

The evidence that short-term fructose intake potentiates exercise capacity by nitric oxide-mediated mechanisms yields an optimal fructose feeding frame in which beneficial effects of fructose have been acquired while detrimental effects have not yet been manifested. This highlights the significance of exercise physiology in providing constructive regimens to improve physical performance.
Keywords: fructose, exercise, oxygen consumption, nitric oxide, shear stress-induced vasodilation
Abstract
To test the hypothesis that high fructose (HF) consumption divergently affects exercise capability as a function of feeding duration, rats were fed a normal (as control) diet or a normal caloric diet with HF for 3, 6, 10, and 30 days, respectively, and then were run on a treadmill. Results show that running distance and work were significantly increased, which was associated with greater exercise oxygen consumption in rats fed HF for 3 (HF-3D) and 6 days, but were decreased in rats fed HF for 30 days (HF-30D) compared with rats in their respective control groups. Shear stress-induced vasodilation (SSID) in isolated plantaris muscle arterioles was significantly greater in the HF-3D group than the control group. The difference in SSID between the two groups was abolished by Nω-nitro-l-arginine methyl ester (L-NAME), suggesting a nitric oxide (NO)-mediated response. Expression of phosphorylated/activated endothelial NO synthase (eNOS) and release of nitrite/NO were significantly increased in vessels of animals in the HF-3D group than controls. In contrast, arterioles isolated from the hypertensive rats in the HF-30D group displayed significantly attenuated NO-mediated SSID accompanied with greater production of superoxide compared with vessels of control animals. Additionally, the NO-dependent modulation of myocardial oxygen consumption (MV̇o2) was also impaired in the HF-30D group, and was prevented by blocking superoxide production with apocynin, an inhibitor that also normalized the reduced SSID in the HF-30D group. In conclusion, short-term (3–6 days) HF feeding enhances exercise potential via an increase in endothelial sensitivity to shear stress, which stimulates eNOS to release NO, leading to better tissue perfusion and utilization of oxygen. However, long-term (30 days) HF feeding initiates endothelial dysfunction by superoxide-dependent mechanisms to compromise exercise performance.
NEW & NOTEWORTHY The evidence that short-term fructose intake potentiates exercise capacity by nitric oxide-mediated mechanisms yields an optimal fructose feeding frame in which beneficial effects of fructose have been acquired while detrimental effects have not yet been manifested. This highlights the significance of exercise physiology in providing constructive regimens to improve physical performance.
since the american heart association recommended an overall reduction of sugar consumption in 2009 (23), concerns have been raised about the adverse effects of fructose consumption and its link to various health problems (38). The increase in fructose consumption has been demonstrated to be proportional to cardiovascular dysfunction; more specifically, the metabolic syndrome, which is characterized as insulin resistance (9, 39), dyslipidemia (27, 35), and hypertension (18, 20), followed by the development of diabetes. In this context, overconsumption of fructose is believed to be an independent risk factor for the development of obesity (4) and cardiovascular diseases (10). Although the specific mechanism or mechanisms responsible for the alterations in cardiovascular dysfunction remain elusive, crucial roles of oxidative stress in mediating the pathological process of cardiovascular dysfunction, cell death, and tissue damage (3, 7, 8, 16, 17, 41) have emerged. Because most of the studies just mentioned were primarily focused on long-term (from weeks to months) consumption of fructose and conclusions drawn were also based on outcomes from chronic fructose intake, it brings up the question about how the body responds to a short-term (several days) high-fructose (HF) diet. Specifically, whether short-term consumption elicits the same effect as long-term intake, or whether it could possibly be beneficial? During our preliminary studies, we found that pregnant rats fed an HF diet performed significantly more work during exercise than pregnant rats that were fed a normal diet. Although pregnancy generates a variety of changes in the body such as hormonal rebalance, metabolic shifts, and adaption of cardiovascular function, this intriguing finding provided the rationale for the present study, which aimed to extend our current understanding of fructose-induced changes in exercise capacity under normal/physiological conditions via testing the duration of fructose feeding and identifying possible mechanisms involved in those changes. Using these questions to search the literature, we found that 1) studies pertaining to the effects of short-term fructose consumption on cardiovascular function and body metabolism are rare (31); 2) despite sufficient studies showing the beneficial effects of exercise on fructose-induced cardiovascular dysfunction (22), few studies focused on whether fructose affects exercise (12), and 3) even though there are a few studies that examined the effects of fructose on exercise capacity, conclusions drawn from those studies are controversial, and underlying mechanisms are even less explored. For instance, in some studies (29, 32), fructose was reported to be potentially capable of improving exercise tolerance, but in others (2, 30), it failed to affect exercise performance. Moreover, the study (6) that was conducted on young rats recovering from fetal protein malnutrition due to a fructose-rich diet consumed by the mother during pregnancy demonstrated that metabolic responses to acute exercise were not affected by fetal fructose overload. Thus, we hypothesized that short-term HF intake potentiates exercise capability, but prolonged feeding compromises exercise capability. To this end, time-course experiments were designed by feeding rats with fructose for 3, 6, 10, and 30 days, respectively, to identify the optimal feeding duration for vigorous exercise performance.
METHODS AND MATERIALS
Animals.
Ten-week-old male Sprague-Dawley rats were obtained from Charles River Laboratories (Wilmington, MA). Rats were fed either a normal diet (as controls) or a HF diet for 3 (HF-3D), 6 (HF-6D), 10 (HF-10D), or 30 (HF-30D) days, respectively. Rats had ad libitum access to food and water. The 60% Fructose Purified Diet (01810078; TestDiet, Richmond, IN) is characterized as a normal caloric diet enriched with fructose and was produced by replacing the same 60% that makes up the carbohydrate fraction (45.65% dextrin, 15% sucrose, and 3% cellulose) in a basal/normal diet with fructose (60% fructose and 1.65% cellulose). The “60% fructose purified diet” maintains an identical percentage of protein, fat, fibers, minerals, and vitamins, creating therefore, the same number of total calories as those of a normal diet. Protocols were approved by the Institutional Animal Care and Use Committee of New York Medical College and conformed to the current guidelines for the care and use of laboratory animals published by the National Institutes of Health and American Physiological Society.
