

Abstract
Characterizing the functional impact of novel mutations linked to autism spectrum disorder (ASD) provides a deeper mechanistic understanding of the underlying pathophysiological mechanisms. Here we show that a de novo Glu183 to Val (E183V) mutation in the CaMKIIα catalytic domain, identified in a proband diagnosed with ASD, decreases both CaMKIIα substrate phosphorylation and regulatory autophosphorylation, and that the mutated kinase acts in a dominant-negative manner to reduce CaMKIIα-WT autophosphorylation. The E183V mutation also reduces CaMKIIα binding to established ASD-linked proteins, such as Shank3 and subunits of l-type calcium channels and NMDA receptors, and increases CaMKIIα turnover in intact cells. In cultured neurons, the E183V mutation reduces CaMKIIα targeting to dendritic spines. Moreover, neuronal expression of CaMKIIα-E183V increases dendritic arborization and decreases both dendritic spine density and excitatory synaptic transmission. Mice with a knock-in CaMKIIα-E183V mutation have lower total forebrain CaMKIIα levels, with reduced targeting to synaptic subcellular fractions. The CaMKIIα-E183V mice also display aberrant behavioral phenotypes, including hyperactivity, social interaction deficits, and increased repetitive behaviors. Together, these data suggest that CaMKIIα plays a previously unappreciated role in ASD-related synaptic and behavioral phenotypes.
SIGNIFICANCE STATEMENT Many autism spectrum disorder (ASD)-linked mutations disrupt the function of synaptic proteins, but no single gene accounts for >1% of total ASD cases. The molecular networks and mechanisms that couple the primary deficits caused by these individual mutations to core behavioral symptoms of ASD remain poorly understood. Here, we provide the first characterization of a mutation in the gene encoding CaMKIIα linked to a specific neuropsychiatric disorder. Our findings demonstrate that this ASD-linked de novo CAMK2A mutation disrupts multiple CaMKII functions, induces synaptic deficits, and causes ASD-related behavioral alterations, providing novel insights into the synaptic mechanisms contributing to ASD.
Keywords: autism, behavior, CaMKII, synaptic transmission
Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disorder that presents with multiple core symptoms, including social interaction and communication deficits, as well as enhanced repetitive behaviors (Lord et al., 2000a). Several hundred genetic mutations have been linked to ASD. Many of these mutations disrupt synaptic communication in the CNS, but the core molecular mechanisms responsible for ASD symptoms remain unclear (State and Šestan, 2012; Krumm et al., 2014; Ronemus et al., 2014; Bourgeron, 2015). The identification and characterization of novel ASD-linked genetic variations have the potential to provide new insights that will facilitate development of a more unified mechanistic understanding of ASD. This study characterizes the functional impact of a de novo mutation in CAMK2A, the gene encoding the calcium/calmodulin-dependent protein kinase II (CaMKII) α isoform, that was recently identified in an ASD proband, the first identified link between a naturally occurring CAMK2A mutation and a specific neuropsychiatric disorder.
CaMKII is an abundant multifunctional serine/threonine kinase with critical roles in synaptic plasticity, learning, and memory (Lisman et al., 2012; Hell, 2014; Shonesy et al., 2014). The major neuronal CaMKIIα and/or β subunits each contain a catalytic kinase domain, a Ca2+/calmodulin-binding regulatory domain containing modulatory autophosphorylation sites, and an association domain that is necessary for assembly of 12-subunit holoenzymes (Lisman et al., 2012; Hell, 2014; Shonesy et al., 2014). Increases of intracellular Ca2+ result in Ca2+/calmodulin binding to the regulatory domains and CaMKII activation. Simultaneous activation of adjacent subunits in the holoenzyme stimulates intersubunit autophosphorylation at Thr286, which generates autonomous (Ca2+-independent) CaMKII activity. CaMKIIα phosphorylates NMDA (Omkumar et al., 1996; Leonard et al., 1999; Strack et al., 2000) and AMPA (Barria et al., 1997b; Mammen et al., 1997; Coultrap et al., 2014) receptor subunits to enhance excitatory synaptic transmission, and facilitates Ca2+ influx via voltage-gated Ca2+ channels (Welsby et al., 2003; Yasuda et al., 2003; Hudmon et al., 2005; Lee et al., 2006; Abiria and Colbran, 2010; Jenkins et al., 2010; Tavalin and Colbran, 2016). Activated CaMKIIα is targeted to dendritic spines and the postsynaptic density via interactions with various CaMKII associated proteins (CaMKAPs) (Hell, 2014), including GluN2B NMDA receptor subunits (Strack and Colbran, 1998; Leonard et al., 2002; Bayer et al., 2006; Halt et al., 2012).
Genetically engineered mice carrying knock-out, knock-in, or other transgenic CaMKII alleles exhibit varied behavioral abnormalities. For example, CaMKIIα knock-out mice have deficits in long-term potentiation, learning, and memory, increased activity, and aggressive behaviors (Silva et al., 1992; Chen et al., 1994). Furthermore, mice deficient in autophosphorylation at either Thr286 or Thr305/Thr306 have impaired synaptic plasticity and deficits in spatial learning and reversal learning in the Morris water maze task, respectively (Giese et al., 1998; Elgersma et al., 2002). The loss of Thr286 autophosphorylation in adolescent mice disrupts synaptic targeting of CaMKIIα in vivo and also reduces anxiety in the elevated plus maze (Gustin et al., 2011). More recently, multiple studies have uncovered novel roles for CaMKII in amphetamine (Loweth et al., 2010), cocaine (Robison et al., 2013), and alcohol (Easton et al., 2013) addiction and maladaptive behaviors. Moreover, abnormal CaMKII signaling has been linked to a variety of neuropsychiatric disorders (Robison, 2014).
Here we show that a de novo CAMK2A missense mutation identified in an ASD proband in the Simons Simplex Collection (Fischbach and Lord, 2010; Iossifov et al., 2014) (Family SSC ID: 14620) decreases CaMKIIα kinase activity and interactions with several synaptic CaMKAPs, and increases CaMKIIα turnover in cells. Neuronal expression of CaMKIIα-E183V disrupts dendritic morphology and basal synaptic transmission. Furthermore, a knock-in mutant mouse engineered to harbor this mutation using CRISPR-Cas9 technology displays core behaviors associated with ASD, including social interaction deficits and repetitive behaviors. Biochemical analyses revealed alterations in CaMKII autophosphorylation, synaptic localization, and protein levels in the brains of CaMKIIα-E183V mice. Together, our findings suggest that CaMKIIα can play an important role in the pathogenesis of ASD-related behaviors.
Materials and Methods
Animals
All mice were on a mixed B6D2 (C57BL/6J (B6) x DBA/2J (D2)) background and were housed (2–5 per cage) on a 12 h light-dark cycle with food and water ad libitum. Breeding cages (HETXHET) used to generate wild-type (Camk2aWT/WT; E/E), heterozygous (Camk2aWT/E183V; E/V), and homozygous (Camk2aE183V/E183V; V/V) mice for experiments were maintained on a high-fat diet. Approximately equal numbers of male and female mice were used for all experiments. Behavioral studies were performed at postnatal days 42–84 during the light cycle (9:00 A.M. to 4:00 P.M.), and multiple experiments were conducted on each mouse in the following order: (1) open field, (2) 3-chamber social test, and then (3) marble burying. All mouse experiments were approved by the Vanderbilt University Institutional Animal Care and Use Committee and were performed in accordance with the National Institutes of Health Guide for the care and use of laboratory animals.
Antibodies used
The following antibodies were used for immunoblotting at the indicated dilutions: mouse anti-CaMKIIα 6G9 (Thermo Fisher Scientific catalog #MA1–048 RRID:AB_325403, 1:5000), mouse anti-CaMKIIβ (Thermo Fisher Scientific catalog #13–9800 RRID:AB_2533045, 1:4000), rabbit anti-phospho-Thr286 CaMKIIα (Santa Cruz Biotechnology catalog #sc-12886-R RRID:AB_2067915, 1:3000), mouse anti-GST (University of California at Davis/National Institutes of Health NeuroMab Facility catalog #73–148 RRID:AB_10671817, 1:2000), rabbit anti-phospho-Ser831 GluA1 (Thermo Fisher Scientific catalog #36–8200 RRID:AB_2533279, 1:1000), polyclonal goat CaMKII antibody (RRID: AB_2631234, 1:5000) (McNeill and Colbran, 1995), rabbit anti-GFP (Invitrogen catalog #A-11122 RRID:AB_221569, 1:3000–4000), HRP-conjugated anti-rabbit (Promega catalog #W4011 RRID:AB_430833, 1:6000), HRP-conjugated anti-mouse (Promega catalog #W4021 RRID:AB_430834, 1:6000), and HRP-conjugated anti-goat (Santa Cruz Biotechnology catalog #sc-2056 RRID:AB_631730, 1:6000), IR dye-conjugated donkey anti-rabbit 800CW (LI-COR Biosciences catalog #926–32213 RRID:AB_621848, 1:10,000), and IR dye-conjugated donkey anti-mouse 680LT (LI-COR Biosciences catalog #926–68022 RRID:AB_10715072, 1:10,000).
