantly with hypoxia or BAPN treatment in either mouse strain. We also measured wall thickness in these vessels. As shown in Fig. 6, hypoxia caused wall thickening (or luminal narrowing) of these small PAs (P < 0.05), and BAPN treatment only slightly limited the remodeling in both mouse genotypes.
Table 2.
Collagen content in distal pulmonary arteries
| Strain | Experimental Group | Total Collagen* | Type I Collagen* | Type I Collagen Concentration† |
|---|---|---|---|---|
| Col1a1+/+ | Normoxia | 28 ± 5, n = 8 | 95 ± 2 | 0.19 ± 0.02, n = 8 |
| Hypoxia | 35 ± 7, n = 7 | 93 ± 3 | 0.16 ± 0.02, n = 8 | |
| Hypoxia +BAPN | 31 ± 5, n = 6 | 91 ± 2 | 0.12 ± 0.02, n = 8 | |
| Col1a1R/R | Normoxia | 36 ± 5, n = 8 | 96 ± 1 | 0.30 ± 0.04, n = 4 |
| Hypoxia | 34 ± 8, n = 7 | 93 ± 3 | 0.20 ± 0.03, n = 7 | |
| Hypoxia +BAPN | 24 ± 3, n = 8 | 95 ± 3 | 0.19 ± 0.04, n = 7 |
Values are presented as means ± SE.
Total collagen and type I collagen values (%) were measured via histological examination. †Measured as µg·ml−1·mg−1 and normalized by mouse right lung tissue wet weight. There were no significant changes between the groups.
Fig. 6.

Morphological changes in distal pulmonary artery wall thickness (A) and degree of narrowing (B; 2 × wall thickness/outer diameter). C: representative PicroSirius Red-stained images of distal PAs (indicated by arrows, scale bar = 100 µm). Results are presented as means ± SE. Col1a1+/+ mice, n = 8 for the normoxia, hypoxia, and hypoxia+BAPN groups; Col1a1R/R mice, n = 7 for the normoxia, hypoxia, and hypoxia+BAPN groups. *P < 0.05 vs. normoxia.
Histopathological changes in RV cardiomyocyte.
Next, we examined the histopathological changes in RV tissues by quantifying the degree of cardiomyocyte hypertrophy and ventricular fibrosis. The cross-section area of cardiomyocyte was used as an alternative indicator of RV hypertrophy, and the histological measurement was consistent the findings with the Fulton index or RV/BW index. That is, with BAPN treatment, RV cardiomyocyte enlargement was limited in Col1a1+/+ mice but it remained significant (P < 0.05) in Col1a1R/R mice (Fig. 7, A and B).
Fig. 7.

Histopathological changes in right ventricle (RV) tissues in all experimental groups. A: representative images of RV cardiomyocyte (stained with hematoxylin and eosin; H&E) in cross-sectional plane. Scale bar = 25 µm. B: cross-sectional area of RV cardiomyocyte obtained from H&E stains as a measure of myocyte hypertrophy. C: RV collagen area percentage obtained from PicroSirius Red stains as a measure of RV fibrosis. Results are presented as means ± SE. Col1a1+/+ mice, n = 6 for the normoxia, hypoxia, and hypoxia+BAPN groups. Col1a1R/R mice, n = 4 for normoxia, n = 6 for hypoxia, and n = 5 for hypoxia+BAPN groups. *P < 0.05 vs. normoxia, †P < 0.05 vs. hypoxia.
RV collagen accumulation, or RV fibrosis, was observed only in the Col1a1+/+ hypoxia group (P < 0.05, Fig. 7C). BAPN treatment prevented collagen accumulation in Col1a1+/+ mice (P < 0.05). There were no significant changes in RV collagen in Col1a1R/R mice, but an increase in RV collagen was observed in the BAPN-treated group (P = 0.08, Fig. 7C). More importantly, our data indicate that RV fibrosis is not necessarily present with the enlargement of the myocyte (such as shown in the Col1a1R/R hypoxia group, Fig. 7, B and C), suggesting that RV hypertrophy is primarily due to increased myocyte size.
Effect of BAPN treatment alone on changes in hemodynamics, arteries, and cardiac function.