Blood pressure.
Blood pressure was recorded before and during (every other day) the fructose-feeding period using a tail cuff connected to a noninvasive blood pressure monitor.
Treadmill protocol.
After recording body weight, rats ran on a treadmill at a constant 10° incline at 4 m/min, followed by an increase of 2 m/min every 2 min until exhaustion. Exhaustion was defined as spending 10 s or more on the grid plate without an attempt by the rat to reengage in running. During the running period, oxygen consumption (V̇o2) was automatically recorded. The maximal value of the work was calculated as the product of the total running distance and body weight of rats.
Shear stress-induced dilation (SSID).
After rats were killed, the plantaris muscle, a muscle located in the calf that is heavily engaged during running, was dissected. Plantaris arterioles were isolated, and then cannulated in a vessel chamber perfused with physiological salt solution (PSS) containing the following (in mmol/l): NaCl, 117; KCl, 4.7; MgSO4, 1.1; KH2PO4, 1.2; glucose, 5.5; CaCl2, 2.5; pyruvate, 2.0; ascorbate, 0.1; and l-arginine, 0.1. The PSS was gassed in a water-jacketed (37°C) reservoir with 95% O2-5% CO2 and equilibrated with NaHCO3 to pH 7.4. The procedure of isolating and cannulating arterioles has been described in detailed previously (24, 25). Isolated arterioles were equilibrated under 80 mmHg without flow for 1 h, during which all vessels developed spontaneous tone that reached to approximately 50–60% of their maximal diameter (passive diameter). After equilibration, calculated flow rates (µl/min) that generate required shear stress values of 5, 10, 15, 20, and 25 dyne/cm2, respectively, on vessels were sequentially applied to vessels via a syringe pump (Harvard Apparatus, Holliston, MA), and changes in diameter in response to each dose of flow rate/shear stress were recorded (16). The change in diameter of arterioles in response to each dose of flow rate was kept stable for ~2 min before a second dose was applied. After control SSID, vessels were incubated with Nω-nitro-l-arginine methyl ester (L-NAME, an inhibitor of NO synthase, 3 × 10−4 mol/l) for 45 min to evaluate the role of NO in the mediation of SSID, and then SSID was once again recorded. In separate experiments, SSID was recorded before (control) and after exposure of vessels to apocynin (APO, an inhibitor of superoxide production, 10−5 mol/l). At the end of experiments, vessels were incubated in a Ca2+-free solution with 10−3 mol/l EGTA for 10 min, followed by recording their passive diameter at 80 mmHg.
Shear stress-stimulated release of NO/nitrite and endothelial NO synthase (eNOS) phosphorylation.
To measure shear stress-induced release of NO, shear stress (2 and 10 dyne/cm2, respectively) was continuously applied to isolated second-order mesenteric arteries for 3 min, and perfusate was collected. Nitrite formation in the perfusate was assessed using 2,3-diaminonaphthalene (DAN) and an HPLC/fluorescence detector-based assay to determine 1-(H)-naphthotriazole, a fluorescent product upon the reaction of nitrite and DAN. Thus, the perfusate sample was incubated with DAN (5 µg/ml), and then 20 µl of the sample was injected into the HPLC detector to obtain the fluorescent signal of 1-(H)-naphthotriazole. The final concentration of nitrite/NO in the perfusate was normalized to the internal surface area of the vessel (πdl), and expressed as picomoles of nitrite per millimeter squared of the internal vessel surface area per minute (pmol/mm2/min) (17). At the end of experiments, shear stress-stimulated vessels were collected to determine eNOS phosphorylation. Mesenteric arteries were selected for the assessments of shear stress-stimulated release of nitrite, as well as eNOS phosphorylation, because of their abundance and sufficient length. Mesenteric arteries exhibit the same patterns of vascular response to shear stress and endothelial mediators as those in skeletal muscle arteries (14, 36).
Vascular superoxide production.
As described previously (42), isolated plantaris arteries were incubated with dihydroethidium (DHE, 10−5 mol/l) for 1 h, during which the superoxide in the vessels reacted with DHE to form 2-hydroxyethidium (EOH), which was detected with an HPLC/fluorescence detector. After incubation with DHE, the vessels were washed and homogenized in acetonitrile/water (1:1), and then centrifuged for 10 min. After centrifuging, the supernatant fraction was collected for HPLC analysis and the precipitate was used for protein measurement using a Bio-Rad Protein Assay (Bio-Rad, Hercules, CA). The final concentration of superoxide in each sample was normalized to the protein contents of their corresponding vessels, and expressed as picomoles of superoxide (EOH) per microgram of protein.
In vitro myocardial oxygen consumption (MV̇o2).
As described previously (33), rats were killed in a chamber containing CO2. Following thoracotomy, hearts were removed and placed in a MOPS-PSS solution. The left ventricle (LV) was cut into pieces (~25 mg/piece) and placed in an oxygenated chamber to monitor MV̇o2. After recording the basal MV̇o2, increasing concentrations of bradykinin (BK, 10−7 to 10−4 mol/l; an eNOS activator) were cumulatively added to the chamber, and changes in MV̇o2 were then recorded. In separate experiments, LV tissues were incubated with either L-NAME (10−4 mol/l) or apocynin (APO 10−5 mol/l) for 60 min. After that, BK (10−4 mol/l) was administered and MV̇o2 was once more recorded to evaluate the action of NO in the regulation and role of superoxide in the modulation of MV̇o2, respectively.
Western blot analysis.