Cloning, transfection, and cell lysate preparation
The mouse CaMKIIα-E183V mutant cDNA was generated in the pcDNA3 vector by site-directed mutagenesis using the following primers (5′-3′): forward, GGATACCTCTCCCCAGTGGTGCTGAGGAAGGACC; and reverse, GGTCCTTCCTCAGCACCACTGGGGAGAGGTATCC, and verified by sequencing. The HA-ubiquitin plasmid was generously provided by Dr. Brian Wadzinski (Vanderbilt University) (Watkins et al., 2012). Dr. Craig Garner (Stanford University) generously provided the GFP-Shank3 plasmid (Arons et al., 2012). The CaMKII-WT insert was subcloned into the mApple expression vector (a generous gift from Dr. David Piston, Vanderbilt University) and used as a template to create the mApple-CaMKII-E183V construct using the primers defined above. The eGFP plasmid was purchased from Clontech. Constructs for GFP-CaMKIIα (Abiria and Colbran, 2010), CaMKIIα (Jiao et al., 2008), CaMKIIβ (Baucum et al., 2012), GST-GluN2B (Strack et al., 2000), GST-Densin IN (Jiao et al., 2011), GST-β2a (Abiria and Colbran, 2010), and GST-GluA1 (Barria et al., 1997a) were previously described. The GST-mGlu5 construct was made by cloning residues 827–964 of rat mGlu5 (a generous gift from Dr. Jeffrey P. Conn, Vanderbilt University) into the pGEX6P-1 vector BamHI and EcoR1 using primers (5′ to 3′): forward, CTGGAAGTTCTGTTCCAGGGGCCCGGATCCAAACCGGAGAGAAAT; and reverse, GCCGCAAGCTTGTCGACGGAGCTCGAATTCTTAGGTCCCAAAGCGCTT.
HEK293T cells (ATCC catalog #CRL-3216, RRID:CVCL_0063) used for cell-based assays were cultured and maintained in DMEM containing 10% FBS and 1% penicillin/streptomycin at 37°C at 5% CO2. Unless indicated otherwise, transfections were performed by incubating cells (50%–70% confluency; passages 3–18) plated on 10 cm cell culture dishes with PEI and plasmid DNA (2:1 PEI/DNA). Unless indicated otherwise, after 24–48 h, cells were scraped into 500–1000 μl of lysis buffer (1% Triton X-100, 150 mm NaCl, 25 mm HEPES, pH 7.5, 0.2 mm PMSF, 1 mm benzamidine, 10 μg/ml leupeptin, 10 μm pepstatin, and 1 μm microcystin), and lysates were mixed end-over-end at 4°C for 20 min. After centrifugation (12,200 × g, 20 min), soluble fractions were used for experiments as indicated.
Western blot analysis
Samples were resolved on 10% SDS-PAGE gels and transferred to nitrocellulose membrane (Protran). The membrane was blocked in blot buffer containing 5% nonfat dry milk, 0.1% Tween 20, in Tris-buffered saline (19.98 mm Tris, 136 mm NaCl) at pH 7.4 for 30 min at room temperature. The membrane was incubated with primary antibody (see dilutions above) in blot buffer for 1 h at room temperature or overnight at 4°C. After washing, membranes were incubated with HRP-conjugated secondary antibody for 30 min at room temperature, washed again, and then visualized via enzyme-linked chemiluminescence using the Western Lightening Plus-ECL, enhanced chemiluminescent substrate (PerkinElmer) and visualized using Premium x-ray Film (Phenix Research Products). Images were quantified using ImageJ software (RRID:SCR_003070). Secondary antibodies conjugated to infrared dyes (LI-COR Biosciences) were used for development with an Odyssey system (LI-COR Biosciences).
GST-GluA1 and syntide phosphorylation assays
HEK293FT cells expressing CaMKIIα (WT or E183V) were lysed 24–48 h after transfection in cold low ionic strength modified lysis buffer (2 mm Tris-HCl pH 7.5, 2 mm EDTA, 2 mm EGTA, 1% v/v Triton X-100, 1 mm DTT, 0.2 mm PMSF, 1 mm benzamidine, 10 μg/ml leupeptin, 10 μm pepstatin, and 1 μm microcystin). Soluble fractions were adjusted to equal CaMKIIα-WT and CaMKIIα-E183V subunit concentrations using soluble fractions of nontransfected cells (by immunoblot comparison with purified CaMKIIα standards), and then incubated at 30°C with 2 mm CaCl2, 1 μm calmodulin, 5 mm MgCl2, 0.4 mm ATP, 1 μm GST-GluA1 (827–906), and 0.5 μm GO6983 (Tocris Bioscience). At the indicated times, aliquots were mixed with LDS sample buffer (Invitrogen) and analyzed by immunoblotting for total and pT286 CaMKIIα, GST and pSer831-GluA1. Secondary antibodies conjugated to infrared dyes (LI-COR Biosciences) were used for development with an Odyssey system (LI-COR Biosciences). Signal intensities for pS831 and pT286 were normalized to GST and CaMKIIα signals, respectively. HEK293FT cells expressing CaMKIIα (WT or E183V) were lysed 24–48 h after transfection in cold low ionic strength modified lysis buffer (2 mm Tris-HCl, pH 7.5, 2 mm EDTA, 2 mm EGTA, 1% v/v Triton X-100, 1 mm DTT, 0.2 mm PMSF, 1 mm benzamidine, 10 μg/ml leupeptin, 10 μm pepstatin, and 1 μm microcystin). Lysates were incubated with 500 μm CaCl2, 1 μm calmodulin, 20 μm syntide at 30°C for 2 min in assay buffer (50 mm HEPES, 10 mm MgCl, 1 μm dithiothreitol, 400 μm [γ-32P]ATP, 700–1000 cpm/pmol). Reactions were stopped on P82 Whatman paper, which were then washed, and phosphorylation was determined using a scintillation counter.
Size-exclusion chromatography
The FPLC Superose 6 column (10 × 400 mm) was equilibrated at 1 ml/min in 50 mm Tris, pH 7.5, 150 mm NaCl, 0.5 mm TCEP. Soluble fractions (1.0 ml) of transfected HEK293T cells (see above) were applied to the column, and 1 ml fractions were collected at a flow rate of 0.25 ml/min for analysis by immunoblotting for CaMKIIα.
Analysis of Thr286 autophosphorylation in lysates
Soluble fractions of HEK293T cells (24–48 h after transfection) were supplemented with 2 mm CaCl2, 1 μm calmodulin, 5 mm MgCl2, 0.4 mm ATP, and incubated at 30°C for 2 min. Reactions were mixed with SDS sample buffer and analyzed by immunoblotting for total and pT286 CaMKIIα. Levels were analyzed by immunoblot analysis (above) and normalized to protein levels from Ponceau staining of nitrocellulose membranes as described previously (Gustin et al., 2010).
Cycloheximide chase
HEK293T cells expressing CaMKIIα-WT, CaMKIIα-E183V, CaMKIIα-K42R, and/or mApple-CaMKIIα-E183V were divided to ∼75% confluency in 6-well cell culture dishes 24 h after transfection. After an additional ∼24 h, cycloheximide (50 μg/μl; Cell Signaling Technology) was added and after the indicated time cells were washed with ice-cold PBS and lysed in 200 μl of lysis buffer. Levels of CaMKIIα were analyzed by immunoblot (above) and normalized to total protein loading determined by analyses of Ponceau-S-stained nitrocellulose membranes, as described previously (Gustin et al., 2010).
Immunoprecipitation
Soluble fractions of transfected HEK293T cells were incubated at 4°C for 1 h with rabbit anti-GFP (Thermo Fisher Scientific A-11222) or mouse anti-CaMKIIα (Pierce, MA1–048) (3 μl each) and either protein A or G magnetic beads (Invitrogen), respectively (30 μl each). After washing 3 times with lysis buffer, beads were suspended in 40 μl of 2× SDS sample buffer and heated at 95°C for 10 min. Inputs and immune complexes were immunoblotted as indicated and analyzed by Western blot analysis (above).
