

Abstract
Purpose
Vascular endothelial growth factor receptor 2 (VEGFR2) plays a key role in VEGF-induced angiogenesis. The goal of this project was to test the hypothesis that editing genomic VEGFR2 loci using the technology of clustered regularly interspaced palindromic repeats (CRISPR)-associated DNA endonuclease (Cas)9 in Streptococcus pyogenes (SpCas9) was able to block VEGF-induced activation of Akt and tube formation.
Methods
Four 20 nucleotides for synthesizing single-guide RNAs based on human genomic VEGFR2 exon 3 loci were selected and cloned into a lentiCRISPR v2 vector, respectively. The DNA fragments from the genomic VEGFR2 exon 3 of transduced primary human retinal microvascular endothelial cells (HRECs) were analyzed by Sanger DNA sequencing, surveyor nuclease assay, and next-generation sequencing (NGS). In the transduced cells, expression of VEGFR2 and VEGF-stimulated signaling events (e.g., Akt phosphorylation) were determined by Western blot analyses; VEGF-induced cellular responses (proliferation, migration, and tube formation) were examined.
Results
In the VEGFR2-sgRNA/SpCas9–transduced HRECs, Sanger DNA sequencing indicated that there were mutations, and NGS demonstrated that there were 83.57% insertion and deletions in the genomic VEGFR2 locus; expression of VEGFR2 was depleted in the VEGFR2-sgRNA/SpCas9–transduced HRECs. In addition, there were lower levels of Akt phosphorylation in HRECs with VEGFR2-sgRNA/SpCas9 than those with LacZ-sgRNA/SpCas9, and there was less VEGF-stimulated Akt activation, proliferation, migration, or tube formation in the VEGFR2-depleted HRECs than those treated with aflibercept or ranibizumab.
Conclusions
The CRISPR-SpCas9 technology is a potential novel approach to prevention of pathologic angiogenesis.
Keywords: CRISPR, Cas9, VEGF, VEGFR2, tube formation
Vascular endothelial growth factor, originally known as vascular permeability factor, is a critical regulator of both physiologic angiogenesis during embryogenesis, skeletal growth, and reproductive functions and the pathologic angiogenesis associated with tumors, intraocular neovascular disorders, and other conditions.1,2 Angiogenesis is the process through which new blood vessels grow from preexisting vasculature.2–4 In mammals, the VEGF family is comprised of five members, VEGF-A, -B, -C, and -D, and placenta growth factor (PlGF). In this family, the first discovered, VEGF-A (also called VEGF), can stimulate endothelial cell mitogenesis and migration, and it is a key regulator of blood vessel growth.1,2 The human VEGF-A gene is organized as eight exons separated by seven introns, and their alternative exon splicing can result in the generation of four different isoforms (VEGF121, VEGF165, VEGF189, and VEGF206).1,2,5 Native VEGF is a heparin-binding homodimeric glycoprotein of 45 kDa, and its properties closely correspond to those of VEGF165.1,6
In the family of VEGF receptors (VEGFRs), there are three members (VEGFR1, -2, and -3), and VEGFR1 and -2 consist of seven immunoglobulin-like domains in their extracellular domains, a single transmembrane region, and a consensus tyrosine kinase sequence that is interrupted by a kinase-insert domain.1,7,8 On VEGF binding, VEGFR2 undergoes dimerization and autophosphorylation on several tyrosine residues, activating downstream signaling pathways including phosphoinositide-3 kinase (PI3K)/Akt9 and Raf-Mek/Erk.10 It is currently believed that VEGFR2 (also known as kinase domain region [KDR] or Flk-1) is the major mediator of the known VEGF-induced output including microvascular permeability and neovascularization.1,11
Abnormal angiogenesis is associated with proliferative diabetic retinopathy (PDR), retinopathy of prematurity (ROP), and neovasular AMD.12 Without timely treatment, the fragile new vessels leak blood into the vitreous, blur vision, destroy the retina, and can lead to blindness. Preventing VEGF-stimulated activation of its receptors with neutralizing VEGF antibodies (e.g., ranibizumab) and the fusion of extracellular domains of VEGFR1 and -2 (aflibercept) has become an important therapeutic approach to angiogenesis in these eye diseases.13–15 Although these drugs can lessen vessel leakage and angiogenesis, continuous intraocular injections are necessary. Even though the overall risk of endophthalmitis and retinal detachment during the repeat injection appears to be low,16,17 these repetitive injections increase burden to both of physicians and patients.
The clustered regularly interspaced palindromic repeats (CRISPR)-associated DNA endonuclease (Cas)9 in Streptococcus pyogenes (SpCas9) processes pre-crRNA transcribed from the repeat spacer into CRISPR RNA (crRNA) and cleaves invading nucleic acids at the direction of crRNA and transactivating crRNA (tracrRNA).18 In SpCa9, there are two active domains (HNH and RuvC), each of which can cleave one strand of the foreign double-stranded DNA (dsDNA). The site-specific cleavage relies on both base-pairing complementarity of the crRNA with the target protospacer DNA and the protospacer adjacent motif (PAM). A short single guide RNA (sgRNA) consisting of the crRNA and tracrRNA can guide SpCas9 specifically to cleave dsDNA. This CRISPR/Cas9 system is an efficient tool for generation of mutations in eukaryotic genomes and subsequent protein depletion and provides a novel opportunity for therapeutic genome editing in diseased cells and tissues.19–22
In this study, we successfully delivered a CRISPR/Cas9 system into primary human retinal microvascular endothelial cells (HRECs) for editing the genomic VEGFR2 exon 3 and found that the CRISPR/Cas9-mediated depletion of VEGFR2 was able to block VEGF-induced activation of Akt, cell proliferation, migration, and tube formation, suggesting that editing the genomic VEGFR2 locus by the CRISPR/Cas9 is a potentially powerful approach to preventing pathologic angiogenesis.
Materials and Methods
Major Reagents
Vascular endothelial growth factor was purchased from R&D systems (Minneapolis, MN, USA). Antibodies against phosphor-VEGFR2 (p-VEGFR2, Y1175), VEGFR2, Akt, p-Akt (S473), Caspase (cysteine-aspartic proteases) 9, and p-BAD (Cell Signaling Technology; Danvers, MA, USA). Aflibercept (40 μg/μL) and ranibizumab (10 μg/μL) were from the pharmacy of Massachusetts Eye and Ear (Boston, MA, USA). The primary antibody against β-actin and secondary antibodies of the horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG and anti-mouse IgG were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Enhanced chemiluminescent substrate for detection of HRP was obtained from Thermo Fisher Scientific (Waltham, MA, USA).
DNA Constructs
The four 20-nt target DNA sequences preceding a 5′-NGG PAM sequence at exon 3 in the genomic VEGFR2 locus (NC_000004.12)19 were selected for generating single-guide RNA (sgRNA) for SpCas9 targets using the CRISPR design website (http://crispr.mit.edu, in the public domain). The four target sequences were 5′-AGCCTACAAGTGCTTCTACC-3′ (K11), 5′-TTCCCGGTAGAAGCACTTGT-3′ (K12), 5′-CTTCTACCGGGAAACTGACT-3′ (K13), and 5′-GTGTCATTTCCGATCACTTT-3′ (K14). The control sgRNA sequence (5′-TGCGAATACGCCCACGCGATGGG-3′) was designed to target the lacZ gene from Escherichia coli.19 The lentiCRISPR v2 vector23 was purchased from Addgene (Cat. 52961; Cambridge, MA, USA).