The shear stress-stimulated mesenteric arteries were pulverized in liquid nitrogen. An equal amount of protein from each sample (10 µg of vessels) was separated on a 10% SDS-PAGE and transferred to a polyvinylidene fluoride membrane. Membranes were probed with primary antibodies of eNOS (BD Transduction Laboratories), phospho-eNOS (ser1177; Cell Signaling), and β-actin (Sigma-Aldrich), followed by appropriate secondary antibodies conjugated with horseradish peroxidase. Immunoreactive bands were visualized with a chemiluminescent signal, and the densitometry was normalized to β-actin.
Tissue glycogen content.
By using an established phenol-sulfuric acid colorimetric measurement of glycogen (28), plantaris muscles and the liver were isolated and cut into small pieces (10 mg for each sample). The tissue was completely digested with 30% KOH and the glycogen was precipitated by adding ethanol. Diluted glycogen was determined spectrophotometrically (490 nm) using the phenol-sulfuric acid method (28) to a standard curve of glycogen (0–100 µg), and final concentrations were normalized to the protein content of samples.
Statistical analysis.
Data are expressed as means ± SE. The number of rats is identified as n. The equation 4ηQ/πr3 = value of shear stress (dyne/cm2) was used to calculate applied flow rates necessary for generating required shear stress values, where η is the viscosity of PSS (0.007 poise at 37°C), Q is the flow rate (µl/min), and r is the vessel radius. Statistical analysis was performed using repeated-measures ANOVA, followed by the Tukey-Kramer post hoc test and Student’s t-test. Statistical significance was accepted as P < 0.05.
RESULTS
Fructose and blood pressure.
Rats were randomly divided into two groups, and changes in body weight were recorded before (zero day) and after receiving their specific (normal or HF) diet for 3, 6, 10, or 30 days, respectively. Figure 1A shows that both groups of rats displayed comparable weight gain in an age-dependent manner, suggesting that the normal caloric diet enriched with fructose does not nutriologically affect the body weight in a 30-day feeding period. Blood pressures, including systolic, diastolic, and mean arterial, were not different in rats fed HF up to 10 days (Fig. 1B shows an example of HF-3D), but they were significantly increased in rats in the HF-30D group (Fig. 1C) compared with their time-matched controls, indicating that long-term consumption of fructose imposes detrimental effects on systemic blood pressure.
Fig. 1.

Changes in body weight (A) and blood pressure (B and C) in rats fed a normal diet or a high-fructose (HF) diet for 3 days (HF-3D) or 30 days (HF-30D) (n = 10 in each group). *Significant difference from normal diet rats. SBP, systolic blood pressure; DBP, diastolic blood pressure; MAP, mean arterial pressure.
Fructose and exercise capability.
Parameters of exercise are summarized in Fig. 2, showing that running distance (Fig. 2A) and maximal work (running distance of each rat was normalized to its body weight; Fig. 2B) were significantly increased in rats in the HF-3D and HF-6D groups compared with time-matched control rats fed a normal diet. However, the increments became smaller and eventually disappeared when HF was given for 10 days. Based on these findings, in the mechanistic-based studies that followed, only rats in the HF-3D group were selected as the definitive example of short-term HF. Rats in the HF-30D group displayed significantly shorter running distances and less maximal work than their control counterparts. The longer running distance and greater work output in rats in the HF-3D group (shown in Fig. 2) were paralleled with an increase in oxygen consumption (Fig. 3A), suggesting that greater exercise capability correlated with more efficient oxygen utilization. This adaptation disappeared in response to a long-term HF diet (HF-30D, Fig. 3B). As such, a short-term HF diet benefits exercise capability, but a long-term HF diet compromises it. It was also noticed that rats in the long-term groups, including those fed the normal and HF diets (Fig. 3B), exhibited much more work than rats in the short-term groups (Fig. 3A), mainly due to the age-related increase in body weight.
Fig. 2.
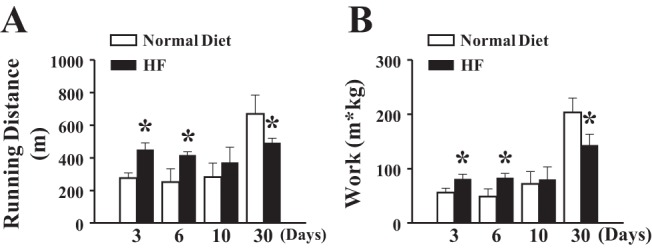
Divergent effects of fructose on exercise capability of rats as a function of feeding duration. HF feeding for 3 and 6 days enhances running distance, whereas 30 days reduces running distance (A) and maximal work (B) on a treadmill (n = 10 in each group). *Significant difference from rats fed a normal diet.
Fig. 3.
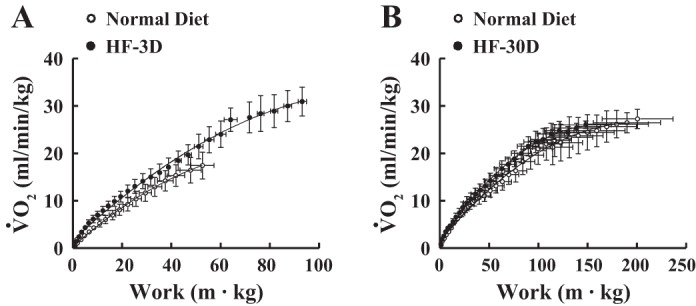
Summarized data for maximal work as a function of oxygen consumption (V̇o2) in rats in the HF-3D (A, n = 10) and HF-30D (B, n = 7) groups, respectively, and rats fed a time-matched, normal diet. Rats in the HF-3D group increased their maximal work and oxygen utilization compared with rats in the normal-diet control group.