To examine whether BAPN alone could induce changes in systemic or pulmonary hemodynamics or arterial remodeling, we performed in vivo pulmonary vascular impedance measurements on additional C57BL6 male mice with 8 wk of BAPN treatment. We also examined the structural and geometrical changes in the aorta and proximal PAs of these mice. These results are summarized in Table 3. Neither systemic pressure nor pulmonary pressure was altered by BAPN treatment alone. There were no changes in aortic or pulmonary arterial diameters. Finally, we did not observe significant changes in Z0, Zc, PP, PWV, or RV mass (by Fulton index) with BAPN treatment alone (Table 3).
Table 3.
Arterial structure and function and hemodynamic changes in C57BL6 mice with or without BAPN treatment alone
| C56BL6 Mice | Aorta OD | Aortic Pressure | Left PA OD | mPAP | Left PA Collagen* | Z0 | ZC | PP | PWV | Fulton Index |
|---|---|---|---|---|---|---|---|---|---|---|
| Control | 543 ± 47 | 73 ± 6 | 510 ± 51 | 15 ± 3 | 62 ± 5% | 1.4 ± 0.3 | 0.29 ± 0.03 | 12 ± 2 | 3.1 ± 0.4 | 28 ± 1% |
| BAPN | 539 ± 19 | 75 ± 3 | 513 ± 4 | 13 ± 1 | 59 ± 8% | 1.0 ± 0.1 | 0.31 ± 0.06 | 15 ± 1 | 4.1 ± 0.8 | 29 ± 1% |
Values are means ± SE. There were no significant changes between mouse groups in large systemic and pulmonary artery geometry, left PA collagen concentration, or pulmonary vascular impedance. Aorta OD, aorta outer diameter (µm); BAPN, β-aminopropionitrile; left PA OD, left pulmonary artery outer diameter (µm); mPAP, mean pulmonary arterial pressure; Z0, total pulmonary vascular resistance (mmHg·min/ml); ZC, characteristic impedance (mmHg·min/ml); PP, pulse pressure (mmHg); PWV, pulse wave velocity (m/s). For aorta and left PA ODs, n = 4 per group. For all functional parameters including Fulton index, n = 6 for control and n = 4 for BAPN-treated group.
Measured as area percentage.
To examine whether BAPN alone impairs cardiac function including CO, we performed in vivo RV function measurements on additional C57BL6 mice. First, we found that BAPN alone did not affect RV or LV mass, and no RV hypertrophy was evident (Table 4). Second, BAPN alone did not affect SV, RV afterload (Ea), or RV contractility (Ees) (Table 4). BAPN did decrease the end-diastolic pressure-volume relationship (EDPVR), an index of ventricular stiffness (P < 0.05, Table 4), as expected. Although the change was not significant, it is worth noting that BAPN tended to decrease CO (P = 0.10) in these wild-type mice.
Table 4.
RV structure and function changes in C57BL6 mice with or without 10 days of BAPN treatment alone
| C56BL6 Mice | RV | LV+S | Fulton Index | CO | SV | Ea | Ees | Ees/Ea | EDPVR |
|---|---|---|---|---|---|---|---|---|---|
| Control | 21 ± 1 | 78 ± 2 | 26 ± 1 | 8.1 ± 0.6 | 15 ± 1 | 1.8 ± 0.1 | 1.3 ± 0.1 | 0.8 ± 0.1 | 0.08 ± 0.01 |
| BAPN | 22 ± 1 | 86 ± 5 | 26 ± 1 | 6.5 ± 0.7 | 14 ± 2 | 1.9 ± 0.4 | 1.5 ± 0.4 | 0.9 ± 0.2 | 0.04 ± 0.004* |
Values are means ± SE. RV, right ventricle (g); LV+S, left ventricle with septum (g); CO, cardiac output (ml/min); SV, stroke volume (ml); Ea, effective arterial elastance (mmHg/µl); Ees, end-systolic pressure-volume relationship or contractility (mmHg/µl); EDPVR, end-diastolic pressure-volume relationship (mmHg/µl). Controls, n = 9, BAPN-treated group, n = 5.
P < 0.05 vs. controls.