To express SpGuides in the targeted cells, the oligos of top oligos 5′-CACCG-20 nt (target VEGFR2 DNA sequences K11, 12 13, and 14 or the lacZ sgRNA sequence)-3′ and bottom oligos: 5′-AAAC-20 nt (20 nt: complimentary target VEGFR2 DNA sequences or lacZ sgRNA sequence)-C-3′ were annealed and cloned into the lentiCRISPR v2 vector by BsmB1 (New England Biolabs, Boston, MA, USA), respectively. All clones were confirmed by DNA sequencing using a primer 5′-GGACTATCATATGCTTACCG-3′ from the sequence of U6 promoter that drives expression of sgRNAs. Both synthesis of primers and oligos and sequencing of PCR products and clones were performed at the Massachusetts General Hospital (MGH) DNA Core Facility (Cambridge, MA, USA).
Cell Culture and Transfection
Porcine aortic endothelial (PAE) cells overexpressing human VEGFR2 (PAE-KDR cells)24,25 were cultured in a 1:1 mixture of DMEM and Ham's F-12 Nutrient Mixture (Thermo Fisher Scientific, Grand Island, NY, USA), which was supplemented with 10% fetal bovine serum (FBS; Lonza, Walkersville, MD, USA), 100 U/mL penicillin G, and 100 mg/mL streptomycin (Gemini BioProducts, West Sacramento, CA, USA). Primary human retinal microvascular endothelial cells (HRECs) were purchased from Cell Systems (Kirkland, WA, USA) and cultured in an endothelial growth medium (EGM)-2 kit (Lonza). Human embryonic kidney (HEK) 293T cells (HEK 293 containing SV40 T-antigen) from The Dana-Farber Cancer Institute/Harvard Medical School (Boston, MA, USA) were cultured in high-glucose (4.5 g/L) DMEM supplemented with 10% FBS. The medium used to harvest the lentiviral supernatant from 293T cells was high-glucose DMEM supplemented with 30% FBS. All cells were cultured at 37°C in a humidified 5% CO2 atmosphere.26
Production of Lentivirus
The lentiCRISPR v2 vector inserted with sgRNA (2000 ng), the packaging plasmid psPAX2 (Addgene: 12260) (900 ng), and the envelope plasmid VSV-G (Addgene: 8454) (100 ng) were mixed together and then added to a mixture of 6 μL lipofectamine 3000 (Thermo Fisher Scientific) with 90 μL OPTI-MEM (Thermo Fisher Scientific). This transfection mix was incubated at room temperature for 30 minutes and then carefully transferred into a 60-mm cell culture dish with HEK 293T cells that were approximately 70% confluent without antibiotics. After 18 hours (37°C, 5% CO2), the medium was replaced with growth medium supplemented with 30% FBS, and at 24 hours after the medium change, lentiviruses were harvested. The viral harvest was repeated at 24-hour intervals three times. The virus-containing media were pooled, centrifuged at 800g for 5 minutes, and the supernatant was used to infect PAEC-KDR cells supplemented with 8 μg/mL polybrene (Sigma-Aldrich Corp., St. Louis, MO, USA). To infect HRECs, the pooled and clarified supernatants from transfected HEK 293T cells with the plasmids described above in a 250-mm cell culture dish were then pelleted in sterile tubes at 25,000g for 90 minutes, to concentrate the viruses. Finally, the viral pellets were resuspended in 300 μL sterile TNE buffer (50 mM Tris, pH 7.8, 130 mM NaCl, 1 mM EDTA), transferred into microtubes, and then dissolved at 4°C with gentle rotation overnight. Next these dissolved retroviruses were titered for infecting HRECs in combination with 8 μg/mL polybrene or kept at −80°C.26–28 The infected cells were selected in media using puromycin (Sigma-Aldrich Corp.; 0.5 μg/mL), and the resulting cells were examined by Western blotting.26–28
Titering of Lentiviruses
The titering of the virus was carried out using a colony forming assay as described previously.29 On day 1, NIH 3T3 TK cells (American Type Culture Collection, Manassas, VA, USA) were plated in a six-well plate (5 × 104 cells per well) overnight in the media of DMEM supplemented with 10% FBS; on day 2, the media were changed into those containing polybrene (8 μg/mL), and serial dilutions (10−1, 10−2, 10−3, 10−4, 10−5, 10−6) of the viral stock in TNE buffer were added into the plates (10 μL/well), respectively; on day 3, the cells were split into two dishes of 10 cm in the media-containing puromycin (2 μg/mL); on day 8, the cells were grown into colonies of 25 to 50 cells, which could be visualized under the microscope, the colonies were stained with 0.1% Coomassie blue, and the titer was calculated as CFU (concentration, expressed as colony forming units per milliliter).
Western Blotting Analysis
Human retinal microvascular endothelial cells at 90% confluence in a 24-well plate were deprived from serum and growth factors for continuous incubation for 7 hours, and then some of these cells were pretreated with aflibercept (0.8 mg/mL) or ranibizumab (0.2 mg/mL) for 15 minutes. Subsequently, the serum-deprived cells were treated for 30 minutes with (1) VEGF (20 ng/mL alone) or (2) VEGF (20 ng/mL) plus aflibercept (0.8 mg/mL) or ranibizumab (0.2 mg/mL). After washing twice with ice-cold PBS, cells were lysed in 1× sample buffer, which was diluted with extraction buffer (10 mM Tris-HCl, pH 7.4, 5 mM EDTA, 50 mM NaCl, 50 mM NaF, 1% Triton X-100, 20 μg/mL aprotinin, 2 mM Na3VO4, and 1 mM phenylmethylsulfonyl fluoride) from the 5× protein sample buffer (25 mM EDTA [pH = 7.0], 10% SDS [Sigma-Aldrich Corp.], 500 mM dithiothreitol, 50% sucrose, 500 mM Tris HCl [pH = 6.8], and 0.5% bromophenol blue). The samples were boiled for 5 minutes and then centrifuged for 5 minutes at 13,000g. Proteins from the centrifuged and heated samples were separated by 10% SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membranes, and subjected to Western blot analyses using the appropriate antibodies. Experiments were repeated at least three times. Signal intensity was determined by densitometry using ImageJ software (National Institutes of Health, Bethesda, MD, USA).26
Surveyor Nuclease Assay and DNA Sequencing