We next aimed to clarify the mechanism or mechanisms responsible for changes (i.e., short-term HF enhanced and long-term reduced) in exercise capability, thus the following studies focused on shear stress-induced vasodilation (SSID) and NO release, vascular eNOS activity and superoxide production, and myocardial oxygen consumption (MV̇o2). Summarized results are depicted in Figs. 4 and 5 (short-term HF) and Figs. 7 and 8 (long-term HF).
Fig. 4.

Wall shear stress (WSS)-diameter relationships in vessels isolated from rats fed a normal diet and rats in the HF-3D group. A: shear stress-induced vasodilation (SSID) of isolated plantaris muscle arteries was significantly enhanced in the HF-3D group compared with the normal-diet group (n = 8 in each group). Nω-nitro-l-arginine methyl ester (L-NAME, an inhibitor of NO synthase) had less inhibitory effects on SSID of rats fed a normal diet (B) than rats in the HF-3D group (C), resulting in elimination of their difference in the presence of L-NAME (D). *Significant difference between two curves. PD, passive diameter.
Fig. 5.

A: HPLC-based determination of shear stress-stimulated release of nitrite/nitric oxide (NO) in vessels of rats in the HF-3D and normal-diet groups (n = 6–8). Elevation of NO release in vessels of rats in the HF-3D group (A) was associated with an upregulation of endothelial NO synthase (eNOS) phosphorylation (p-eNOS) and unchanged eNOS protein content (B and C; n = 3 blots). *Significant difference from rats in the normal diet group.
Fig. 7.

Shear stress-diameter relationships in vessels isolated from rats in the normal-diet and HF-30D groups. A: SSID of isolated plantaris muscle arteries was significantly reduced in rats in the HF-30D group compared with rats in the normal-diet group. B: L-NAME inhibited the dilator response by ~50% in normal-diet control animals, but (C) less than 10% in the HF-30D group (n = 6–10). *Significant difference between two curves.
Fig. 8.

A: bradykinin (BK) dose-dependently suppressed myocardial oxygen consumption (MV̇o2) in rats fed a normal diet, which was absent in rats in the HF-30D group, but it was restored by apocynin (APO) (n = 6 in each group). B: significantly greater vascular superoxide production in the HF-30D group than in the normal-diet group (n = 5 in each groups). C: APO-dependent normalization of SSID in arterioles of rats in the HF-30D group (n = 6). *Significant difference from normal-diet control animals. #Significant difference from the HF-30D group. +Significant difference between two curves.
Increased SSID and NO release as a function of short-term HF feeding.
Figure 4 shows that shear stress resulted in a dose-dependent increase in the diameter of isolated plantaris arterioles in both groups fed a normal diet and a HF diet, but it was significantly greater in the HF-3D group than rats in the normal-diet control group (Fig. 4A). L-NAME exhibited a greater inhibitory effect on the HF-3D group (Fig. 4C) than it did on rats fed a normal diet (Fig. 4B), leading to an L-NAME-induced abolishment of differences in SSID between the two groups (Fig. 4D). This result suggests that the originally enhanced portion of SSID in arterioles of rats in the HF-3D group (Fig. 4A) was mediated by endothelial NO. Alternately, because SSID in skeletal muscle arterioles is mediated by endothelial NO and prostaglandins (25), rats in the normal-diet and HF-3D groups displayed a comparable remaining portion that was insensitive to L-NAME (Fig. 4D), suggesting that the prostaglandin-mediated component of SSID was not significantly affected by HF.
Furthermore, Fig. 5 provides direct evidence indicating that the enhanced NO-mediated SSID in the HF-3D group (Fig. 4) resulted from a greater release of NO (Fig. 5A). This conclusion was further supported by Western blot analysis (Fig. 5, B and C) showing that although protein expression of eNOS was comparable, shear stress-stimulated eNOS phosphorylation, an index of eNOS activity, was significantly increased in vessels of rats in the HF-3D group compared with their controls. Thus, feeding rats HF for 3 days enables sensitizing endothelial responses to shear stress, which activates eNOS to increase NO release, leading to the greater vasodilation.
Fructose and glycogen.
In the speculation that an increase in glycogen store as a function of HF feeding may be responsible for the enhanced exercise performance in the HF-3D group, the glycogen content in the skeletal muscle and liver was measured. As shown in Fig. 6, tissue glycogen content in rats in the HF-3D group did not differ from the normal-diet controls either in muscle or liver, arguing against the possibility of changes in glycogen storage contributing to enhanced exercise capability.
Fig. 6.
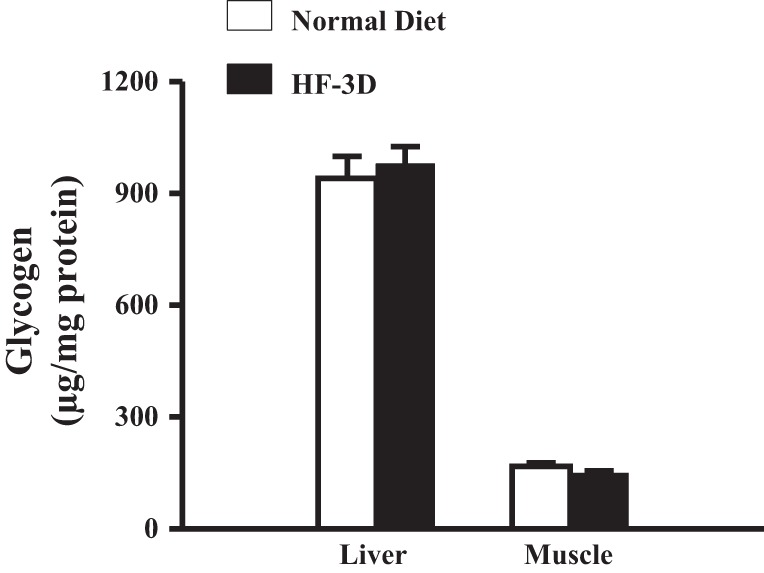
Comparable glycogen contents in plantaris muscles and livers of rats in the normal-diet and HF-3D groups (n = 5 in each group).