Fluorescence plate-binding array
GST-fusion protein (GST, GST-GluN2B 1260–1309), GST-CaV1.3(1–126), GST-β2a (full-length protein), GST-Densin-IN(793–824), and GST-mGlu5 (827–964) were expressed in Escherichia coli and purified essentially as described previously (Robison et al., 2005a). Wells of glutathione-coated 96-well plates (Thermo Fisher Scientific) were coated with the indicated GST protein (200 pmol in 100 μl binding buffer: 50 mm Tris-HCl, pH 7.5, 200 mm NaCl, 0.1 mm EDTA, 5 mm 2-mercaptoethanol, 0.1% v/v Tween 20, 5 mg/ml BSA) at 4°C overnight and then washed to remove unbound protein. Soluble fractions of HEK293FT cells expressing mApple-CaMKIIα-WT or mApple-CaMKIIα-E183V prepared in cold low ionic strength lysis buffer were adjusted to ∼150 nm mApple-CaMKIIα subunit concentration (see above). Where indicated, soluble fractions were supplemented with 2.5 mm CaCl2, 1 μm calmodulin, 10 mm MgCl2, and 400 μm ADP (final concentrations). Our previous studies have shown that binding of Ca2+/calmodulin and adenine nucleotides induces conformational changes that fully support binding to many CaMKAPs, such as GluN2B and the densin-IN domain (Robison et al., 2005b; Jiao et al., 2011), without the need for Thr286 autophosphorylation. Soluble fractions were then added to the precoated wells of glutathione-coated plate at 4°C for 2 h. The wells were washed in wash buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.5% v/v Triton X-100, and 2.4 mm CaCl2) 2 times and bound mApple-CaMKIIα was detected using a fluorescent plate reader at 592 nm. Data from quadruplicate wells were averaged to provide 1 data point. Analyses were repeated 3 times using independently prepared HEK293 cell-soluble fractions.
Primary hippocampal neuronal culture and transfection
Dissociated hippocampal neurons were prepared from E18 Sprague Dawley rat embryos, as previously described (Sala et al., 2003; Shanks et al., 2010). Neurons were transfected at DIV 7–8 (analysis of dendritic arborization) or DIV 14–15 (analysis of spine density and dendritic morphology) using Lipofectamine 2000 following the manufacturer's directions (Thermo Fisher Scientific). A total 1 μg of DNA was transfected for each well of a 12-well plate. Neurons were fixed with 4% paraformaldehyde in 0.1 m phosphate buffer.
Confocal microscopy and image analysis
Images were collected using an LSM 710 META Inverted confocal microscope using 63× (1.40 Plan-apochromat oil) or 20× (0.80 Plan-Apochromat) objectives. Transfection conditions were coded to blind the experimenter. Cells were selected for analysis based on GFP expression. Laser settings were slightly adjusted to generate images with a similar overall dynamic range in each channel so that differences in protein expression levels between cells did not impact image analysis. We noted that mAp-CaMKIIα-E183V fluorescence was typically somewhat lower than mAp-CaMKIIα-WT fluorescence, perhaps reflecting increased turnover of the mutated protein (see below), although this was not assessed quantitatively. Dendritic spine measurements were performed using 10–14 images of cells transfected at DIV 14–15 in the z-plane using a z-step size of ∼0.3 μm. Images were imported into IMARIS software (RRID:SCR_007370) for quantitative analysis. Nonprimary dendritic segments (20–50 μm long) at least 60 μm from the cell body and devoid of intersecting dendrites were selected. Dendritic spine density was analyzed as number of spines/10 μm, and the average spine density for each cell was calculated from 2–4 dendritic segments. Spines were classified using the Imaris XTension software based on predefined parameters for mushroom spines, stubby spines, long/thin spines, or filopodia (Swanger et al., 2011). Overall dendritic morphology of cells transfected at DIV 7–8 was performed using the Filament Tracer (RRID:SCR_007366) tool for semiautomatic tracing of dendrites in images collected with the 20× objective, followed by an automated Sholl analysis, and other measures of dendritic morphology (e.g., dendritic branch points, total dendritic length). Data were statistically analyzed by two-way repeated-measures ANOVA (Sholl) or one-way ANOVA (morphology).
Analysis of synaptic targeting of mApple-CaMKIIα
Image files (see above) were renamed to blind the experimenter to the specific transfection condition before analysis using ImageJ. Only mushroom spines, visually identified as having a larger spine head than neck, were analyzed. Briefly, for each spine, three regions of interest (ROI) were defined, encompassing the spine head, adjacent dendritic shaft, and a nearby extracellular background region with no fluorescence (see Fig. 2C, circles 1, 2, and 3, respectively). GFP and mApple fluorescence intensities in each ROI were quantified. After subtraction of the background signal for each channel (circle 3), mApple/GFP fluorescence ratios in the spine head and shaft were separately calculated to normalize for differences in intracellular volumes sampled in each ROI. The mApple/GFP ratios were then expressed as a spine/shaft ratio for each spine analyzed, providing an index of the relative targeting of the mApple protein to the dendritic spine. Spine/dendrite ratios of ∼1.00 were obtained for soluble mApple, which should exactly codistribute with soluble GFP, validating this approach (see Fig. 2C).
Figure 2.

CaMKIIα-E183V associates with CaMKIIα-WT to reduce Thr286 autophosphorylation. A, Soluble fractions of HEK293T cells expressing similar levels of CaMKIIα-WT, CaMKIIα-E183V, or CaMKIIα-K42R were resolved by size-exclusion chromatography. CaMKIIα immunoblots of the eluted fractions are shown below a graph showing the elution profiles. Data are representative of three independent analyses. Dotted lines indicate the elution positions of two molecular weight marker proteins. B, Soluble fractions of HEK293T cells coexpressing GFP or GFP-CaMKIIα-WT with untagged CaMKIIα (WT or E183V) were immunoprecipitated using GFP antibodies. Inputs and immune complexes were immunoblotted for CaMKIIα, as indicated. Data are representative of three independent analyses. C, Immunoblots of myc-CaMKIIβ complexes isolated from HEK293T cells coexpressing myc-CaMKIIβ with empty vector control, CaMKIIα-WT, or CaMKIIα-E183V. Data are representative of three independent analyses. D, Soluble fractions of HEK293T cells coexpressing GFP-CaMKIIα-WT with untagged CaMKIIα-E183V or CaMKIIα-WT were incubated with Ca2+/calmodulin and ATP (see Materials and Methods) and then immunoblotted using antibodies to total and pT286-CaMKIIα. Representative immunoblot (left) and graph summarizing ratios of pT286- to total-CaMKIIα-GFP from five independent experiments (right). **p = 0.0028 (one-sample t test, t(8) = 3.747). Data are mean ± SEM; n = 5.
Lentiviral production
The FSGW plasmid, which contains a synapsin promoter, a sequence for eGFP, and a woodchuck hepatitis virus posttranscriptional regulatory element, was digested with BamHI and AgeI, and ligated with a DNA linker that contains P2A self-cleavage site using the following oligos: forward, GATCCGGAAGCGGAGCTACTAACTTCAGCCTGCTGAAGCAGGCTGGAGACGTGGAGGAGAACCCTGGACCTCCT; and reverse, CCGGAGGAGGTCCAGGGTTCTCCTCCACGTCTCCAGCCTGCTTCAGCAGGCTGAAGTTAGTAGCTCCGCTTCCG. Primers used for production of mApple-CaMKIIα and control lentiviral constructs are as follows: forward, GCGCAGTCGAATTCAAGCTGCTAGCGCCACCATGGTGAGCAAGGGCGAGG; and mApple control: forward, AGTTAGTAGCTCCGCTTCCGGATCCCTTGTACAGCTCGTCCAT; with reverse primer, AGTTAGTAGCTCCGCTTCCGGATCCCTTGTACAGCTCGTCCAT. Lentivirus was generated in HEK293T/17 (ATCC catalog #CRL-11268, RRID:CVCL_1926) suspension cells cultured in 293Freestyle expression medium containing 0.1% Pluronic F68 (Invitrogen). Cells were transfected at a density of 2 × 106 cell/ml in 200 ml fresh media containing no supplements. The transfer plasmid, pRSV-REV, pVSVG and pMDLp/g plasmids (Nakagawa et al., 2004) were transfected in a 2:1:1:1 ratio using PEI. Two days after transfection, the cells were centrifuged and filtered through a 0.45 μm syringe filter. The conditioned media was ultracentrifuged (82,700 × g; 2 h; 4°C) through a 2 ml cushion of 20% sucrose in sterile PBS. The viral pellet was resuspended in ∼100 μl of ice-cold PBS and titered using the Lenti-X p24 Rapid Titer Kit (Clontech) (titers: >1 × 107 TU/ml).