Some transduced cells were pelleted for genomic DNA extraction using the QuickExtract DNA Extraction Solution (Epicenter, Chicago, IL, USA) following the manufacturer's protocol. In brief, the pelleted cells were resuspended in the QuickExtract solution, vortexed for 15 seconds, incubated at 65°C for 6 minutes, vortexed for 15 seconds, and then incubated at 98°C for 5 minutes. The genomic region approximately 200 bp around the PAM was PCR amplified with high-fidelity Herculase II DNA polymerases (Agilent Technologies, Santa Clara, CA, USA). The PCR primers for PAEC-KDR cells were P13F (forward 5′-GAGGGACTTGGACTGGCTTT-3′) and P13R (reverse 5′-CCACTGAACCGGCCCAATC-3′) and for HRECs were P12F2 (forward 5′-TAACACGGCCCAAGCTCCAG-3′) and P12R (reverse 5′-GGGACTTGGACTGGCTTTGG-3′). The PCR products were separated in 2% agarose gel and purified with a gel extraction kit (Thermo Fisher Scientific) for Sanger DNA sequencing and next-generation sequencing (NGS). DNA sequencing was performed by the MGH DNA core facility, and a surveyor nuclease assay was performed according to the manufacturer's instructions (Integrated DNA Technologies, Coralville, IA, USA). Briefly, the purified PCR products (300 ng) were incubated with the surveyor nuclease and surveyor enhancer S with an additional 1/10 MgCl2 (0.15 M) for 30 minutes at 42°C following the manufacturer's recommended protocol.30
Cell Proliferation Assay
Human retinal microvascular endothelial cells were seeded into 24-well plates at a density of 30,000 cells per well in an endothelial growth medium kit. After attaching the plates, the cells were starved for growth factors for 7 hours. Some of these cells were pretreated with aflibercept (0.8 mg/mL) or ranibizumab (0.2 mg/mL) for 15 minutes and then VEGF (20 ng/mL) was added into the pretreated wells. The treatment was repeated daily. After 48 hours, the cells were trypsin detached and then counted in a hemocytometer under a light microscope. A minimum of three independent experiments were performed.25,26
Wound Healing Assay
The wound healing assay was performed as previously described31 with minor modifications. Once cells reached 80% confluence in 48-well plates, they were starved for growth factors for 7 hours. After the cell monolayer was scraped with a sterile pipette tip (200 μL), the cells were washed twice to remove detached cells. The cells were pretreated with aflibercept (0.8 mg/mL) or ranibizumab (0.2 mg/mL) for 15 minutes before VEGF (20 ng/mL) was added the wells. The wound was photographed at 0 and 18 hours after wounding with an EVOS FL Auto microscope (Thermo Fisher Scientific). Quantification was done by measuring the number of pixels in the wound area using Adobe Photoshop (Adobe Systems, San Jose, CA, USA) and analyzed using ImageJ software.25
Tube Formation Assay
This assay was performed as previously described.32 Briefly, 5 mL of a collagen mixture was prepared on ice as follows: 0.5 mL of 10× Roswell Park Memorial Institute medium 1640 (RPMI) (a porch of RPMI powder [Thermo Fisher Scientific] dissolved in 90 mL distilled H2O and brought to 100 mL as a total volume), 0.4 ml of NaHCO3/NaOH solution (2 g of NaHCO3 and 4 mL 10 N NaOH in 70 ml distilled H2O), 0.1 mL (4-(2-hydroxyethyl)-1-piperazineethane sulfonic acid) (HEPES) (Thermo Scientific), 2.5 μL fibronectin (Sigma-Aldrich Corp.), 2.5 μL laminin (Sigma-Aldrich Corp.), and 4.0 mL PureCol type I bovine collagen solution (Advanced BioMatrix, San Diego, CA, USA). The pH of the collagen mixture was adjusted to the range of 7.0 to 7.5 using 6 N HCl. This collagen gel mixture was added to a 96-well plate (70 μL/well), which was then incubated for about 60 minutes at 37°C to let the collagen gel polymerize. After polymerization, HRECs (4.5 × 104) were seeded in each well with their cultured medium maintained in a 37°C incubator. This day was considered day 1. On day 2, the medium was removed, and 30 μL gel mixture was added to each well. After incubation for about 60 minutes at 37°C, the collagen gel was polymerized, and the medium (100 μL) (endothelial cell basal medium [EBM] supplemented with 0.5% horse serum, 0.1% bovine brain extract supplemented with VEGF [20 ng/mL] or VEGF [20 ng/mL] plus aflibercept [0.8 mg/mL] or ranibizumab [0.2 mg/mL]) was added. On days 3 and 4, three different fields per well were randomly chosen and photographed using the EVOS FL Auto microscope. Images were captured using Adobe Photoshop, and the data were imported as a TIFF file into ImageJ software. After calibration with a stage micrometer, the total length of all tubing with each field was measured, and the data were analyzed using Prism 6 software (GraphPad Software, San Diego, CA, USA). Experiments were performed a minimum of three times.25,33,34
Results
Depletion of VEGFR2 Expression in PAE-KDR Cells and HRECs Using CRISPR/Cas9
Vascular endothelial growth factor receptor 2 plays a critical role in pathologic angiogenesis.1,11 To screen sgRNAs for guiding SpCas9 to edit the genomic VEGFR2 locus, we cloned four DNA targets (K11, 12, 13, and 14; Fig. 1A) into a lentiCRISPR v2 vector that expressed both a CRISPR cassette and SpCas9. The resulting lentiviruses were used to infect PAE-KDR cells because these cells are relatively easy to manage.25 Western blot analysis showed that VEGFR2 was depleted about 90% in the PAE-KDR cells transduced by K12-sgRNA and SpCas9 (Fig. 1B), indicating that K12-sgRNA was able to direct SpCas9 to cleave the VEGFR2 exon 3, resulting in depletion of VEGFR2 in the transduced PAE-KDR cells. A nuclease survey assay indicated that there were mutations in the VEGFR2 exon 3 amplified by PCR from the K12-sgRNA/SpCas9 transduced PAE-KDR cells but not from the lacZ-sgRNA/SpCas9 transduced cells (Fig. 1C). As expected, Sanger DNA sequencing of the PCR products demonstrated that there were mutations beginning from the third position prior to the PAM in the K12-sgRNA/SpCas9 transduced PAE-KDR cells but not in the lacZ-sgRNA PAE-KDR cells (Fig. 1D). The NGS indicated that there were 77.22% insertions/deletions (indels) in and around the PAM region from the K12-sgRNA/SpCas9 edited PAE-KDR cells (Fig. 1E) and that there was only a single normal DNA sequence from the lacZ-sgRNA/SpCas9 transduced PAE-KDR cells. These results demonstrate that the K12-sgRNA is able to guide SpCas9 to cleave the genomic VEGFR2 locus at the expected site, resulting in nonhomologous end-joining (NHEJ) repair and subsequent depletion of VEGFR2 in the PAE-KDR cells.
Figure 1.