Superoxide-dependent impairment of SSID as a function of long-term HF feeding.
SSID data from the long-term HF groups are depicted in Fig. 7. In contrast to arteriolar responses in the HF-3D group, vessels of rats in the HF-30D group exhibited a significantly attenuated SSID compared with their time-matched normal-diet controls (Fig. 7A). L-NAME resulted in significantly reduced SSID (by ~50%) in arterioles of rats fed a normal diet (Fig. 7B), but it elicited a much smaller inhibition (less than 10%) in vessels of rats in the HF-30D group (Fig. 7C), suggesting an attenuation of NO-mediated fraction.
Changes in NO bioavailability were further verified by measuring MV̇o2, a response that, in normal conditions, is modulated/suppressed by NO (33). As shown in Fig. 8A, BK suppressed MV̇o2 in a dose-dependent manner in rats fed a normal diet, but it failed to affect MV̇o2 in rats in the HF-30D group, responses that were similar to those observed in normal hearts treated with L-NAME (data not shown). This result indicates an absence of (or impaired) NO bioactivity in the myocardium of rats in the HF-30D group. Nevertheless, in the presence of APO, the BK-stimulated NO suppression of MV̇o2 in rats in the HF-30D group was restored to normal (Fig. 8A), implying a superoxide-inactivation of NO as a function of long-term HF feeding. This was confirmed by a threefold increase in vascular superoxide production in the HF-30D group compared with its corresponding control group (Fig. 8B). Moreover, the reduction in NO-mediated SSID (shown in Fig. 7A) was normalized when the vessels were treated with APO (Fig. 8C), further confirming that superoxide plays a crucial role in the impairment of NO-mediated responses.
Overall, data in Figs. 7 and 8 indicate that long-term intake of HF promotes vascular production of superoxide, which inactivates NO to barricade cardiac utilization of oxygen. When superoxide is inhibited, NO bioavailability is recovered, leading to the normalization of SSID and functional modulation of MV̇o2.
DISCUSSION
HF alters exercise capacity in a time-dependent manner.
In addressing the issue of whether fructose consumption on exercise is a “friend or foe,” the present study reveals an interesting phenomenon that shows a dual role for fructose in regulation of exercise capability. Running distance and maximal work were significantly increased in rats fed HF for 3–6 days. Over time, this potentiation of exercise capability diminished, and then disappeared once the rats reached their 10th day of fructose feeding. Further feeding for up to 30 days resulted in impairment in exercise capability (Figs. 2 and 3). A similar phenotype of changes in systemic blood pressure was also presented as the development of hypertension in rats fed a HF diet for 30 days (Fig. 1). The results support our working hypothesis, and have led to further questions about why and how it happened.
Short-term HF intake enhances eNOS activity to increase shear stress-stimulated release of NO and -mediated vasodilation leading to adaptation of tissue perfusion during exercise.
It has been well accepted that during aerobic exercises such as running, tissue blood supply increases significantly to provide sufficient oxygen to working muscles. The increase in blood flow creates greater shear stress on the surface of the vascular endothelium, leading to stimulation/activation of eNOS, which releases NO to further dilate vessels in a positive feedback loop (24, 37). As a result, enhanced vasodilation provides increased blood supply to cardiac and skeletal muscles to meet their metabolic needs during exercise (26). In this regard, NO promotes cardiomyocytes and working muscles to use oxygen more efficiently to maintain the elevated cardiac work required for exercise and to improve exercise potential and tolerance. This is accomplished by NO-dependent regulation of mitochondrial metabolism via modulating tissue oxygen consumption (43). In the present study, an augmented SSID (Fig. 4, A–C) and great release of NO (Fig. 5A) are indeed responsible for the potentiation of exercise capacity (Fig. 2A) and optimal oxygen utilization (Fig. 3A) as a function of a short-term HF diet. To identify the source of the increased NO production in rats in the HF-3D group, eNOS expression was assessed by Western blotting. As shown in Fig. 5, B and C, in the presence of comparable eNOS protein contents, expression of phosphorylated eNOS in response to stimulation with shear stress was much higher in rats in the HF-3D group than in controls. Phosphorylation of eNOS is indispensable for the shear stress-stimulated activation of the enzyme (11); a higher expression of eNOS phosphorylation is an indicator of greater enzyme activity. Our data demonstrate that in vivo HF feeding for 3 days seems insufficient to change eNOS protein content, but it can trigger an increase in eNOS activity by sensitizing endothelial response to shear stress. Consequently, the greater NO-mediated vasodilation, better tissue perfusion, and adaptation of tissue oxygen utilization by NO-mediated regulation of oxygen consumption initiate the promotion of exercise capability. Noteworthy, an increase in eNOS phosphorylation (Fig. 5, B and C), as a function of in vivo short-term HF feeding is a novel finding, which provides molecular evidence to support the results obtained from functional experiments (SSID in Fig. 4) and biochemistry analysis (perfusate nitrite/NO in Fig. 5A). Alternatively, this activation of eNOS may not necessarily be initiated by fructose per se, but rather mediated by its metabolites from hepatic metabolism (in vivo). In this regard, human studies have provided evidence indicating that a bolus injection of glucose did (but fructose did not) initiate an insulin-dependent increase in calf blood flow (40). This implies the in vivo presence of interactions among glucose, fructose, and insulin during the process of carbohydrate metabolism, by which fructose may indirectly activate eNOS/NO signaling to elicit vasodilation in skeletal muscles via, perhaps, its metabolite-mediated pathway. Of note, the finding of comparable glycogen content in tissues of rats in the normal-diet and HF-3D groups (Fig. 6) negates the possibility that a promotion of glycogen accumulation in skeletal muscles is responsible for the increase in exercise potential by short-term HF feeding.