Electrophysiology
Cultured hippocampal neurons (DIV 14) were transduced with lentiviruses expressing GFP alone, GFP, and mApple-CaMKII-WT, or GFP and mApple-CaMKII-E183V (MOI = 10). Typical neuronal transduction efficiencies were ∼80%-90%. On the day of recordings (DIV 20–22), neurons were transferred to oxygenated ACSF containing the following (in mm): 92 NaCl, 2.5 KCl, 2.0 CaCl2, 2.0 MgSO4, 1.25 NaH2PO4, 30 NaHCO3, 25 glucose, 3 Na-pyruvate, 5 Na-ascorbate, 5 N-acetyl-l-cysteine, 0.05 picrotoxin, and 0.001 tetrodotoxin. Recording pipettes (resistance: 5–8 mΩ) were filled with the following (in mm): 140 K-gluconate, 4 NaCl, 2 MgCl2, 10 HEPES, 4 Mg-ATP, 0.3 Na-GTP, 10 Na-phosphocreatine, pH 7.2. GFP-positive neurons were selected for miniature EPSC (mEPSC) recordings at −70 mV in the presence of tetrodotoxin to block spontaneous firing and 50 μm picrotoxin to inhibit IPSCs. Following break-in of the cells with the patch pipette, the cells were allowed to stabilize for 2 min before events were recorded. The first 500 miniature events (or 2 min traces if there were not 500 events total) were individually selected and analyzed using MiniAnalysis (Synaptosoft, RRID:SCR_014441) with a 7 pA amplitude threshold.
Production of E183V-KI mice
Guide RNA design was guided by software developed in the laboratory of Feng Zhang (Massachusetts Institute of Technology, http://crispr.mit.edu). A specific guide Cas9 CRISPR target (CTCCCCAGAAGTGCTGAGGAAAG) was chosen due to relative position overlapping the E183 codon and lack of strongly predicted off-target sequences; notably, no off-target sequences were found on chromosome 18. The guide RNA was inserted into the pX330 plasmid, a generous gift from Dr. Robert Macdonald (Vanderbilt University), which encodes Cas9. A donor DNA for homology-directed repair was used to insert a double mutation at the second and third bases of codon 183 to encode valine instead of the natural glutamate, also introducing a novel restriction endonuclease site (TspRI). Donor DNA sequence is as follows: CTGCCTGTCTGTGTGTCTGGGGAGCAGGGTTCGCAGGGACACCCGGGTACCTCTCCCCAGTGGTGCTGAGGAAAGACCCGTACGGGAAGCCCGTGGACCTGTGGGCCTGTGGTAAGTCCAAT. Coin-jection of donor DNA and pX330 plasmid in hybrid C57BL6J/DBA2J (B6/D2) embryos was performed in the Vanderbilt Transgenic Mouse Core. From these injections, one male pup was identified as having the E183V mutation with no additional insertions or deletions by Sanger sequencing (Genewiz). PCR amplification of the region in exon 8, including the E183 codon, was performed using the primers: forward, GTTCTCCAGCTCCAGGGCTATCCTCGTCC; and reverse, GGTCTTCATCCCAGAACGGGGGATACCC, and the forward primer was used as the sequencing primer. The male E183V mouse (RRID:MGI MGI: 5811610) (F1) was mated with 2 WT B6D2 hybrid females (The Jackson Laboratory; B6D2F1/J stock #100006), yielding two litters (F2 generation) with a total of six confirmed heterozygous E183V-KI pups (3 male, 3 female). These were used for HETxHET breeding to produce WT, heterozygous KI, and homozygous KI pups (F3 generation) for the experiments reported herein.
Mouse brain homogenization
Mice were anesthetized with isofluorane, decapitated, and forebrains were quickly dissected and immediately frozen on dry ice. Half of a forebrain (cut alone the mid-line) was homogenized using a Dounce homogenizer in 150 mm KCl, 50 mm Tris-HCl, 1 mm DTT, 0.2 mm PMSF, 1 mm benzamidine, 10 μg/ml leupeptin, 10 μm pepstatin, and 1 μm microcystin. Levels of CaMKIIα (6G9) and CaMKIIβ (goat antibody) were analyzed by immunoblot analysis (above) and normalized to protein levels from Ponceau staining of nitrocellulose membranes as described previously (Gustin et al., 2010).
Subcellular fractionation of mouse forebrain
Forebrains were dissected and fractionated as previously described (Baucum et al., 2013, 2015). Briefly, mice were anesthetized with isofluorane, decapitated, and forebrains were quickly dissected, cut in half down the midline, and a half brain was immediately homogenized in an isotonic buffer (150 mm KCl, 50 mm Tris HCl, pH 7.5, 1 mm DTT, 0.2 mm PMSF, 1 mm benzamidine, 1 μm pepstatin, 10 mg/L leupeptin, 1 μm microcystin). The homogenate (∼2 ml) was rotated end-over-end at 4°C for 30 min and then centrifuged at 10,000 × g for 1 h. After removing the supernatant (cytosolic S1 fraction), the pellet was resuspended in the isotonic buffer containing 1% (v/v) Triton X-100, triturated until homogeneous, and then rotated end-over-end at 4°C for 30 min. Lysates were then centrifuged at 10,000 × g, and the supernatant (Triton-soluble membrane S2 fraction) was removed. The second pellet (Triton-insoluble synaptic P2 fraction) was resuspended in isotonic buffer containing 1% Triton X-100 and 1% deoxycholate and then sonicated. The S1, S2, and P2 fractions were mixed with 4× SDS-PAGE sample buffer. Levels of CaMKIIα (6G9), CaMKIIβ (Thermo Fisher Scientific, 13–9800), and phosphorylation of Thr286/Thr287 were analyzed by immunoblot (above) and normalized to protein loading on each lane, as determined from Ponceau-S-stained nitrocellulose membranes (Gustin et al., 2010).
Behavioral assays
Age-matched (7–13 weeks) male and female mice generated as described above were tested sequentially in the order listed below during the light cycle (9:00 A.M. to 4:00 P.M.) with ≥1 d off between tests. The experimenter was blinded to genotypes during testing.
Open-field locomotor activity.
Mice were placed in the center of a novel open field box with dimensions of 40 × 40 × 40 cm. Locomotor activities were recorded for 30 min and analyzed using activity monitors (Med Associates). The center zone was defined as ≥10 cm from the wall.
Three-chambered assay.
Social interaction was measure in a 3-chamber polycarbonate apparatus with 4-inch sliding gates separating the 7 × 9-inch chambers. Both side chambers contained an inverted metal wire pencil cup in the corner. The mouse was prevented from climbing on the cup by placing a plastic cup full of water on it. There were three phases to the testing: (1) A 10 min habituation session allowed the test mouse to freely explore all three chambers. (2) After guiding the test mouse into the center chamber, a WT novel stranger mouse of the same gender was placed under one of the two metal cups. The test mouse was then allowed to freely explore all three chambers for 10 min. (3) The test mouse was again moved to the center chamber while a second WT stranger mouse was placed under the remaining empty metal cup. The subject mouse again freely explored all three chambers for 10 min. Stranger mice were gender matched and habituated to the metal cup the previous day for ∼30 min. The time that test mice spent interacting with stranger mice was scored as the amount of time the nose of the mouse was located within 2 cm of the wire cup using ANY-maze software (Stoelting, RRID:SCR_014289). Rotational behavior was analyzed during the entire 30 min in all three chambers, scored as rotations of the body in ANY-maze software (Stoelting, RRID:SCR_014289).
Marble burying assay.
Mice were placed in a novel cage with 12 exposed marbles on ∼3 inches of Diamond Soft bedding (Harlan #7089). After 30 min, mice were removed. Marbles were scored as buried if >75% was covered by the bedding.
Molecular graphics
Molecular graphics and analyses were performed with the UCSF Chimera package (RRID:SCR_004097) (Pettersen et al., 2004). Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco.
Statistics
Statistical tests and parameters are indicated in figure legends. Differences were considered significant if p < 0.05. Sample sizes for each experiment are based on previously published studies from our laboratories and standards in the field. Statistical outliers were identified by the ROUT method in Prism software (GraphPad Prism, RRID:SCR_002798) set at 1%. The experimenter was blind to the genotype or mutant being tested for analyses of CaMKII localization in dendritic spines, dendritic spine density, and arborization and for all behavioral testing. Data are mean ± SEM with individual data points indicated, or as box plots; the box limits show first and third quartile, the center line indicates the median, and whiskers indicate minimum and maximum values.
Results
E183V mutation in CaMKIIα disrupts catalytic activity
Glutamate 183 is a highly conserved residue in helix H of the “substrate-binding” C-terminal lobe of the CaMKIIα catalytic domain (Fig. 1A). Because the negatively charged Glu183 side chain is not surface exposed in any available crystal structure (Chao et al., 2011), we predicted that replacement with a hydrophobic valine side chain (E183V), detected in a whole exome sequencing study of ASD individuals (Iossifov et al., 2014), would affect catalytic function. We first noted that transfection of equal amounts of plasmid DNA (3 μg) into HEK293T cells resulted in lower levels of CaMKIIα-E183V expression relative to CaMKIIα-WT (33 ± 7%) (Fig. 1B). Therefore, to equalize protein expression levels for subsequent analyses, we transfected cells using an ∼1:3 ratio of cDNA expressing CaMKIIα-WT or CaMKIIα-E183V.