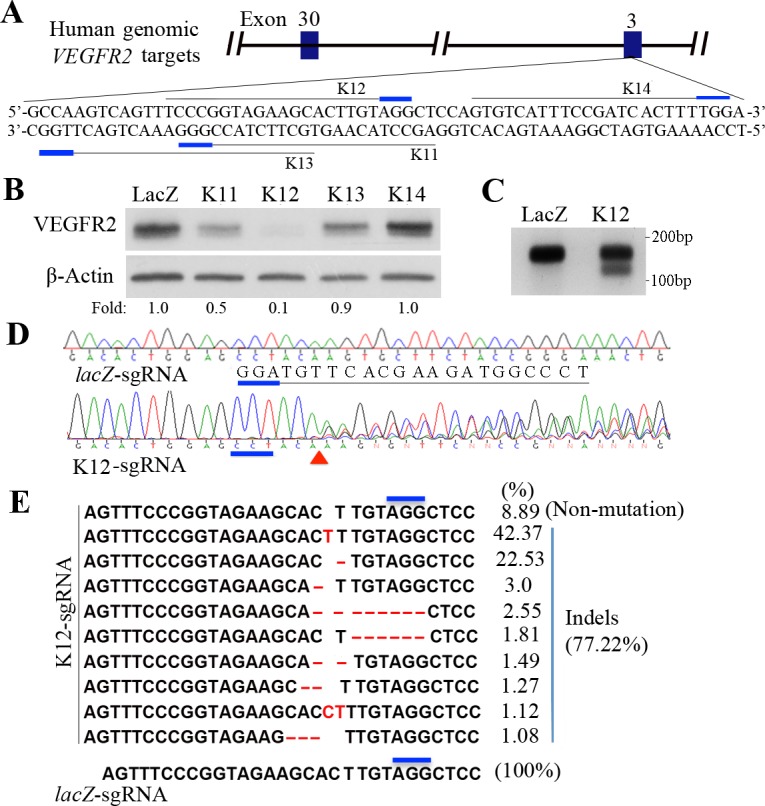
Screening sgRNAs for guiding SpCas9 to edit human VEGFR2 in PAE-KDR cells (A) Schematic of human genomic VEGFR2 targets. Four targets for generating respective sgRNAs were selected at the human genomic VEGFR2 exon 3 and were named K11, K12, K13, and K14. The PAM sequences are marked in short thick blue lines. (B) Western blot analysis of VEGFR2 expression in the CRISPR/Cas9-engineered PAE-KDR cells using indicated antibodies. lacZ-sgRNA served as a negative control sgRNA. Lanes of LacZ, K11, 12, 13, and 14 denote the protein samples from PAE-KDR cells, which were transduced by SpCas9 together with lacZ-, K11-, K12-, K13-, or K14-sgRNAs. “Fold” was calculated by first normalizing to the level of β-actin and then calculating the ratio of the K11, K12, K13, and K14 over the LacZ lane, respectively. This is representative of two independent experiments. (C) Purified PCR products (300 ng) from the PAE-KDR cells transduced by SpCas9 together with lacZ-sgRNA (LacZ) or K12-sgRNA (K12) were subjected to a survey nuclease assay. The purified DNA fragments were incubated with surveyor nuclease and surveyor enhancer S with an additional 1/10 MgCl2 (0.15 M) for 30 minutes at 42°C and then separated in a 2% agarose gel stained with ethidium bromide. The PCR primers used were P13F (5′-GAGGGACTTGGACTGGCTTT-3′ [forward]) and P13R (5′-GACCGAGGCCAAGTCAGTTT-3′ [reverse]). (D) Purified PCR products from the CRISPR/Cas9-edited PAE-KDR cells in C were subjected to Sanger DNA sequencing. The middle line indicates the locus of human genomic VEGFR2 DNA sequence. The DNA sequencing results, indicated by lacZ-sgRNA and K12-sgRNA, were derived from the PAE-KDR cells transduced by SpCas9 together with lacZ-sgRNA or K12-sgRNA. The PAMs are indicated below a thick blue line, and the expected cleavage site of SpCas9 is indicated by a red triangle. (E) Purified PCR products from the PAE-KDR cells transduced by SpCas9 with K12-sgRNA or lacZ-sgRNA were subjected to NGS. There were 10 different DNA sequences in the NGS results from the K12-sgRNA/SpCas9-transduced PAEC-KDR cells. These included a normal DNA sequence (8.89%) and indel sequences (77.22%). Only one normal DNA sequence (100%) resulted from the lacZ-sgRNA/SpCas9–transduced PAE-KDR cells. The thick blue lines indicate the PAM sequence. Red letters (C, T) denote inserted nucleotides, and red lines indicate deleted nucleotides.
To test whether the CRISPR/Cas9 system worked in HRECs, the lentiviruses, which were used to infect PAE-KDR cells, were used to infect HRECs. However, these lentiviruses harvested from the cultured medium of transfected HEK293T cells failed to infect HRECs. Thus, high-speed centrifuging (25,000g for 90 minutes) was used to pellet the lentiviruses. These concentrated lentiviruses infected HRECs (5 CFUs/cell) successfully. Subsequently, a DNA fragment within the genomic VEGFR2 exon 3 was amplified from the transduced HRECs by the K12-sgRNA (or lacZ-sgRNA) together with SpCas9. Sanger DNA sequencing indicated that there were mutations around the PAM in the K12-sgRNA/SpCas9–transduced HRECs but not in the control lacZ-sgRNA/SpCas9–transduced cells (Figs. 2A and 2B); NGS showed that there were 83.57% indels around the PAM in the K12-sgRNA/SpCas9 transduced HRECs, but none in the lacZ-sgRNA–transduced cells (Fig. 2C). Importantly, expression of mature VEGFR2 was depleted almost completely in the K12-sgRNA/SpCas9–transduced cells, but not in the lacZ-sgRNA/SpCas9–transduced HRECs (Fig. 2D). These experiments further demonstrate that the K12-sgRNA is capable of guiding SpCas9 to cleave the genomic VEGFR2 locus at the expected position, leading to a subsequent NHEJ DNA repair, resulting in indels and depletion of VEGFR2 expression in the HRECs.
Figure 2.
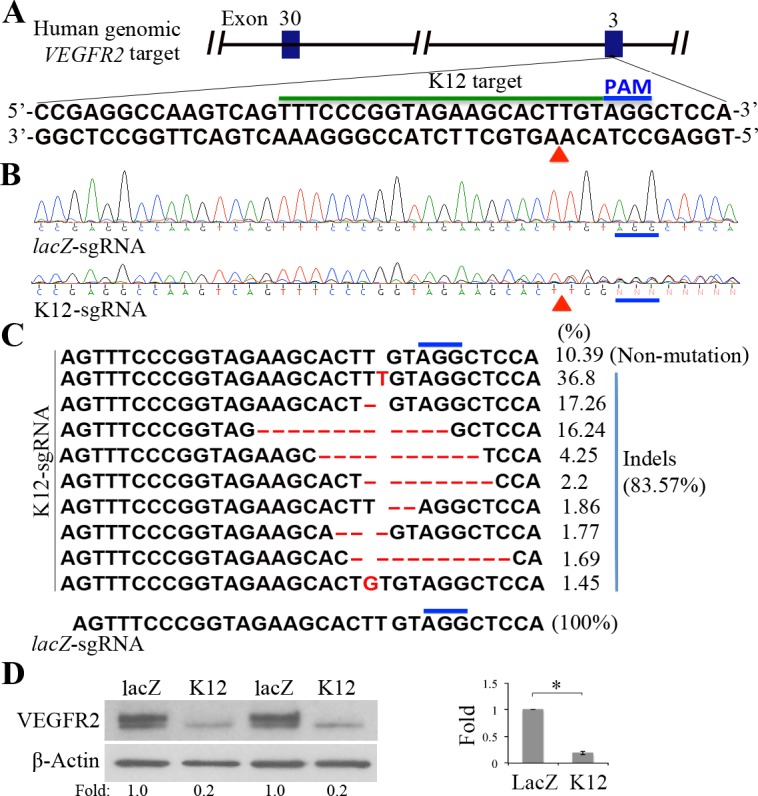
Editing the genomic VEGFR2 using CRISPR/Cas9 in HRECs. (A) Schematic of a K12-sgRNA target at the human genomic VEGFR2 exon 3. The K12 target DNA sequence and the PAM sequence are indicated below a black line and blue line, respectively. The red triangle points to an expected cleavage site of SpCas9 at the human genomic VEGFR2 locus. (B) Purified PCR products from the CRISPR/Cas9-engineered HRECs were subjected to Sanger DNA sequencing. The DNA sequencing results, indicated by lacZ-sgRNA and K12-sgRNA, were derived from the HRECs transduced by SpCas9 together with lacZ-sgRNA or K12-sgRNA. The PAMs are indicated above a thick blue line and the expected cleavage site of SpCas9 is indicated by a red triangle. The PCR primers were P12F2 (forward: TAACACGGCCCAACTCCAG-3′) and P12R (reverse: 5′-GGGACTTGGACTGGCTTTGG-3′). (C) Purified PCR products from HRECs transduced by SpCas9 with K12-sgRNA or lacZ-sgRNA were subjected to NGS. There were 10 different DNA sequences found in the NGS results from K12-sgRNA/SpCas9–transduced HRECs, including a normal DNA sequence (10.39%) and indel sequences (83.57%). Only one normal DNA sequence (100%) resulted from the lacZ-sgRNA–transduced HRECs. Thick blue lines indicate the PAM sequence; red letters (G, T) denote inserted nucleotides; and red lines indicate deleted nucleotides. (D) Western blot analysis of VEGFR2 expression in the CRISPR/Cas9-edited HRECs using indicated antibodies. lacZ-sgRNA served as a negative sgRNA control. “Fold” was calculated by first normalizing to the level of β-actin and then calculating the ratio of the K12 over the LacZ lane. Each bar graph indicates mean ± SD of three independent experiments (folds). *Significant difference in results between the two compared groups. P < 0.05 using an unpaired t-test.