Long-term HF intake increases vascular superoxide production to impair NO-mediated SSID and NO modulation of MV̇o2, resulting in compromised exercise capability.
The beneficial effects of HF on exercise capacity seem to be transient; they were declining over time, and eventually vanished. As indicated, HF feeding for 30 days caused hypertension (Fig. 1), compromised exercise capability as evidenced by shorter running distances and less work output (Figs. 2 and 3), and moreover, impaired vascular function characterized as an attenuated NO-mediated SSID (Fig. 7) and loss of NO-dependent modulation of MV̇o2 (Fig. 8A). In agreement with previous studies (33, 34, 43), administration of BK to normal hearts stimulated NO release to suppress oxygen consumption, a response that spares oxygen to potentiate exercise tolerance. However, the BK-induced NO-mediated suppression of MV̇o2 was basically indiscernible in myocardium of rats in the HF-30D group (Fig. 8A), suggesting a failure of NO-dependent regulation of MV̇o2 and, consequently, less efficient oxygen utilization. APO normalized the alterations in both MV̇o2 (Fig. 8A) and SSID (Fig. 8C), further confirming a superoxide-driven inactivation of NO. As such, pivotal roles for superoxide emerged during the process of development of hypertension (Fig. 1), endothelial dysfunction (Fig. 7), and blunted exercise capability (Fig. 3) as a function of long-term HF consumption. In the vasculature, superoxide is generated primarily by NADPH oxidase, xanthine oxidase, and even uncoupled eNOS (41). Previous studies demonstrated a correlation between superoxide and cardiovascular diseases such as hypertension (15), diabetes (16), and aging (19), as well as superoxide and chronic fructose intake (3, 8). Superoxide scavenges NO to form peroxynitrite, a highly reactive molecule that further impairs vascular function (5, 13). Our data are in line with those of studies in which APO prevented hepatic dysfunction in rats fed a HF diet via an antioxidant mechanism (8), and alternately, rats with a long-term fructose-rich diet displayed a significant reduction in aerobic exercise capacity, as a consequence of enhanced oxidative stress (3).
In conclusion, first, fructose indeed provides beneficial effects on exercise capability in a time-dependent manner up to 6 days. This finding is of significance because one aim of exercise physiology is to identify optimal exercise regimens that can improve physical performance (1, 21). In this context, it is plausible to speculate that consumption of dietary fructose daily or on a short-term basis might serve as an energy source before sporting competitions. Second, the increase in vascular eNOS activity in response to short-term HF intake was reported for the first time, which provides a mechanistically based explanation for the promotion of exercise capacity via improving NO-mediated cardiovascular responses to adapt working muscle performance. Third, results highlighting the detrimental effects of a long-term HF diet in a superoxide-dependent manner were consistent with results from previous studies, although the present study was focused specifically on fructose-induced alterations in the cardiovascular system to affect exercise capability, whereas others focused mainly on changes in the digestive and metabolic systems. Fourth, short-term fructose consumption did not significantly affect glycogen content in the liver and skeletal muscles. Nevertheless, in consideration of actions of mitochondrial enzymes, such as cytochrome-c oxidase or citrate synthase that serve as markers of muscular oxidative potential in the regulation of exercise capability, fructose-induced changes in their activities need to be clarified in future studies.
Alternatives and perspectives.
Several limitations should be pointed out. First, the study lacked in vivo evidence specifically showing changes in skeletal muscle NO levels and blood flow during treadmill exercise. Although we provided evidence indicating that in vitro changes in eNOS phosphorylation, NO production, and SSID in skeletal muscle arterioles were strongly correlated with running distance and oxygen consumption in rats fed HF on a short-term and long-term basis, respectively, in vitro findings cannot necessarily be completely extrapolated to in vivo conditions. Second, it is important to note that although using a normal caloric diet enriched with HF was aimed to identify pure fructose-dependent changes, the HF-initiated responses observed in the present study may not agree with the standard terms of translational biology regarding physiologically related fructose-induced alterations in natural populations. That issue has always been challenged and addressed among animal studies involving, for instance, smoking- and angiotensin II-induced pathological processes, or some pathogeneses developed specifically in genetically engineered animal models. Alternatively, in a viewpoint of pros and cons, respectively, the present study has shed light on a time-dependent regulatory mechanism in terms of an optimal fructose feeding frame, in which beneficial effects of fructose have been acquired while detrimental effects have not yet been manifested. In particular, fructose-induced adaptation to exercise capacity as a function of short-feeding duration may clarify or lessen some of the negative connotation surrounding fructose consumption.
GRANTS
Support for this work was provided by National Heart, Lung, and Blood Institute Grants HL-43023, HL-129797, and HL-070653.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.S., A.H., E.K., S.S., T.H.H., and D.S. conceived and designed research; A.S., A.H., E.K., S.S., and D.S. performed experiments; A.S., A.H., and D.S. analyzed data; A.S., A.H., and D.S. interpreted results of experiments; A.S., A.H., and D.S. prepared figures; A.S., A.H., and D.S. drafted manuscript; A.H., E.K., S.S., T.H.H., and D.S. edited and revised manuscript; A.S., A.H., E.K., S.S., T.H.H., and D.S. approved final version of manuscript.