Figure 1.

The CaMKIIα-E183V mutation reduces catalytic activity. A, Top, Crystal structure of single human CaMKIIα subunit (extracted from a holoenzyme structure: PDB: 3SOA) with catalytic domain (blue), regulatory domain (yellow), and association domain (gray). Positions of Glu183 (E183) and Thr286 autophosphorylation site (T286) are indicated. Bottom, Sequence alignment of residues 168–192 of human CaMKIIα with the corresponding residues of CaMKIIα from the indicated species, with E183 highlighted in red. B, Top, CaMKIIα immunoblots of soluble fractions from HEK293T cells transfected with 3 μg plasmid DNA to express either CaMKIIα-WT or CaMKIIα-E183V. Middle, Ponceau stain of nitrocellulose membrane showing total protein. Bottom, Relative expression levels of CaMKIIα-WT and CaMKIIα-E183V. **p = 0.0025 (one-sample t test, t(3) = 9.505). Data are mean ± SEM; n = 4. C, GST-GluA1 was incubated with soluble fractions of HEK293T cells expressing empty vector, CaMKIIα-WT, or about equal levels of CaMKIIα-E183V in the presence of Ca2+/CaM/ATP. After the indicated time, samples were immunoblotted for GST, pS831-GluA1, and total CaMKIIα. Top, Representative immunoblot. Bottom, Summary of pS831-GluA1/GST ratios normalized to CaMKIIα-WT at 5 min. **p = 0.0002 (one-way ANOVA with Tukey's post hoc analysis, F(2,6) = 51.07). D, Specific kinase activity of HEK293T cell-soluble fractions after expression of empty vector control, CaMKIIα-WT, or CaMKIIα-E183V using a peptide substrate (20 μm syntide-2). **p = 0.0040 (one-way ANOVA with Tukey's post hoc analysis, F(2,3) = 57.57; n = 2).
We next tested for effects of E183V mutation on CaMKIIα activity using a physiological substrate that is important for LTP induction, the AMPA receptor GluA1 subunit. CaMKIIα phosphorylates serine 831 on GluA1 to enhance AMPA receptor conductance (Barria et al., 1997b). Incubation of HEK293T cell-soluble fractions expressing CaMKIIα-WT with purified GST-GluA1, Ca2+/calmodulin, Mg2+, and ATP at 30°C resulted in robust time-dependent phosphorylation of Ser831 on GST-GluA1. GluA1 phosphorylation, using HEK293T cell-soluble fractions containing similar levels of CaMKIIα-E183V, was significantly reduced (by 68.3 ± 12% at 5 min; one-way ANOVA with Tukey's post hoc, p = 0.0002, n = 3) (Fig. 1C), but there was no detectable GluA1 phosphorylation using control HEK293T cell-soluble fractions. Thus, the CaMKIIα-E183V mutant retains only a low level of kinase activity toward GluA1. Interestingly, CaMKIIα-E183V appears to be essentially inactive in similar experiments performed using the model peptide substrate syntide-2 (Fig. 1D), indicating a possible substrate-selective effect of E183V mutation on kinase activity. In combination, these data establish that E183V mutation severely compromises the catalytic function of CaMKIIα.
CaMKIIα-E183V forms a holoenzyme
To determine whether E183V mutation affects holoenzyme formation, we analyzed soluble fractions of HEK293T cells expressing CaMKIIα (WT or mutated) by size-exclusion chromatography on a calibrated Superose 6 column. As expected based on prior studies, the peak of CaMKIIα-WT elution was in fraction 18, the same fraction in which thyroglobulin (∼600 kDa) eluted (Fig. 2A). CaMKIIα-E183V consistently eluted in fraction 16, indicative of a holoenzyme structure with slightly larger Stoke's radius, perhaps due to an altered (more open) conformation and/or post-translational modifications. However, a CaMKIIα-K42R mutant, which is inactive due to the mutation in the nucleotide binding site, eluted in the same fractions as CaMKIIα-WT. These data suggest that CaMKIIα-E183V can assemble into a holoenzyme-like structure, and that it is slightly larger Stoke's radius is not a direct result of the lack of kinase activity.
To assess whether CaMKIIα-WT and CaMKIIα-E183V can coassemble to form a mixed holoenzyme, GFP-tagged CaMKIIα-WT or GFP alone was coexpressed with untagged CaMKIIα-WT or CaMKIIα-E183V in HEK293T cells. Similar amounts of CaMKIIα-WT or CaMKIIα-E183V were coimmunoprecipitated with GFP-CaMKIIα-WT from the soluble fractions, but the untagged kinases were not coprecipitated with GFP alone (Fig. 2B). Similarly, CaMKIIα-WT and CaMKIIα-E183V formed mixed holoenzymes with myc-CaMKIIβ (Fig. 2C). Together, these data indicate that the ASD-associated E183V mutation does not affect the ability of CaMKIIα to form holoenzymes with WT kinase subunits.
CaMKIIα-E183V has dominant-negative activity toward CaMKIIα-WT
Next, we tested the hypothesis that the coexpression of CaMKIIα-E183V interferes with Thr286 autophosphorylation of CaMKIIα-WT. GFP-CaMKIIα-WT was expressed in HEK293T cells with untagged CaMKIIα-WT or CaMKIIα-E183V. Addition of the GFP tag reduces electrophoretic mobility, allowing for independent assessment of the levels of Thr286 autophosphorylation of the tagged WT enzyme and the untagged proteins. HEK293T cell-soluble fractions were incubated with saturating concentrations of Ca2+/calmodulin, Mg2+, and ATP at 30°C and then immunoblotted for both total and pT286 CaMKIIα. Coexpression of untagged CaMKIIα-E183V significantly reduced the pT286/total ratio for GFP-CaMKIIα-WT by 51.4 ± 7.4% (one-sample t test, p = 0.0028, n = 5) compared with coexpression of similar levels of untagged CaMKIIα-WT (Fig. 2D). Thus, CaMKIIα-E183V subunits can exert dominant-negative activity to reduce Thr286 autophosphorylation of CaMKIIα-WT subunits in the same holoenzyme.
CaMKIIα-E183V disrupts interactions with Shank3 and known CaMKAPs
CaMKII interacts with CaMKAPs to regulate multiple synaptic functions. Recent proteomic analyses in our laboratory detected several ASD-linked proteins associated with synaptic CaMKII holoenzymes, including the Shank protein family (Baucum et al., 2015). Because SHANK3 is one of the most frequently mutated genes associated with ASD (Moessner et al., 2007; Jiang and Ehlers, 2013), we examined the effects of E183V mutation on CaMKIIα interaction with GFP-Shank3 in soluble fractions of cotransfected HEK293T cells. Antibodies to the GFP tag readily coprecipitated WT-CaMKIIα from cell-soluble fractions containing GFP-Shank3, but not from control soluble fractions expressing GFP alone; the E183V mutation abolished coprecipitation with GFP-Shank3 (Fig. 3A). To determine whether CaMKIIα-E183V mutation disrupts interactions with other CaMKAPs, we developed a fluorescence-based binding assay. GST-tagged CaMKII-binding domains from various CaMKAPs were immobilized in glutathione-coated 96-well plates and then incubated with mApple(mAp)-tagged CaMKIIα-WT or mAp-CaMKIIα-E183V. The mAp-CaMKIIα-WT robustly binds to the NMDA receptor GluN2B subunit, the voltage-gated calcium channel β2a subunit, the internal CaMKII-binding domain of densin (Den-IN), and the mGlu5 metabotropic glutamate receptor, but E183V mutation significantly reduced all of these interactions (two-way ANOVA with Tukey's post hoc; percentage reduction compared with WT: GluN2B 84.0 ± 2.9%, p < 0.0001; β2a subunit 84.4 ± 5.2%, p < 0.0001; Den-IN 81.0 ± 5.2%, p < 0.0001; mGlu5 88.1 ± 5.2%, p < 0.0001; n = 3 biological replicates) (Fig. 3B). Together, these data indicate that CaMKIIα-E183V mutation disrupts interactions with a variety of known CaMKAPs, including ASD-associated proteins, such as Shank3, GluN2B, and mGlu5.
Figure 3.