Editing VEGFR2 Using CRISPR/Cas9 Blocked VEGF-Induced Activation of Akt
Vascular endothelial growth factor binding to VEGFR2 triggers its downstream signaling transduction (e.g., PI3K/Akt).9,35,36 Akt, also known as protein kinase B, is a serine/threonine kinase that plays a key role in multiple cellular responses such as cell proliferation and migration, and angiogenesis.37–39 Aflibercept and ranibizumab, both of which can block VEGF binding to VEGFR2, are important drugs for certain eye diseases (e.g., neovascular AMD and PDR).40,41 To determine the effective concentrations of aflibercept and ranibizumab in blocking VEGF-stimulated phosphorylation of VEGFR2, we pretreated HRECs with aflibercept or ranibizumab with a serial of concentrations and then VEGF was added. As shown in Figures 3A and 3B, aflibercept and ranibizumab blocked VEGF-induced phosphorylation of VEGFR2 in concentrations of 1 and 2 μg/mL, respectively; in addition, molecular ratios of aflibercept and ranibizumab to VEGF were 1.65 and 7.92, respectively, in blocking 80% of VEGF-induced phosphorylation of VEGFR2.
Figure 3.
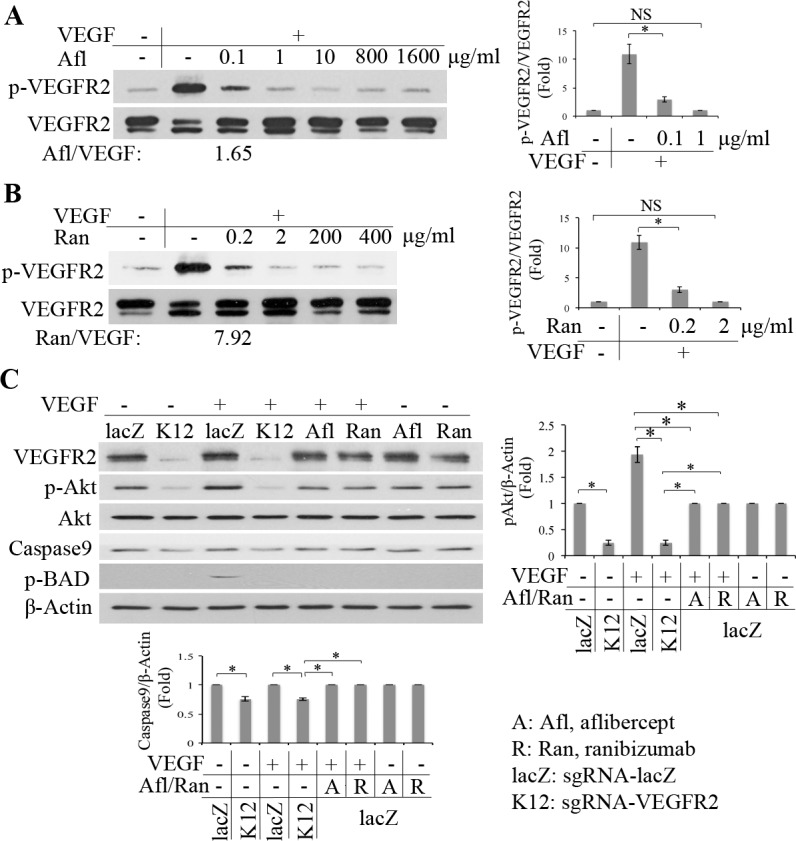
Editing VEGFR2 with use of CRISPR/Cas9 blocked VEGF-induced activation of Akt. (A, B) After HRECs were starved for growth factors overnight in wells of a 24-well plate, some of them were pretreated with a serial concentration of Afl:aflibercept (A) or Ran:ranibizumab (B) as indicated for 15 minutes, and then VEGF (20 ng/mL) was added into the cells. When the cells were treated with VEGF for 30 minutes, their lysates were subjected to Western blot using the indicated antibodies. The ratios of molecular numbers of Afl (aflibercept, 0. 1 μg/mL) or Ran (ranibizumab, 0.2 μg/mL) to VEGF (20 ng/mL) in the treated HRECs are 1.65 and 7.92, respectively. (C) The serum-deprived HRECs were pretreated with Afl:aflibercept (0.8 mg/mL) or Ran:ranibizumab (0.2 mg/mL) for 15 minutes, and then VEGF (20 ng/mL) was added. When the cells were treated with VEGF for 30 minutes, their lysates were subjected to Western blot using the antibodies indicated. The lanes of LacZ and K12 represent the cell lysates from HRECs transduced by SpCas9 together with lacZ-sgRNA or K12 sgRNA. In bar graphs of this figure, “Fold” was calculated by first normalizing to the level of β-actin and then calculating the ratio of the K12 and other lanes over the LacZ lane. Each bar graph indicates mean ± SD of three independent experiments (folds). *Significant difference in results between the two compared groups. P < 0.05 using an unpaired t-test.
We next compared the efficiency of editing the genomic VEGFR2 locus using CRISPR/Cas9 with aflibercept (800 μg/mL) or ranibizumab (200 μg/mL) in blocking VEGF-induced activation of Akt and its downstream effectors (e.g., p-BAD and Caspase 9). As shown in Figure 3C, VEGF induced phosphorylation of Akt and BAD in the lacZ-sgRNA/SpCas9 transduced cells, but failed to stimulate phosphorylation of Akt and BAD in the K12-sgRNA/SpCas9–transduced HRECs. In addition, depletion of VEGFR2 using the K12-sgRNA/SpCas9 suppressed constitutive activation of Akt and expression of Caspase 9 (Fig. 3C), indicating generation of more cleaved Caspase 9 and enhanced apoptosis in the HRECs with K12 than those with lacZ. Furthermore, as expected, both aflibercept and ranibizumab blocked VEGF-induced activation of Akt, but this blocking was less effective than that with K12-sgRNA/SpCas9–mediated depression of Akt activation (Fig. 3C). These data indicate that the CRISPR/Cas9-mediated depletion of VEGFR2 expression is alternative to that of either aflibercept or ranibizumab in blocking VEGF-induced activation of Akt in HRECs.