REFERENCES
- 1.Achten J, Jeukendrup AE. Optimizing fat oxidation through exercise and diet. Nutrition 20: 716–727, 2004. doi: 10.1016/j.nut.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 2.Björkman O, Sahlin K, Hagenfeldt L, Wahren J. Influence of glucose and fructose ingestion on the capacity for long-term exercise in well-trained men. Clin Physiol 4: 483–494, 1984. doi: 10.1111/j.1475-097X.1984.tb00134.x. [DOI] [PubMed] [Google Scholar]
- 3.Botezelli JD, Cambri LT, Ghezzi AC, Dalia RA, Voltarelli FA, de Mello MA. Fructose-rich diet leads to reduced aerobic capacity and to liver injury in rats. Lipids Health Dis 11: 78, 2012. doi: 10.1186/1476-511X-11-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bray GA, Nielsen SJ, Popkin BM. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am J Clin Nutr 79: 537–543, 2004. [Erratum in Am J Clin Nutr 80: 1090, 2004]. [DOI] [PubMed] [Google Scholar]
- 5.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87: 840–844, 2000. doi: 10.1161/01.RES.87.10.840. [DOI] [PubMed] [Google Scholar]
- 6.Cambri LT, de Araujo GG, Ghezzi AC, Botezelli JD, Mello MA. Metabolic responses to acute physical exercise in young rats recovered from fetal protein malnutrition with a fructose-rich diet. Lipids Health Dis 10: 164, 2011. doi: 10.1186/1476-511X-10-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cannizzo B, Luján A, Estrella N, Lembo C, Cruzado M, Castro C. Insulin resistance promotes early atherosclerosis via increased proinflammatory proteins and oxidative stress in fructose-fed ApoE-KO mice. Exp Diabetes Res 2012: 941304, 2012. doi: 10.1155/2012/941304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castro MC, Francini F, Schinella G, Caldiz CI, Zubiría MG, Gagliardino JJ, Massa ML. Apocynin administration prevents the changes induced by a fructose-rich diet on rat liver metabolism and the antioxidant system. Clin Sci (Lond) 123: 681–692, 2012. doi: 10.1042/CS20110665. [DOI] [PubMed] [Google Scholar]
- 9.Elliott SS, Keim NL, Stern JS, Teff K, Havel PJ. Fructose, weight gain, and the insulin resistance syndrome. Am J Clin Nutr 76: 911–922, 2002. [DOI] [PubMed] [Google Scholar]
- 10.Esposito K, Giugliano D. Diet and inflammation: a link to metabolic and cardiovascular diseases. Eur Heart J 27: 15–20, 2006. doi: 10.1093/eurheartj/ehi605. [DOI] [PubMed] [Google Scholar]
- 11.Fisslthaler B, Dimmeler S, Hermann C, Busse R, Fleming I. Phosphorylation and activation of the endothelial nitric oxide synthase by fluid shear stress. Acta Physiol Scand 168: 81–88, 2000. doi: 10.1046/j.1365-201x.2000.00627.x. [DOI] [PubMed] [Google Scholar]
- 12.Ghezzi AC, Cambri LT, Ribeiro C, Botezelli JD, Mello MA. Impact of early fructose intake on metabolic profile and aerobic capacity of rats. Lipids Health Dis 10: 3, 2011. doi: 10.1186/1476-511X-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gryglewski RJ, Palmer RM, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 320: 454–456, 1986. doi: 10.1038/320454a0. [DOI] [PubMed] [Google Scholar]
- 14.Huang A, Sun D, Jacobson A, Carroll MA, Falck JR, Kaley G. Epoxyeicosatrienoic acids are released to mediate shear stress-dependent hyperpolarization of arteriolar smooth muscle. Circ Res 96: 376–383, 2005. doi: 10.1161/01.RES.0000155332.17783.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang A, Sun D, Kaley G, Koller A. Superoxide released to high intra-arteriolar pressure reduces nitric oxide-mediated shear stress- and agonist-induced dilations. Circ Res 83: 960–965, 1998. doi: 10.1161/01.RES.83.9.960. [DOI] [PubMed] [Google Scholar]
- 16.Huang A, Yang YM, Feher A, Bagi Z, Kaley G, Sun D. Exacerbation of endothelial dysfunction during the progression of diabetes: role of oxidative stress. Am J Physiol Regul Integr Comp Physiol 302: R674–R681, 2012. doi: 10.1152/ajpregu.00699.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang A, Yang YM, Yan C, Kaley G, Hintze TH, Sun D. Altered MAPK signaling in progressive deterioration of endothelial function in diabetic mice. Diabetes 61: 3181–3188, 2012. doi: 10.2337/db12-0559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iyer SN, Katovich MJ, Raizada MK. Changes in angiotensin AT1 receptor density during hypertension in fructose-fed rats. Adv Exp Med Biol 396: 49–58, 1996. doi: 10.1007/978-1-4899-1376-0_6. [DOI] [PubMed] [Google Scholar]
- 19.Jacobson A, Yan C, Gao Q, Rincon-Skinner T, Rivera A, Edwards J, Huang A, Kaley G, Sun D. Aging enhances pressure-induced arterial superoxide formation. Am J Physiol Heart Circ Physiol 293: H1344–H1350, 2007. doi: 10.1152/ajpheart.00413.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jalal DI, Smits G, Johnson RJ, Chonchol M. Increased fructose associates with elevated blood pressure. J Am Soc Nephrol 21: 1543–1549, 2010. doi: 10.1681/ASN.2009111111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeukendrup AE. Carbohydrate intake during exercise and performance. Nutrition 20: 669–677, 2004. doi: 10.1016/j.nut.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 22.Johnson RJ, Murray R. Fructose, exercise, and health. Curr Sports Med Rep 9: 253–258, 2010. doi: 10.1249/JSR.0b013e3181e7def4. [DOI] [PubMed] [Google Scholar]