DISCUSSION
This study was motivated by our prior finding showing that collagen cross-link and content differentially affect proximal PA stiffening during 10 days of hypoxia exposure (39). Our experimental approach of using transgenic mutant mice and a cross-link inhibitor allows us to decouple changes in collagen content and cross-linking, and thus identify their individual roles in PA remodeling (Fig. 1). In this study, we further investigated the consequences of this altered collagen metabolism for whole lung hemodynamics including steady and dynamic components, and assessed their effects on RV overload in early HPH. Unexpectedly, we did not observe reduced distal PA narrowing (evidenced by Z0) with BAPN treatment in either mouse genotype, which suggests negligible effects of both collagen content and cross-linking on the steady hemodynamic component. However, the increase in dynamic hemodynamic component (evidenced by PP) and RV hypertrophy (evidenced by the Fulton index and cardiomyocyte size) were prevented by BAPN in Col1a1+/+ mice, and this effect was absent in Col1a1R/R mice. These results indicate an important role for collagen content, but not cross-linking, in the pulsatile hemodynamic component. Moreover, we observed a significant correlation between PP and RV hypertrophy index, indicating that the dynamic hemodynamic component is an important contributor to RV overload and RV hypertrophy in early HPH.
Lack of effect of collagen on steady RV afterload.
We have previously found that BAPN limits the proximal PA stiffening during 10 days of hypoxia by preventing collagen cross-linking (38, 39). Because collagen accumulation occurs also in distal PA remodeling during HPH (6, 11, 20, 27), we expected similar effects of BAPN on distal PAs, and consequently, limited increases in PVR with the experimental strategy used here. Interestingly, we found that in both mouse strains, the increase in total PVR (Z0) was not prevented by BAPN during HPH progression. These results suggest that the steady RV afterload is not affected by collagen cross-linking or content. Our lung histological results also show that PA medial thickening was slightly but not significantly limited by BAPN treatment (Fig. 6) in both strains, which supports the suggestion that collagen-mediated extracellular matrix changes are not responsible for the increase in Z0 in early HPH.
Our results do not agree with the previous findings that BAPN treatment partially limits the increase in pulmonary pressure in HPH rats (11) or mice (15). We have also observed a significant reduction in mPAP and Z0 in a rat model of pulmonary arterial hypertension (SU5416 + hypoxia) with BAPN treatment (unpublished data). Because these three studies [(11), the present mouse study, and our ongoing rat study] use similar doses of BAPN (300–400 mg·kg−1 day−1) and the prior mouse study (15) used a dose of 150 mg·kg−1 day−1, we suspect that the discrepancy is related to different responses between species (rat vs. mouse) as a function of dose. The duration of hypoxia might be another factor because we focused on early HPH here. Nevertheless, the goal of our study is not to investigate the therapeutic effect of BAPN in HPH development. Instead, we used the combination of the Col1a1 mouse strain and BAPN to examine the effect of decoupled PA collagen content and cross-linking on pulmonary hemodynamics and RV afterload in early HPH. Potential dose-dependent, time-dependent, or species-dependent differences warrant separate investigation.
Collagen content, not cross-linking, contributes to pulsatile RV afterload.
To examine the changes in dynamic hemodynamic components, we investigated the whole lung hemodynamics through PP, PWV, and total pulmonary arterial compliance (C and Ca). In general, PP is considered the dynamic component of the pulsatile pressure and it has been found universally that PP and PWV are correlated in systemic circulations (3). In the pulmonary circulation, we observed a similar, significant correlation between PP and PWV (Fig. 4D). Furthermore, PP and PWV are often simplified as indicators of main artery stiffness because both are affected by the conduit artery compliance. However, it is important to note that they are also influenced by other cardiac and vascular factors: for example, arteriolar constriction could result in earlier wave reflections and hence an increase in PP (16). Thus, PP and PWV are more holistic measures of the dynamic components of the pulsatile blood pressure (such as wave reflections) and do not reflect only changes in proximal PA compliance.
An important observation of this study is the different PP (or PWV) changes between Col1a1+/+ and Col1a1R/R mice: whereas hypoxia-induced PP (PWV) elevation was prevented by BAPN in Col1a1+/+ mice, this effect was absent in Col1a1R/R mice (Fig. 4, A and B). These changes in pulsatile RV afterload are consistent with the changes in PA collagen content but not collagen cross-linking. The biomechanical mechanisms for the changes in PP, including the role of collagen content, are not completely understood. We speculate that longitudinal differences in PA stiffening, from proximal to distal, possibly differentially mediated by collagen content and cross-linking, are key to pulmonary vascular wave reflections. As shown in Fig. 5, a mismatch exists between proximal and distal PA compliance in BAPN-treated Col1a1R/R mice, which likely increases wave reflections and contributes to elevated PP or PWV, whereas this is absent in BAPN-treated wild-type mice. The mismatched proximal and distal PA stiffening in BAPN-treated Col1a1R/R mice may be associated with the decoupled changes in PA collagen content and cross-linking, whereas the concomitant changes in proximal and distal PA stiffening in wild-type mice are likely due to "undisturbed" collagen metabolism that enables synchronous and coincident collagen content and cross-linking changes.