Editing VEGFR2 Using CRISPR/Cas9 Prevented VEGF-Induced Proliferation and Migration
Vascular endothelial growth factor–induced autophosphorylation of VEGFR2 stimulates its downstream signaling, which in turn triggers cellular responses including cell proliferation and migration.37–39 To examine VEGF-induced proliferation of HRECs that had been transduced by SpCas9 together with K12-sgRNA or lacZ-sgRNA, these HRECs were deprived of the growth factors for 7 hours and then treated with VEGF (20 ng/mL) for 48 hours.26,42 As expected, VEGF stimulated proliferation of HRECs transduced by SpCas9 with lacZ-sgRNA but failed to induce that of HRECs transduced by SpCas9 with K12-sgRNA (Fig. 4A). Also, as expected, aflibercept and ranibizumab prevented VEGF-induced cell proliferation, but more proliferation resulting from treatment with aflibercept or ranibizumab than that of HRECs transduced by SpCas9 withK12-sgRNA (Fig. 4A). These results indicate that editing genomic VEGFR2 using CRISPR/Cas9 is an alternative strategy to aflibercept or ranibizumab in blocking proliferation of HRECs.
Figure 4.
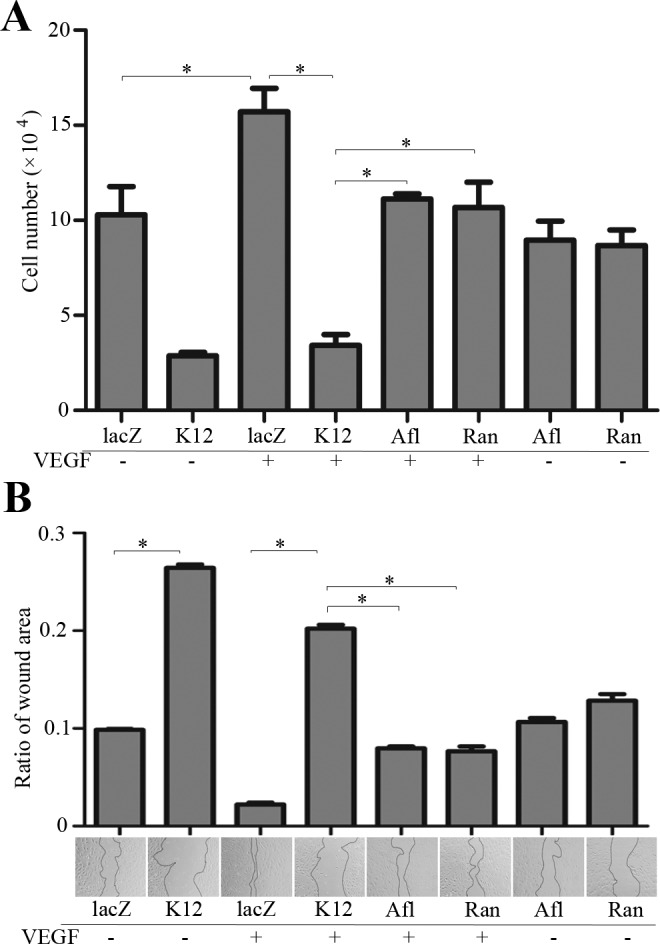
Editing genomic VEGFR2 using CRISPR/Cas9 prevented VEGF-induced proliferation and migration. (A) the HRECs, as described in Figure 3, were seeded in wells of 48-well plates (30,000 cells per well). After attaching the plates, the cells were starved for growth factors for 7 hours, and then some of the cells were pretreated with treated aflibercept or ranibizumab (20 μL/mL) for 15 minutes. Subsequently, VEGF (20 ng/mL) was added into the cells as indicated. On the following day, the treatment was repeated. After 48 hours, the cells were trypsin detached and then counted in a hemocytometer under a light microscope. (B) When HRECs, as described in Figure 3, grew to near confluence in a 48-well plate, they were starved for growth factors for 7 hours and then scratched with a sterile 200-μL pipette tip. After they were washed twice with PBS, cells were treated as described in A. After 16 hours, the scratched area was photographed, the boundaries of scratches were outlined with the dotted lines, and the scratched areas were analyzed using ImageJ software. Afl, aflibercept; Ran, ranibizumab. Each bar graph indicates mean ± SD of three independent experiments. *Significant difference in results between the two compared groups. P < 0.05 using an unpaired t-test. One representative experiment is shown below the bar graphs.
Next, we compared the efficiency of editing VEGFR2 using CRISPR/Cas9 to treatment with aflibercept or ranibizumab in VEGF-induced cell migration,31 a key event in angiogenesis.11 As shown in Figure 4B, VEGF induced cell migration, whereas both aflibercept and ranibizumab blocked this action; however, this blockage was less efficient than that resulting from editing the genomic VEGFR2 locus using K12-sgRNA/SpCas9.
Editing VEGFR2 Using CRISPR/Cas9 Blocked VEGF-Induced Tube Formation
To evaluate the ability of the HRECs transduced by SpCas9 together with K12-sgRNA or lacZ-sgRNA to form tubes, an important in vitro model for angiogenesis,33,34,42,43 we treated HRECs with or without VEGF plus or minus the anti-VEGF drugs. As shown in Figure 5, VEGF stimulated tube formation in the HRECs transduced by SpCas9 with lacZ-sgRNA, but failed to induce this reaction in those that were transduced by K12-sgRNA/SpCas9. In addition, both aflibercept and ranibizumab blocked the VEGF-induced tube formation as expected, but the blockage was less efficient than that resulting from editing the genomic VEGFR2 locus with K12-sgRNA/SpCas9 (Fig. 5). These results indicate that editing genomic VEGFR2 using CRISPR/Cas9 is an alternative strategy to either aflibercept or ranibizumab in blocking VEGF-induced tube formation, suggesting that editing genomic VEGFR2 with K12-sgRNA/SpCas9 is a potentially powerful therapeutic approach to treating abnormal angiogenesis.
Figure 5.