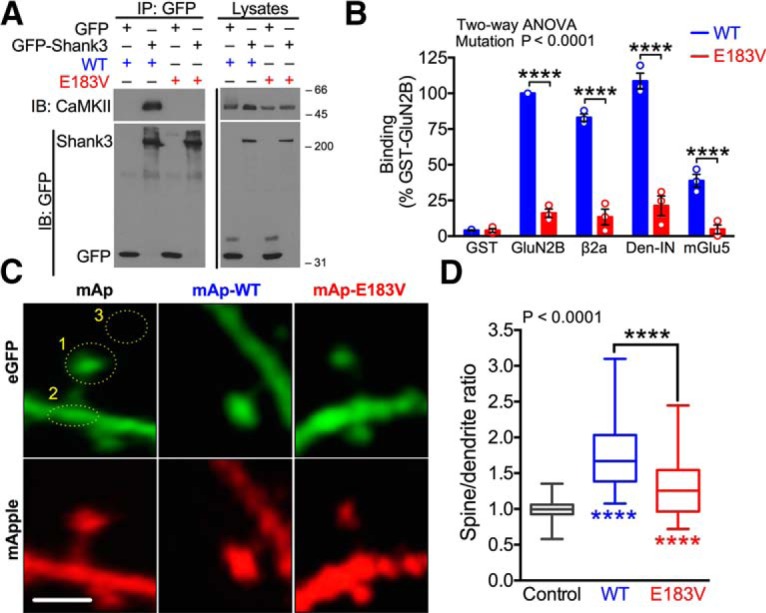
E183V mutation disrupts interactions of CaMKIIα with CaMKAPs and CaMKIIα synaptic targeting. A, Soluble fractions of HEK293T cells expressing GFP or GFP-Shank3 with CaMKIIα (WT or E183V) were immunoprecipitated using an anti-GFP antibody. GFP and CaMKII immunoblots of cell-soluble fractions and GFP immune complexes representative of three independent replicates. B, Soluble fractions of HEK293FT cells expressing mApple(mAp)-CaMKIIα (WT or E183V) were incubated with GST, GST-GluN2B, GST-β2a, GST-Densin-IN, or GST-mGlu5 in glutathione-coated 96-well plates. All incubations included excess Ca2+, calmodulin, and ADP to induce an activated conformation of CaMKIIα, except for the GST-mGlu5 wells. Graph summarizes data from three independent replicates, with binding normalized to the WT interaction with GST-GluN2B. ****p < 0.0001(Tukey's post hoc). p < 0.0001, mutation effect (two-way ANOVA; F(4,20) = 84.33). Data are mean ± SEM. C, Primary hippocampal neuron cultures (DIV 14) were transfected to express soluble eGFP with either soluble mApple (mAp) or mAp-CaMKIIα (WT or E183V). Dendritic arbors were imaged at DIV 20–22 by confocal microscopy. Fluorescence levels of mApple and eGFP were quantified in dendritic spines (circle 1), the underlying dendritic shaft (circle 2), and a nearby background region (circle 3). Scale bar, 1 μm. D, Ratios of mAp to GFP fluorescence were quantified in spines and in the underlying dendritic shafts. Box plots represent spine/shaft ratios; box limits show first and third quartiles. Centerline indicates the median. Whiskers represent minimum and maximum values (n = 54 mAp, n = 56 mAp-WT, n = 53 mAp-E183V spines from 6 or 7 neurons from three separate cultures/transfections). p < 0.0001 (one-way ANOVA with Tukey's post hoc analysis, F(2,160) = 60.55). Colored asterisks indicate difference from control. ****p < 0.0001.
CaMKIIα-E183V disrupts synaptic targeting
Given the impact of E183V mutation on CaMKIIα activity and its interactions with CaMKAPs, we tested whether E183V mutation disrupts synaptic targeting of CaMKIIα in primary cultures of rat hippocampal neurons. Neurons were transfected after 14 DIV to express a soluble GFP marker of morphology along with soluble mAp, mAp-CaMKIIα-WT, or mAp-CaMKIIα-E183V, and then fixed at DIV 20–22. Fluorescence images (Fig. 3C) were collected by confocal microscopy, and the relative mApple and eGFP fluorescence levels in dendritic spines and the underlying dendritic shaft were compared (see Materials and Methods). Both mAp-CaMKIIα-WT and mAp-CaMKIIα-E183V were significantly targeted to synapses (spine/dendrite ratios of 1.77 ± 0.06, n = 56 spines, and 1.31 ± 0.06, n = 53, respectively, compared with the expected 1.00 ± 0.02 (n = 54) for mApple alone. However, synaptic targeting of mAp-CaMKIIα-E183V mutation was significantly reduced compared with mAp-CaMKIIα-WT (p < 0.0001) (Fig. 3D). Thus, E183V mutation reduces CaMKIIα targeting to spines under basal culture conditions, presumably due to disrupted binding to synaptic CaMKAPs (Figs. 2D, 3B).
CaMKIIα-E183V decreases dendritic spine density but does not affect spine morphology
ASD-associated mutations in several synaptic proteins can affect dendritic spine density and morphology (Penzes et al., 2011). To determine the effect of CaMKIIα-E183V mutation, we reconstructed dendritic arbors of neurons transfected as in Figure 3C using Imaris software (Fig. 4A). Neurons expressing mAp-CaMKIIα-E183V had a significantly lower spine density compared with neurons expressing mAp alone or mAp-CaMKIIα-WT (mAp, 14.5 ± 2.1 spines/10 μm, n = 6 neurons; mAp-WT, 14.8 ± 1.7, n = 7; mAp-E183V, 6.4 ± 0.6, n = 7; one-way ANOVA with Tukey's post hoc; p < 0.005 in both cases) (Fig. 4B). However, there was no significant difference in the distribution of spines between the major morphological subtypes (stubby, mushroom, or long/thin) (Swanger et al., 2011) (Fig. 4C). These data indicate that the CaMKIIα-E183V mutant significantly reduces dendritic spine density in cultured hippocampal neurons but does not affect spine morphology.
Figure 4.

Expression of CaMKIIα-E183V reduces spine density but does not alter spine morphology. A, Rat hippocampal neurons were transfected at DIV 14 with soluble GFP plus soluble mAp or mAp-CaMKIIα (WT or E183V). Confocal images (63×) were collected at DIV 20–22. Scale bar: Top, 20 μm. Representative images of GFP in transfected neurons are shown, along with a zoomed-in view of GFP and mAp fluorescence in the indicated dendritic segments. Imaris 3D reconstructions used to quantitatively analyze dendritic spines are shown below. Scale bar: Bottom, 5 μm. B, Dendritic spine densities (one-way ANOVA with Tukey's post hoc analysis, F(2,17) = 9.462, p = 0.0017). C, Percentages of stubby, mushroom, or thin spines (two-way ANOVA with Tukey's post hoc analysis, transfection F(2,54) = 8.2e-14, p > 0.9999). Summary data (mean ± SEM) were quantified from 2–4 nonprimary dendritic segments per neuron from 6–8 neurons (three independent cultures/transfections). **p < 0.01.
CaMKIIα-E183V enhances dendritic arborization
Psychiatric and neurodevelopmental disorders, including ASD, are often associated with aberrant dendritic arborization (Penzes et al., 2011). Furthermore, CaMKII has been shown to regulate dendritic branching in cultured neurons (Fink et al., 2003) and in vivo (Wu and Cline, 1998; Klug et al., 2012). To determine the effects of E183V mutation on dendritic development, we transfected cultured hippocampal neurons at DIV 8, during the normal robust postnatal increase of endogenous CaMKIIα expression (Bito et al., 1996). Images of GFP fluorescence in whole neurons (10–12 DIV) were traced to analyze morphology (Fig. 5A). A Sholl analysis revealed that expression of mAp-CaMKIIα-E183V significantly enhanced dendritic arborization relative to expression of mAp control or mAp-CaMKIIα-WT (mAp, n = 22 neurons; mAp-WT, n = 24; mAp-E183V, n = 24; two-way ANOVA, transfection, p = 0.0021) (Fig. 5B). This was accompanied by significant increases in numbers of dendritic branch points (mAp, 120 ± 8, p = 0.0024; mAp-WT, 95 ± 10, p < 0.0001; mAp-E183V, 178 ± 16; one-way ANOVA with Tukey's post hoc) (Fig. 5C), dendritic segments (mAp, 246 ± 15, p = 0.0023; mAp-WT, 196 ± 97, p < 0.0001; mAp-E183V 364 ± 31; one-way ANOVA with Tukey's post hoc) (Fig. 5D) and terminal points (Fig. 5E). Interestingly, expression of mAp-CaMKIIα-WT significantly decreased total dendritic length compared with control and mAp-CaMKIIα-E183V, yet no significant difference was observed between control mAp and mAp-CaMKIIα-E183V (mAp, 6853 ± 409 μm, p = 0.0041; mAp-WT, 4894 ± 267 μm; mAp-E183V, 8179 ± 487 μm, p < 0.0001; one-way ANOVA with Tukey's post hoc) (Fig. 5F). Concomitantly, the expression of mAp-CaMKIIα-E183V significantly decreased the length of individual dendritic segments compared with control mAp and mAp-CaMKIIα-WT (mAp, 28.4 ± 0.9 μm, p = 0.0088; mAp-WT, 28.9 ± 1.2 μm, p = 0.0022; mAp-E183V, 23.8 ± 1.0 μm, one-way ANOVA with Tukey's post hoc) (Fig. 5G). However, no overall difference was seen in diameter and volume of individual dendrites (Figs. 5H,I). Together, these data indicate that overexpression of the CaMKIIα-E183V mutant in cultured neurons enhances dendritic arborization.