Linkage between pulsatile RV afterload and RV hypertrophy.
Traditionally, PVR is considered to be the main determinant of RV afterload, but the pulsatile component may be also important for the adaptation of RV, which has been discussed in depth recently (24, 25, 37). Our results here provide novel evidence that the pulsatile RV afterload (e.g., PP) is responsible for RV hypertrophy independent of changes in steady afterload in early HPH. In particular, we found that even with persistently elevated PVR (Z0) and mPAP, RV hypertrophy was limited by BAPN treatment in Col1a1+/+ mice. Moreover, in both mouse strains, RV hypertrophy was moderately but significantly related to PP, a dynamic hemodynamic component (Fig. 4C). The mechanobiological mechanisms of this relationship remain to be elucidated.
Effect of BAPN alone on RV hypertrophy and function.
Finally, we found that BAPN treatment alone did not induce RV (or LV) hypertrophy or change systemic pressure, CO, or stroke volume, or other hemodynamic metrics in healthy, wild-type mice (Tables 3 and 4); these results are consistent with prior reports (5, 10, 23). We also confirmed minimal effects of BAPN alone on systemic and pulmonary arteries. BAPN did reduce RV stiffness (evidenced by EDPVR; Table 4). The effect of decreasing myocardial stiffness has been similarly observed in normal pig LVs after treatment at the same dose for 6 wk, and these changes were associated with decreased total collagen, decreased cross-linked collagen, and disrupted collagen integrity (10). In pathological remodeling such as pressure overload-induced LV hypertrophy, collagen accumulation leads to increased LV stiffness and diastolic dysfunction, and the effect is due to collagen cross-linking rather than collagen concentration (2, 17). Because BAPN is a cross-linking inhibitor and the mutation in Col1a1R/R mice affects collagen degradation only, we expect similar effects of BAPN on the RVs of wild-type and mutant mice during the hypertrophic remodeling. Moreover, in a normal heart ~2 to 4% of the myocardium is collagen (2) and with RV hypertrophy, the collagen content can increase by ~50% (26), which leads to only ~3 to 6% of heart weight. Our histological measurement indicates that the maximal RV collagen area percentage did not exceed 8% in our mice, and the cardiomyocyte hypertrophy was not always accompanied by collagen accumulation such as was shown in hypoxic Col1a1R/R mice (Fig. 7, B and C). Therefore, we expect a limited effect of BAPN itself on RV (or myocyte) hypertrophy and conclude that the RV hypertrophy is mainly a result of myocyte hypertrophy secondary to RV afterload changes.
Limitations.
First, we focused our investigation on the pulmonary vascular changes responsible for RV hypertrophy in an early stage of HPH (due to the time duration of hypoxia and the degree of pulmonary pressure elevation). Future investigation of the role of steady and pulsatile afterload in RV remodeling in established, severe HPH will provide more insights into the mechanisms of RV adaptation to chronic pressure overload. Second, we did not measure the total collagen content or cross-linking in the lungs for distal PAs as was performed previously in the proximal PAs (38). Measuring arterial collagen content and cross-linking in the lung parenchyma is challenging due to the presence of veins and airways that also contain collagen. Instead, we assume that BAPN has a similar effect on PA collagen content and cross-linking throughout the pulmonary vasculature and thus used the harvested lungs for histological and ELISA measurements. Using histological analysis and type I collagen ELISA assay in this study allowed us to quantify the relative amount of collagen or its isoform in the lungs. Finally, we were not able to quantify the anatomical distribution of collagen accumulation including intermediate levels of PA, which are possibly key to changes in RV afterload.