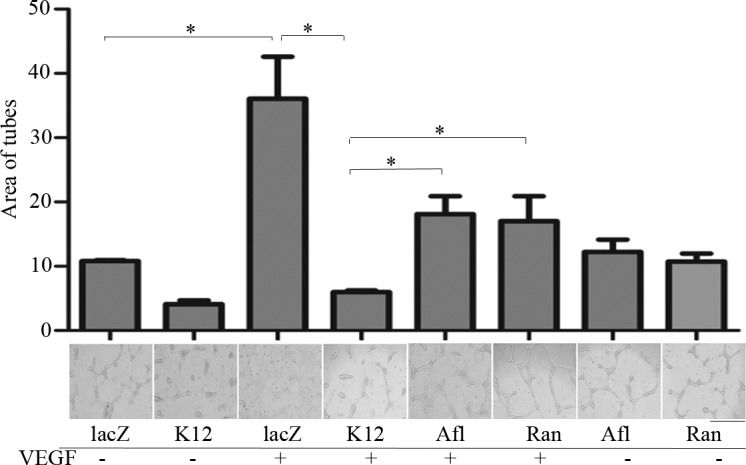
Editing genomic VEGFR2 with use of CRISPR/Cas9 blocked VEGF-induced tube formation. The HRECs, as described in Figure 3, were placed on a polymerized collagen gel in wells of a 96-well plate (45,000 cells per well). After 16 hours, a collagen gel layer was placed on top of the cells. After the top collagen gel was solidified, the EBM-2 medium was added into the wells with or without aflibercept or ranibizumab (20 μL/mL) for 15 minutes. Subsequently, VEGF (20 ng/mL) was added into the wells as indicated. After 24 hours, three different fields on each well were photographed under a light microscope, and the tubes were marked in Photoshop and analyzed using ImageJ software. Afl, aflibercept; Ran, ranibizumab. Each bar graph indicates mean ± SD of three independent experiments. *Significant difference in results between the two compared groups. P < 0.05 using an unpaired t-test. One representative experiment is shown below the bar graphs.
Discussion
In this article, we report that the K12-sgRNA can efficiently guide SpCas9 to cleave the double DNA strands in the human genomic VEGFR2 exon 3, leading to a NHEJ repair and resulting in indels in the VEGFR2 locus and depletion of VEGFR2 expression in HRECs. Not all sgRNAs designed using the online tool could have the same efficiency in guiding SpCas9,30,44 so we used PAE-KDR cells to identify which sgRNA worked best among the four sgRNAs, because PAE-KDR cells could easily be infected by unconcentrated lentiviruses, whereas primary HRECs could only be infected by the concentrated lentiviruses. The pioneering use of PAE-KDR cells as a cell line saved us time while we screened for an effective sgRNA to edit the human genomic VEGFR2 locus.
In this research, we also improved the cloning of sgRNAs, based on a published protocol44 and an Addgene plasmid (catalog number 52961) lentiCRISPR v2 online cloning protocol. Initially, we had one phosphorylated pair of 20 oligonucleotides synthesized, performed dephosphorylation of linearized lentiCRISPR v2 vector by restriction endonuclease BsmB1, and ligation of the two DNA fragments with sticky ends by following the published protocols.44 As a result, the annealed oligonucleotides were successfully cloned into the lentiCRISPR v2 vector. Considering the time we saved on the dephosphorylation of the vector and synthesis of the phosphorylated oligonucleotides, we found that there was at least the same efficiency in ligation of the annealed oligonucleotides (synthesized without phosphorylation) and the BsmB1-linearized lentiCRISPR v2 (without dephosphorylation). In addition, transforming the ligated plasmids into competent bacteria greatly increased the opportunity to obtain the expected clones without continuously culturing the transformed bacteria for 30 minutes as suggested by the published protocols.44 These improvements lessened the burden of discovering the effectiveness of using K12-sgRNA to direct SpCas9 to edit the genomic VEGFR2 locus.
Technologies used for genome editing based on programmable nucleases such as meganucleases,45 zinc finger nucleases,46 transcription activator-like effector nucleases,47 and CRISPR/Cas9 are opening up the possibility of achieving therapeutic genome editing in diseased cells and tissues.21 CRIPSR/Cas9 technology enables especially precise genome editing by introducing DNA double-strand breaks at specific genomic loci. In this research, we cloned four sgRNAs into a lentiCRISPR v2 vector, respectively, to target the human genomic VEGFR2 exon 3 and found that one of them efficiently directed SpCas9 to cleave the DNA at the third position prior to the PAM, resulting in indels created by NHEJ, as well as subsequent depletion of VEGFR2 expression. However, depletion of VEGFR2 in HRECs suppressed constitutive activation of Akt, which is critical for cell survival, suggesting that there is an intracellular mechanism to the activation of VEGFR2/Akt pathway and that this suppression might affect cell proliferation, migration, and angiogenesis. Therefore, the disadvantage of editing genomic VEGFR2 by CRISPR/Cas9 may impair normal endothelial cell growth in young patients.
Currently prevention of abnormal angiogenesis-associated eye diseases such as neovascular AMD or PDR using anti-VEGF drugs (e.g., aflibercept and ranibizumab) is an important therapeutic approach to treating these eye diseases.13–15 Although these drugs can reduce vessel leakage and neovascularization, continuous intraocular injections are required, and multiple injections might cause some side effects such as development of geographic atrophy in the eye.48–51
In summary, with this research, we showed that editing genomic VEGFR2 in HRECs using the CRISPR/Cas9 technology is a very efficient strategy in blocking VEGF-induced activation of Akt and cell proliferation, migration, and tube formation, suggesting editing the genomic VEGFR2 is a potentially novel powerful therapeutic approach to combating pathologic neovascularization.
Acknowledgments
Supported by National Institutes of Health, National Eye Institute Grants R01 EY012509 (HL) and in part by Core Grant P30EY003790.
Disclosure: X. Huang, None; G. Zhou, None; W. Wu, None; G. Ma, None; P.A. D'Amore, None; S. Mukai, None; H. Lei, None
References
- 1. Ferrara N,, Gerber HP,, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003; 9: 669–676. [DOI] [PubMed] [Google Scholar]
- 2. Senger DR,, Galli SJ,, Dvorak AM,, Perruzzi CA,, Harvey VS,, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983; 219: 983–985. [DOI] [PubMed] [Google Scholar]
- 3. Muller YA,, Christinger HW,, Keyt BA,, de Vos AM. The crystal structure of vascular endothelial growth factor (VEGF) refined to 1.93 A resolution: multiple copy flexibility and receptor binding. Structure. 1997; 5: 1325–1338. [DOI] [PubMed] [Google Scholar]
- 4. Muller YA,, Li B,, Christinger HW,, Wells JA,, Cunningham BC,, de Vos AM. Vascular endothelial growth factor: crystal structure and functional mapping of the kinase domain receptor binding site. Proc Natl Acad Sci U S A. 1997; 94: 7192–7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tischer E,, Mitchell R,, Hartman T,, et al. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J Biol Chem. 1991; 266: 11947–11954. [PubMed] [Google Scholar]
- 6. Houck KA,, Leung DW,, Rowland AM,, Winer J,, Ferrara N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem. 1992; 267: 26031–26037. [PubMed] [Google Scholar]
- 7. Shibuya M,, Yamaguchi S,, Yamane A,, et al. Nucleotide sequence and expression of a novel human receptor-type tyrosine kinase gene (flt) closely related to the fms family. Oncogene. 1990; 5: 519–524. [PubMed] [Google Scholar]
- 8. Terman BI,, Carrion ME,, Kovacs E,, Rasmussen BA,, Eddy RL,, Shows TB. Identification of a new endothelial cell growth factor receptor tyrosine kinase. Oncogene. 1991; 6: 1677–1683. [PubMed] [Google Scholar]
- 9. Gerber HP,, McMurtrey A,, Kowalski J,, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998; 273: 30336–30343. [DOI] [PubMed] [Google Scholar]
- 10. Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J. 2000; 351 Pt 2: 289–305. [PMC free article] [PubMed] [Google Scholar]
- 11. Holmes K,, Roberts OL,, Thomas AM,, Cross MJ. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007; 19: 2003–2012. [DOI] [PubMed] [Google Scholar]