Figure 5.
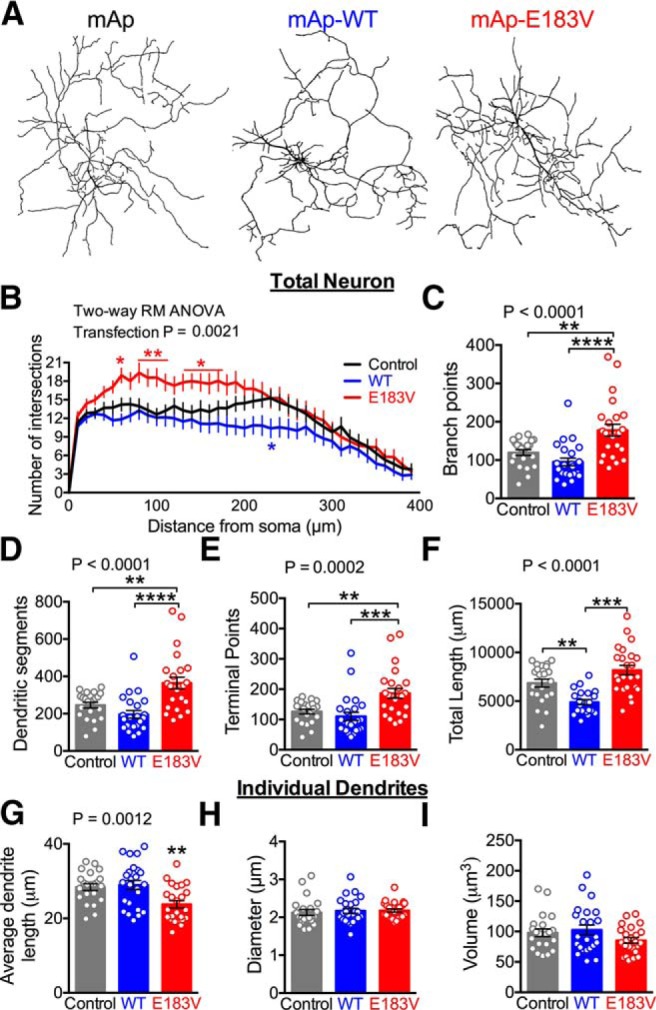
Expression of CaMKIIα-E183V enhances dendritic arborization but decreases individual dendrite length. A, Rat hippocampal neurons were transfected at DIV 8 with soluble GFP, plus soluble mAp or mAp-CaMKIIα (WT or E183V). Confocal images were collected at DIV 10–12; representative traces of the dendritic arbors generated using Imaris for quantitative analyses are shown. A total of 22–24 cells from three separate cultures/transfections were analyzed. B, Sholl analyses of dendritic arbor traces (two-way repeated-measures ANOVA with Tukey's post hoc analysis, F(78,2613) = 2.032, p < 0.0001, interaction; F(39,2613) = 53.05, p < 0.0001, distance; F(2,67) = 6.757, p = 0.0021, transfection; F(67,2613) = 28.15, p < 0.0001, subjects). C, Dendritic branch points (one-way ANOVA with Tukey's post hoc analysis, F(2,66) = 13.36, p < 0.0001). D, Numbers of dendritic segments (one-way ANOVA with Tukey's post hoc analysis, F(2,66) = 13.36, p < 0.0001). E, Terminal points (one-way ANOVA with Tukey's post hoc analysis, F(2,67) = 9.584, p = 0.0002). F, Total dendritic length (one-way ANOVA with Tukey's post hoc analysis, F(2,64) = 16.33, p < 0.0001). G, Average dendritic length (one-way ANOVA with Tukey's post hoc analysis, F(2,67) = 7.463, p < 0.0001). H, Average dendritic diameter (one-way ANOVA with Tukey's post hoc analysis, F(2,67) = 0.1876, p = 0.8294). I, Average dendritic volume (one-way ANOVA with Tukey's post hoc analysis, F(2,66) = 2.017, p = 0.1412). *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001. Data are mean ± SEM.
CaMKIIα-E183V reduces AMPAR-mediated synaptic transmission
To investigate the impact of the E183V mutation on synaptic transmission, we compared AMPAR-mediated mEPSCs in cultured hippocampal neurons following viral expression of mAp-CaMKIIα-WT or mAp-CaMKIIα-E183V together with soluble eGFP (Fig. 6A,B). Neurons were transduced at DIV 14, and mEPSCs were recorded from GFP-positive cells at DIV 20–22. Expression of mAp-CaMKIIα-WT significantly increased both the amplitude and frequency of mEPSCs relative to the control (expression of GFP alone) (Fig. 6C--E). In contrast, expression of mAp-CaMKIIα-E183V significantly reduced mEPSC frequency compared with both mAp-CaMKIIα-WT and mAp control, but significantly reduced mEPSC amplitude only compared with mAp-CaMKIIα-WT (amplitude means: mAp = 26.9 ± 1.0 pA, n = 20; mAp-WT = 31.6 ± 1.6 pA, n = 19, mAp-E183V = 24.2 ± 1.4 pA, n = 20; one-way ANOVA, p = 0.0013; frequency means: mAp = 17.5 ± 1.8 Hz, n = 20; mAp-WT = 25.7 ± 2.3 Hz, n = 19; mAp-E183V = 9.0 ± 1.4, n = 20; one-way ANOVA, p < 0.0001) (Fig. 6C–E). Overall, these results indicate that the CaMKIIα-E183V mutation disrupts AMPAR-mediated synaptic transmission. Moreover, the decreased mEPSC frequency is likely related to the observed decreases in spine density due to CaMKIIα-E183V expression. These data indicate that CaMKIIα-E183V can exert a dominant-negative effect on the endogenous CaMKIIα-WT in these hippocampal cultures to modulate excitatory synaptic transmission.
Figure 6.

Expression of CaMKIIα-E183V inhibits basal excitatory synaptic transmission. A, Design of lentiviral constructs. A synapsin promoter and a P2A cleavage site allow for coexpression of mAp-CaMKIIα (WT or E183V) with soluble GFP or GFP alone. B, Rat hippocampal neurons (DIV 14) were transduced at DIV 14, and representative confocal images were collected at DIV 20–22. C, Representative mEPSC recordings from virally transduced rat hippocampal neurons at DIV 20–22, GFP alone (black), mAp-CaMKIIα-WT (mAp-WT; blue), or mAp-CaMKIIα-E183V (mAp-E183V; red). A total of 19 or 20 cells from three separate cultures/transductions were analyzed. D, Cumulative probability distributions of mEPSC amplitudes and average mEPSC amplitudes (inset). Cumulative probability: Kruskal-Wallis with Dunn's post hoc analysis (K-W statistic = 23.03, p < 0.0001). Box plot inset, p = 0.0013 (one-way ANOVA with Tukey's post hoc analysis, F(2,55) = 7.511). Colored asterisks indicate difference from control. E, Cumulative probability distributions of mEPSC interevent intervals and average mEPSC frequencies (inset). Cumulative probability: Kruskal-Wallis with Dunn's post hoc analysis (K-W statistic = 133.7, p < 0.0001). Box plot inset, p < 0.0001 (one-way ANOVA with Tukey's post hoc analysis, F(2,55) = 20.00). Colored asterisks indicate differences from control. *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001. Data are mean ± SEM.
CaMKIIα-E183V knock-in mice have reduced levels of CaMKIIα
To investigate the in vivo impact of the ASD-linked E183V-CaMKIIα mutation, we created a knock-in mouse on a hybrid C57BL6J/DBA2J background using a CRISPR/Cas9 strategy (see Materials and Methods; Fig. 7A). Mice were maintained on the hybrid background and all animals used were bred from F2 E183V heterozygotes (Fig. 7B). Notably, all mice bred normally, were viable, and had no visible anatomical differences. Immunoblots of whole forebrain extracts from mice at approximately P90 revealed that total CaMKIIα levels were significantly reduced to 50 ± 13% (one-sample t test, p = 0.0341, n = 4) and 15 ± 5% (one-sample t test, p = 0.0004, n = 4) in heterozygous (Camk2aWT/E183V) and homozygous (Camk2aE183V/E183V) mutant mice, respectively, compared with WT (Camk2aWT/WT, n = 4), (Fig. 7C, top). Total levels of CaMKIIβ were unchanged (Fig. 7C, bottom).