In summary, the current study examined how collagen-mediated PA remodeling affects steady and pulsatile pulmonary hemodynamics and RV hypertrophy in early HPH. We demonstrated that BAPN did not affect resistance or steady RV afterload for either mouse strain, suggesting a lack of effect of either collagen content or cross-linking on distal PA narrowing. In contrast, the increase in pulse pressure or pulsatile RV afterload was prevented by BAPN in Col1a1+/+ mice, and this change was absent in Col1a1R/R mice, suggesting a role for collagen content, not cross-linking, in the pulsatile RV afterload. Finally, the correlation between pulse pressure and RV hypertrophy independent of changes in resistance suggest that the pulsatile RV afterload is critical to RV overload and RV hypertrophy in early HPH. Thus, our findings are helping to uncover the mechanical mechanisms by which pulsatile vs. steady hemodynamics contribute to RV dysfunction in pulmonary hypertension.
GRANTS
Support for this study was provided by National Heart, Lung, and Blood Institute Grants R01 HL-086939 and R01 HL-115061.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Z.W. and N.C.C. conceived and designed research; Z.W. and T.A.H. performed experiments; Z.W., D.A.S., and H.A. analyzed data; Z.W., H.A., T.A.H., and N.C.C. interpreted results of experiments; Z.W. prepared figures; Z.W. drafted manuscript; Z.W., D.A.S., T.A.H., and N.C.C. edited and revised manuscript; Z.W., D.A.S., H.A., T.A.H., and N.C.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Jens Eickhoff for statistical analysis on the linear correlation, Dr. Guoqing Song for assistance with surgical preparation and data acquisition, and Dr. Diana Tabima for assistance with ex vivo pulmonary vascular impedance measurement.
REFERENCES
- 1.Barberà JA, Peinado VI, Santos S. Pulmonary hypertension in chronic obstructive pulmonary disease. Eur Respir J 21: 892–905, 2003. doi: 10.1183/09031936.03.00115402. [DOI] [PubMed] [Google Scholar]
- 2.Brower GL, Gardner JD, Forman MF, Murray DB, Voloshenyuk T, Levick SP, Janicki JS. The relationship between myocardial extracellular matrix remodeling and ventricular function. Eur J Cardiothorac Surg 30: 604–610, 2006. doi: 10.1016/j.ejcts.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 3.Cecelja M, Jiang B, McNeill K, Kato B, Ritter J, Spector T, Chowienczyk P. Increased wave reflection rather than central arterial stiffness is the main determinant of raised pulse pressure in women and relates to mismatch in arterial dimensions: a twin study. J Am Coll Cardiol 54: 695–703, 2009. doi: 10.1016/j.jacc.2009.04.068. [DOI] [PubMed] [Google Scholar]
- 4.Drexler ES, Bischoff JE, Slifka AJ, McCowan CN, Quinn TP, Shandas R, Ivy DD, Stenmark KR. Stiffening of the extrapulmonary arteries from rats in chronic hypoxic pulmonary hypertension. J Res Natl Inst Stand Technol 113: 239–249, 2008. doi: 10.6028/jres.113.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eberson LS, Sanchez PA, Majeed BA, Tawinwung S, Secomb TW, Larson DF. Effect of lysyl oxidase inhibition on angiotensin II-induced arterial hypertension, remodeling, and stiffness. PLoS One 10: e0124013, 2015. doi: 10.1371/journal.pone.0124013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Estrada KD, Chesler NC. Collagen-related gene and protein expression changes in the lung in response to chronic hypoxia. Biomech Model Mechanobiol 8: 263–272, 2009. doi: 10.1007/s10237-008-0133-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fraser KL, Tullis DE, Sasson Z, Hyland RH, Thornley KS, Hanly PJ. Pulmonary hypertension and cardiac function in adult cystic fibrosis: role of hypoxemia. Chest 115: 1321–1328, 1999. doi: 10.1378/chest.115.5.1321. [DOI] [PubMed] [Google Scholar]