- 23.Johnson RK, Appel LJ, Brands M, Howard BV, Lefevre M, Lustig RH, Sacks F, Steffen LM, Wylie-Rosett J; American Heart Association Nutrition Committee of the Council on Nutrition, Physical Activity, and Metabolism and the Council on Epidemiology and Prevention . Dietary sugars intake and cardiovascular health: a scientific statement from the American Heart Association. Circulation 120: 1011–1020, 2009. doi: 10.1161/CIRCULATIONAHA.109.192627. [DOI] [PubMed] [Google Scholar]
- 24.Koller A, Huang A, Sun D, Kaley G. Exercise training augments flow-dependent dilation in rat skeletal muscle arterioles. Role of endothelial nitric oxide and prostaglandins. Circ Res 76: 544–550, 1995. doi: 10.1161/01.RES.76.4.544. [DOI] [PubMed] [Google Scholar]
- 25.Koller A, Sun D, Huang A, Kaley G. Corelease of nitric oxide and prostaglandins mediates flow-dependent dilation of rat gracilis muscle arterioles. Am J Physiol Heart Circ Physiol 267: H326–H332, 1994. [DOI] [PubMed] [Google Scholar]
- 26.Laughlin MH, Oltman CL, Bowles DK. Exercise training-induced adaptations in the coronary circulation. Med Sci Sports Exerc 30: 352–360, 1998. doi: 10.1097/00005768-199803000-00004. [DOI] [PubMed] [Google Scholar]
- 27.Lê KA, Ith M, Kreis R, Faeh D, Bortolotti M, Tran C, Boesch C, Tappy L. Fructose overconsumption causes dyslipidemia and ectopic lipid deposition in healthy subjects with and without a family history of type 2 diabetes. Am J Clin Nutr 89: 1760–1765, 2009. doi: 10.3945/ajcn.2008.27336. [DOI] [PubMed] [Google Scholar]
- 28.Lo S, Russell JC, Taylor AW. Determination of glycogen in small tissue samples. J Appl Physiol 28: 234–236, 1970. [DOI] [PubMed] [Google Scholar]
- 29.Marchionni N, Moschi G, Di Bari M, Ferrucci L, Paoletti M, Salani B, Fattirolli F. Improved exercise tolerance by i.v. fructose-1,6-diphosphate in chronic, stable angina pectoris. J Clin Pharmacol 28: 807–811, 1988. doi: 10.1002/j.1552-4604.1988.tb03220.x. [DOI] [PubMed] [Google Scholar]
- 30.Myers J, Atwood JE, Forbes S, Evans B, Froelicher V. Effect of fructose 1,6-diphosphate on exercise capacity in patients with peripheral vascular disease. Int J Sports Med 11: 259–262, 1990. doi: 10.1055/s-2007-1024803. [DOI] [PubMed] [Google Scholar]
- 31.Ngo Sock ET, Lê KA, Ith M, Kreis R, Boesch C, Tappy L. Effects of a short-term overfeeding with fructose or glucose in healthy young males. Br J Nutr 103: 939–943, 2010. doi: 10.1017/S0007114509992819. [DOI] [PubMed] [Google Scholar]
- 32.Ripari P, Pieralisi G. Effects of fructose-1,6-diphosphate on heart rate, ventilation, oxygen consumption and endurance performance. Pharmatherapeutica 5: 249–255, 1988. [PubMed] [Google Scholar]
- 33.Shen W, Xu X, Ochoa M, Zhao G, Wolin MS, Hintze TH. Role of nitric oxide in the regulation of oxygen consumption in conscious dogs. Circ Res 75: 1086–1095, 1994. doi: 10.1161/01.RES.75.6.1086. [DOI] [PubMed] [Google Scholar]
- 34.Song S, Kertowidjojo E, Ojaimi C, Martin-Fernandez B, Kandhi S, Wolin M, Hintze TH. Long-term methionine-diet induced mild hyperhomocysteinemia associated cardiac metabolic dysfunction in multiparous rats. Physiol Rep 3: 3, 2015. doi: 10.14814/phy2.12292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stanhope KL, Havel PJ. Fructose consumption: potential mechanisms for its effects to increase visceral adiposity and induce dyslipidemia and insulin resistance. Curr Opin Lipidol 19: 16–24, 2008. doi: 10.1097/MOL.0b013e3282f2b24a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun D, Cuevas AJ, Gotlinger K, Hwang SH, Hammock BD, Schwartzman ML, Huang A. Soluble epoxide hydrolase-dependent regulation of myogenic response and blood pressure. Am J Physiol Heart Circ Physiol 306: H1146–H1153, 2014. doi: 10.1152/ajpheart.00920.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun D, Huang A, Koller A, Kaley G. Enhanced NO-mediated dilations in skeletal muscle arterioles of chronically exercised rats. Microvasc Res 64: 491–496, 2002. doi: 10.1006/mvre.2002.2450. [DOI] [PubMed] [Google Scholar]
- 38.Tappy L. Q&A: ‘toxic’ effects of sugar: should we be afraid of fructose? BMC Biol 10: 42, 2012. doi: 10.1186/1741-7007-10-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thorburn AW, Storlien LH, Jenkins AB, Khouri S, Kraegen EW. Fructose-induced in vivo insulin resistance and elevated plasma triglyceride levels in rats. Am J Clin Nutr 49: 1155–1163, 1989. [DOI] [PubMed] [Google Scholar]
- 40.Vollenweider P, Tappy L, Randin D, Schneiter P, Jéquier E, Nicod P, Scherrer U. Differential effects of hyperinsulinemia and carbohydrate metabolism on sympathetic nerve activity and muscle blood flow in humans. J Clin Invest 92: 147–154, 1993. doi: 10.1172/JCI116542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wolin MS. Reactive oxygen species and the control of vascular function. Am J Physiol Heart Circ Physiol 296: H539–H549, 2009. doi: 10.1152/ajpheart.01167.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang YM, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol 297: H1829–H1836, 2009. doi: 10.1152/ajpheart.00230.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao G, Bernstein RD, Hintze TH. Nitric oxide and oxygen utilization: exercise, heart failure and diabetes. Coron Artery Dis 10: 315–320, 1999. doi: 10.1097/00019501-199907000-00007. [DOI] [PubMed] [Google Scholar]