- 12. Stahl A,, Connor KM,, Sapieha P,, et al. The mouse retina as an angiogenesis model. Invest Ophthalmol Vis Sci. 2010; 51: 2813–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fong AH,, Lai TY. Long-term effectiveness of ranibizumab for age-related macular degeneration and diabetic macular edema. Clin Interven Aging. 2013; 8: 467–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sarwar S,, Clearfield E,, Soliman MK,, et al. Aflibercept for neovascular age-related macular degeneration. Cochrane Database Syst Rev. 2016; 2: CD011346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sarwar S,, Bakbak B,, Sadiq MA,, et al. Fusion proteins: aflibercept (VEGF Trap-Eye). Dev Ophthalmol. 2016; 55: 282–294. [DOI] [PubMed] [Google Scholar]
- 16. Jager RD,, Aiello LP,, Patel SC,, Cunningham ET., Jr. Risks of intravitreous injection: a comprehensive review. Retina. 2004; 24: 676–698. [DOI] [PubMed] [Google Scholar]
- 17. Sampat KM,, Garg SJ. Complications of intravitreal injections. Curr Opin Ophthalmol. 2010; 21: 178–183. [DOI] [PubMed] [Google Scholar]
- 18. Jinek M,, Chylinski K,, Fonfara I,, Hauer M,, Doudna JA,, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337: 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Swiech L,, Heidenreich M,, Banerjee A,, et al. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nature Biotechnol. 2015; 33: 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cong L,, Ran FA,, Cox D,, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cox DB,, Platt RJ,, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med. 2015; 21: 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hsu PD,, Lander ES,, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014; 157: 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sanjana NE,, Shalem O,, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014; 11: 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Waltenberger J,, Claesson-Welsh L,, Siegbahn A,, Shibuya M,, Heldin CH. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem. 1994; 269: 26988–26995. [PubMed] [Google Scholar]
- 25. Ruan GX,, Kazlauskas A. Axl is essential for VEGF-A-dependent activation of PI3K/Akt. EMBO J. 2012; 31: 1692–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lei H,, Qian CX,, Lei J,, Haddock LJ,, Mukai S,, Kazlauskas A. RasGAP promotes autophagy and thereby suppresses platelet-derived growth factor receptor-mediated signaling events, cellular responses, and pathology. Mol Cell Biol. 2015; 35: 1673–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lei H,, Kazlauskas A. Growth factors outside of the PDGF family employ ROS/SFKs to activate PDGF receptor alpha and thereby promote proliferation and survival of cells. J Biol Chem. 2009; 284: 6329–6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lei H,, Velez G,, Cui J,, et al. N-acetylcysteine suppresses retinal detachment in an experimental model of proliferative vitreoretinopathy. Am J Pathol. 2010; 177: 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ikuno Y,, Kazlauskas A. An in vivo gene therapy approach for experimental proliferative vitreoretinopathy using the truncated platelet-derived growth factor alpha receptor. Invest Ophthalmol Vis Sci. 2002; 43: 2406–2411. [PubMed] [Google Scholar]
- 30. Ran FA,, Hsu PD,, Lin CY,, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013; 154: 1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liang CC,, Park AY,, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007; 2: 329–333. [DOI] [PubMed] [Google Scholar]
- 32. Im E,, Venkatakrishnan A,, Kazlauskas A. Cathepsin B regulates the intrinsic angiogenic threshold of endothelial cells. Molec Biol Cell. 2005; 16: 3488–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arnaoutova I,, Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc. 2010; 5: 628–635. [DOI] [PubMed] [Google Scholar]
- 34. Im E,, Kazlauskas A. Regulating angiogenesis at the level of PtdIns-4,5-P2. EMBO J. 2006; 25: 2075–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Abid MR,, Guo S,, Minami T,, et al. Vascular endothelial growth factor activates PI3K/Akt/forkhead signaling in endothelial cells. Arterioscler Thromb Vasc Biol. 2004; 24: 294–300. [DOI] [PubMed] [Google Scholar]
- 36. Kitamura T,, Asai N,, Enomoto A,, et al. Regulation of VEGF-mediated angiogenesis by the Akt/PKB substrate Girdin. Nat Cell Biol. 2008; 10: 329–337. [DOI] [PubMed] [Google Scholar]
- 37. Franke TF,, Kaplan DR,, Cantley LC,, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997; 275: 665–668. [DOI] [PubMed] [Google Scholar]
- 38. Sarbassov DD,, Guertin DA,, Ali SM,, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005; 307: 1098–1101. [DOI] [PubMed] [Google Scholar]
- 39. Chen J,, Somanath PR,, Razorenova O,, et al. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat Med. 2005; 11: 1188–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mintz-Hittner HA,, Kennedy KA,, Chuang AZ;, BEAT-ROP Cooperative Group. Efficacy of intravitreal bevacizumab for stage 3+ retinopathy of prematurity. N Engl J Med. 2011; 364: 603–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chakravarthy U,, Harding SP,, Rogers CA,, et al. Alternative treatments to inhibit VEGF in age-related choroidal neovascularisation: 2-year findings of the IVAN randomised controlled trial. Lancet. 2013; 382: 1258–1267. [DOI] [PubMed] [Google Scholar]
- 42. Ruan GX,, Zhang DQ,, Zhou T,, Yamazaki S,, McMahon DG. Circadian organization of the mammalian retina. Proc Natl Acad Sci U S A. 2006; 103: 9703–9708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Im E,, Kazlauskas A. Src family kinases promote vessel stability by antagonizing the Rho/ROCK pathway. J Biol Chem. 2007; 282: 29122–29129. [DOI] [PubMed] [Google Scholar]
- 44. Ran FA,, Hsu PD,, Wright J,, Agarwala V,, Scott DA,, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013; 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stoddard BL. Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure. 2011; 19: 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Urnov FD,, Rebar EJ,, Holmes MC,, Zhang HS,, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010; 11: 636–646. [DOI] [PubMed] [Google Scholar]
- 47. Bogdanove AJ,, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science. 2011; 333: 1843–1846. [DOI] [PubMed] [Google Scholar]
- 48. Kumar N,, Mrejen S,, Fung AT,, Marsiglia M,, Loh BK,, Spaide RF. Retinal pigment epithelial cell loss assessed by fundus autofluorescence imaging in neovascular age-related macular degeneration. Ophthalmology. 2013; 120: 334–341. [DOI] [PubMed] [Google Scholar]
- 49. Lois N,, McBain V,, Abdelkader E,, Scott NW,, Kumari R. Retinal pigment epithelial atrophy in patients with exudative age-related macular degeneration undergoing anti-vascular endothelial growth factor therapy. Retina. 2013; 33: 13–22. [DOI] [PubMed] [Google Scholar]
- 50. Grunwald JE,, Daniel E,, Huang J,, et al. Risk of geographic atrophy in the comparison of age-related macular degeneration treatments trials. Ophthalmology. 2014; 121: 150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xu L,, Mrejen S,, Jung JJ,, et al. Geographic atrophy in patients receiving anti-vascular endothelial growth factor for neovascular age-related macular degeneration. Retina. 2015; 35: 176–186. [DOI] [PubMed] [Google Scholar]