- 8.Hiestand D, Phillips B. The overlap syndrome: chronic obstructive pulmonary disease and obstructive sleep apnea. Crit Care Clin 24: 551–563, 2008. doi: 10.1016/j.ccc.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 9.Hunter KS, Lee PF, Lanning CJ, Ivy DD, Kirby KS, Claussen LR, Chan KC, Shandas R. Pulmonary vascular input impedance is a combined measure of pulmonary vascular resistance and stiffness and predicts clinical outcomes better than pulmonary vascular resistance alone in pediatric patients with pulmonary hypertension. Am Heart J 155: 166–174, 2008. doi: 10.1016/j.ahj.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kato S, Spinale FG, Tanaka R, Johnson W, Cooper G 4th, Zile MR. Inhibition of collagen cross-linking: effects on fibrillar collagen and ventricular diastolic function. Am J Physiol Heart Circ Physiol 269: H863–H868, 1995. [DOI] [PubMed] [Google Scholar]
- 11.Kerr JS, Riley DJ, Frank MM, Trelstad RL, Frankel HM. Reduction of chronic hypoxic pulmonary hypertension in the rat by beta-aminopropionitrile. J Appl Physiol Respir Environ Exerc Physiol 57: 1760–1766, 1984. [DOI] [PubMed] [Google Scholar]
- 12.Kobs RW, Chesler NC. The mechanobiology of pulmonary vascular remodeling in the congenital absence of eNOS. Biomech Model Mechanobiol 5: 217–225, 2006. doi: 10.1007/s10237-006-0018-1. [DOI] [PubMed] [Google Scholar]
- 13.Kobs RW, Muvarak NE, Eickhoff JC, Chesler NC. Linked mechanical and biological aspects of remodeling in mouse pulmonary arteries with hypoxia-induced hypertension. Am J Physiol Heart Circ Physiol 288: H1209–H1217, 2005. doi: 10.1152/ajpheart.01129.2003. [DOI] [PubMed] [Google Scholar]
- 14.Mitchell GF, Pfeffer MA, Westerhof N, Pfeffer JM. Measurement of aortic input impedance in rats. Am J Physiol Heart Circ Physiol 267: H1907–H1915, 1994. [DOI] [PubMed] [Google Scholar]
- 15.Nave AH, Mižíková I, Niess G, Steenbock H, Reichenberger F, Talavera ML, Veit F, Herold S, Mayer K, Vadász I, Weissmann N, Seeger W, Brinckmann J, Morty RE. Lysyl oxidases play a causal role in vascular remodeling in clinical and experimental pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol 34: 1446–1458, 2014. doi: 10.1161/ATVBAHA.114.303534. [DOI] [PubMed] [Google Scholar]
- 16.Nichols WW, O’Rourke MF, Vlachopoulos C. McDonald’s Blood Flow in Arteries: Theoretical, Experimental and Clinical Principles (6th ed.). Boca Raton, FL: CRC Press, Taylor & Francis Group, LLC, 2011. [Google Scholar]
- 17.Norton GR, Tsotetsi J, Trifunovic B, Hartford C, Candy GP, Woodiwiss AJ. Myocardial stiffness is attributed to alterations in cross-linked collagen rather than total collagen or phenotypes in spontaneously hypertensive rats. Circulation 96: 1991–1998, 1997. doi: 10.1161/01.CIR.96.6.1991. [DOI] [PubMed] [Google Scholar]
- 18.O’Rourke MF. Vascular impedance in studies of arterial and cardiac function. Physiol Rev 62: 570–623, 1982. [DOI] [PubMed] [Google Scholar]
- 19.Ooi CY, Wang Z, Tabima DM, Eickhoff JC, Chesler NC. The role of collagen in extralobar pulmonary artery stiffening in response to hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol 299: H1823–H1831, 2010. doi: 10.1152/ajpheart.00493.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poiani GJ, Tozzi CA, Choe JK, Yohn SE, Riley DJ. An antifibrotic agent reduces blood pressure in established pulmonary hypertension in the rat. J Appl Physiol 68: 1542–1547, 1990. [DOI] [PubMed] [Google Scholar]
- 21.Poiani GJ, Tozzi CA, Yohn SE, Pierce RA, Belsky SA, Berg RA, Yu SY, Deak SB, Riley DJ. Collagen and elastin metabolism in hypertensive pulmonary arteries of rats. Circ Res 66: 968–978, 1990. doi: 10.1161/01.RES.66.4.968. [DOI] [PubMed] [Google Scholar]
- 22.Reddy AK, Li YH, Pham TT, Ochoa LN, Trevino MT, Hartley CJ, Michael LH, Entman ML, Taffet GE. Measurement of aortic input impedance in mice: effects of age on aortic stiffness. Am J Physiol Heart Circ Physiol 285: H1464–H1470, 2003. doi: 10.1152/ajpheart.00004.2003. [DOI] [PubMed] [Google Scholar]
- 23.Rosin NL, Sopel MJ, Falkenham A, Lee TD, Légaré JF. Disruption of collagen homeostasis can reverse established age-related myocardial fibrosis. Am J Pathol 185: 631–642, 2015. doi: 10.1016/j.ajpath.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 24.Saouti N, Westerhof N, Helderman F, Marcus JT, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Right ventricular oscillatory power is a constant fraction of total power irrespective of pulmonary artery pressure. Am J Respir Crit Care Med 182: 1315–1320, 2010. doi: 10.1164/rccm.200910-1643OC. [DOI] [PubMed] [Google Scholar]
- 25.Saouti N, Westerhof N, Postmus PE, Vonk-Noordegraaf A. The arterial load in pulmonary hypertension. Eur Respir Rev 19: 197–203, 2010. doi: 10.1183/09059180.00002210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schreier D, Hacker T, Song G, Chesler N. The role of collagen synthesis in ventricular and vascular adaptation to hypoxic pulmonary hypertension. J Biomech Eng 135: 021018, 2013. doi: 10.1115/1.4023480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schreier DA, Hacker TA, Hunter K, Eickoff J, Liu A, Song G, Chesler N. Impact of increased hematocrit on right ventricular afterload in response to chronic hypoxia. J Appl Physiol 117: 833–839, 2014. doi: 10.1152/japplphysiol.00059.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 99: 675–691, 2006. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 29.Stergiopulos N, Segers P, Westerhof N. Use of pulse pressure method for estimating total arterial compliance in vivo. Am J Physiol Heart Circ Physiol 276: H424–H428, 1999. [DOI] [PubMed] [Google Scholar]
- 30.Tabima DM, Hacker TA, Chesler NC. Measuring right ventricular function in the normal and hypertensive mouse hearts using admittance-derived pressure-volume loops. Am J Physiol Heart Circ Physiol 299: H2069–H2075, 2010. doi: 10.1152/ajpheart.00805.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tabima DM, Roldan-Alzate A, Wang Z, Hacker TA, Molthen RC, Chesler NC. Persistent vascular collagen accumulation alters hemodynamic recovery from chronic hypoxia. J Biomech 45: 799–804, 2012. doi: 10.1016/j.jbiomech.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J 15: 427–438, 2001. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 33.Tozzi CA, Christiansen DL, Poiani GJ, Riley DJ. Excess collagen in hypertensive pulmonary arteries decreases vascular distensibility. Am J Respir Crit Care Med 149: 1317–1326, 1994. doi: 10.1164/ajrccm.149.5.8173773. [DOI] [PubMed] [Google Scholar]
- 34.Tuchscherer HA, Vanderpool RR, Chesler NC. Pulmonary vascular remodeling in isolated mouse lungs: effects on pulsatile pressure-flow relationships. J Biomech 40: 993–1001, 2007. doi: 10.1016/j.jbiomech.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 35.Vanderpool RR, Kim AR, Molthen R, Chesler NC. Effects of acute Rho kinase inhibition on chronic hypoxia-induced changes in proximal and distal pulmonary arterial structure and function. J Appl Physiol 110: 188–198, 2011. doi: 10.1152/japplphysiol.00533.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vanderpool RR, Naeije R, Chesler NC. Impedance in isolated mouse lungs for the determination of site of action of vasoactive agents and disease. Ann Biomed Eng 38: 1854–1861, 2010. doi: 10.1007/s10439-010-9960-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z, Chesler NC. Pulmonary vascular wall stiffness: an important contributor to the increased right ventricular afterload with pulmonary hypertension. Pulm Circ 1: 212–223, 2011. doi: 10.4103/2045-8932.83453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Z, Chesler NC. Role of collagen content and cross-linking in large pulmonary arterial stiffening after chronic hypoxia. Biomech Model Mechanobiol 11: 279–289, 2012. doi: 10.1007/s10237-011-0309-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Z, Lakes RS, Eickhoff JC, Chesler NC. Effects of collagen deposition on passive and active mechanical properties of large pulmonary arteries in hypoxic pulmonary hypertension. Biomech Model Mechanobiol 12: 1115–1125, 2013. doi: 10.1007/s10237-012-0467-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Z, Schreier DA, Hacker TA, Chesler NC. Progressive right ventricular functional and structural changes in a mouse model of pulmonary arterial hypertension. Physiol Rep 1: e00184, 2013. doi: 10.1002/phy2.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westerhof N, Stergiopulos N, Noble MI. Snapshots of Hemodynamics: An Aid for Clinical Research and Graduate Education. New York: Springer Science+Business Media, 2010. [Google Scholar]