Figure 7.
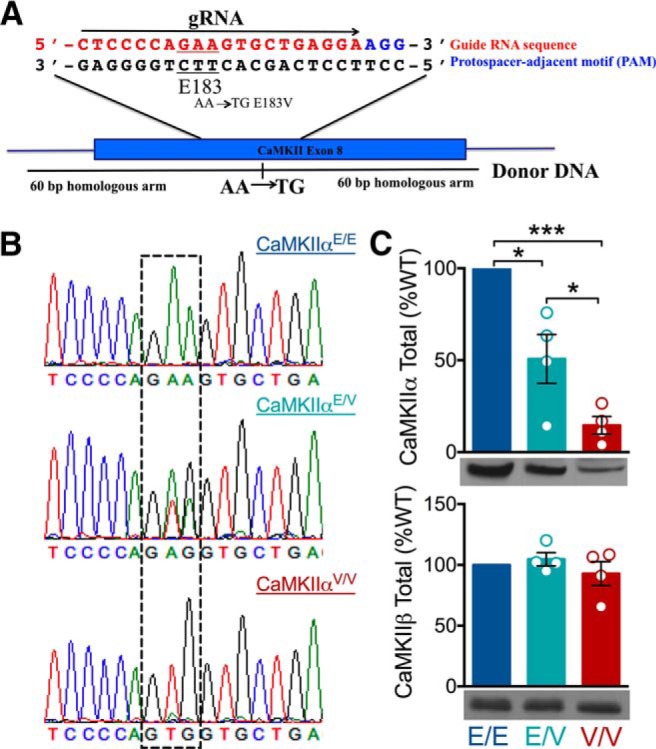
Production of knock-in mutant mice with a CaMKIIα-E183V mutation. A, Partial sequence of exon 8 of the Camk2a gene indicating the selected guide RNA (red) and PAM sequence (blue) used for CRISPR/Cas9, as well as the Glu183 codon (underlined). Donor DNA oligonucleotide was designed to substitute the last two bases of the E183 codon with TG to encode for valine with 60 bp homologous arms on either side of the substitution. B, Representative genomic sequencing of WT (E/E), heterozygous (E/V), and homozygous mice (V/V). Second and third nucleotides of Camk2a codon 183 are highlighted by the dashed box, indicating the WT GAA (Glu) and homozygous KI mutant GTG (Val). C, Immunoblots of mouse forebrain homogenates from E/E, E/V, and V/V mice (n = 4 each) for CaMKIIα (top) and CaMKIIβ (bottom) (CaMKIIα: one-way ANOVA with Tukey's post hoc analysis, F(2,9) = 27.59, p = 0.0001; CaMKIIβ: one-way ANOVA with Tukey's post hoc analysis, F(2,9) = 1.442, p = 0.2905). *p < 0.05. ***p < 0.001. Data are mean ± SEM.
Disruption of CaMKII localization and autophosphorylation in CaMKIIα-E183V knock-in mice
To assess the biochemical impact of E183V mutation in vivo, we prepared cytosolic (S1), Triton-soluble membrane (S2), and Triton-insoluble synaptic (P2) fractions from forebrains of Camk2aWT/WT, Camk2aWT/E183V, and Camk2aE183V/E183V mice, and assessed levels of CaMKIIα and Thr286 autophosphorylation by immunoblotting. Total levels of CaMKIIα were significantly, if modestly, reduced in S1 fractions from Camk2aE183V/E183V mice (73 ± 11%) compared with S1 fractions from Camk2aWT/WT and Camk2aWT/E183V mice (Fig. 8A,B, left). However, total levels of CaMKIIα in the S2 and P2 fractions were significantly reduced in both Camk2aWT/E183V and Camk2aE183V/E183V compared with Camk2aWT/WT (S2: 78 ± 8% and 24 ± 2%, respectively; P2: 88 ± 3% and 18 ± 3%, respectively) (Fig. 8A,B, middle, right). Recalculation of these data revealed genotype-dependent differences in the relative distribution of CaMKIIα between the subcellular fractions expressed as a percentage of total CaMKIIα levels in S1, S2, and P2 combined. Relative CaMKIIα distributions between subcellular fractions from Camk2aWT/WT and Camk2aWT/E183V mice were not significantly different, but a significantly higher percentage of the reduced total CaMKIIα levels was in the cytosolic fraction from Camk2aE183V/E183V mice (Camk2aWT/WT: 15 ± 3%; Camk2aWT/E183V: 15 ± 2%; Camk2aE183V/E183V: 33 ± 6%; mean ± SEM; p = 0.0092, one-way ANOVA), with a significantly reduced percentage in the synaptic fraction (Camk2aWT/WT: 67 ± 3%; Camk2aWT/E183V: 68 ± 2%; Camk2aE183V/E183V: 47 ± 5%; mean ± SEM; p = 0.0023, one-way ANOVA). Interestingly, relative levels of CaMKIIα Thr286 autophosphorylation in the S1 fraction were increased in Camk2aE183V/E183V mice (124 ± 8%) compared with Camk2aWT/WT and Camk2aWT/E183V mice (Fig. 8C, left), whereas levels of Thr286 autophosphorylation were reduced in Camk2aWT/E183V P2 fractions (61 ± 7%) and in Camk2aE183V/E183V S2 and P2 fractions (S2: 58 ± 6%; P2: 56 ± 7%) compared with Camk2aWT/WT (Fig. 8C, middle, right). Together, these data indicate that the E183V mutation interferes with both the subcellular distribution and autophosphorylation of CaMKIIα in vivo.
Figure 8.

Disruption of CaMKII subcellular targeting and autophosphorylation in CaMKIIα-E183V KI mice. A, Cytosolic (S1), membrane (S2), or synaptic fractions (P2) isolated from Camk2aWT/WT, Camk2aWT/E183V, and Camk2aE183V/E183V mice were immunoblotted for total CaMKIIα (top) and phosphorylated Thr286 (pT286, bottom). B, CaMKIIα levels in the S1 (left), S2 (middle), and P2 (right) fractions normalized to WT levels within each fraction (S1: Camk2aWT/E183V, t(6) = 1.484, p = 0.1883, n = 7; Camk2aE183V/E183V, t(6) = 2.498, p = 0.0466, n = 7; S2: Camk2aWT/E183V, t(6) = 2.752, p = 0.0322, n = 7; Camk2aE183V/E183V, t(4) = 31.50, p < 0.0001, n = 5; P2: Camk2aWT/E183V, t(6) = 3.585, p = 0.0116, n = 7; Camk2aE183V/E183V, t(5) = 25.43, p < 0.0001, n = 6; all by one-sample t test compared with theoretical value of 100%). C, The pT286/CaMKIIα ratios in S1, S2, and P2 fractions normalized to WT in each fraction (S1: Camk2aWT/E183V, t(6) = 0.033, p = 0.9744, n = 7; Camk2aE183V/E183V, t(6) = 2.892, p = 0.0276, n = 7; S2: Camk2aWT/E183V, t(6) = 1.882, p = 0.1088, n = 7; Camk2aE183V/E183V, t(5) = 6.908, p = 0.0010, n = 6; P2: Camk2aWT/E183V, t(6) = 5.910, p = 0.0010, n = 7; Camk2aE183V/E183V, t(6) = 6.545, p = 0.0006, n = 7; one-sample t tests). D, Cytosolic (S1), membrane (S2), or synaptic fractions (P2) isolated from Camk2aWT/WT, Camk2aWT/E183V, and Camk2aE183V/E183V mice were immunoblotted for total CaMKIIβ (top) and phosphorylated Thr287 (pT287, bottom). E, CaMKIIβ levels in the S1 (left), S2 (middle), and P2 (right) fractions normalized to WT in each fraction (S1: Camk2aWT/E183V, t(6) = 0.1665, p = 0.8740, n = 7; Camk2aE183V/E183V, t(6) = 0.1335, p = 0.8967, n = 7; S2: Camk2aWT/E183V, t(6) = 0.1958, p = 0.8513, n = 7; Camk2aE183V/E183V, t(6) = 0.0279, p = 0.9786, n = 7; P2: Camk2aWT/E183V, t(6) = 0.5819, p = 0.5818, n = 7; Camk2aE183V/E183V, t(6) = 1.679, p = 0.1443, n = 7; one-sample t tests). F, The pT287/CaMKIIβ ratios in S1, S2, and P2 fractions normalized to WT in each fraction (S1: Camk2aWT/E183V, t(6) = 0.7554, p = 0.4786, n = 7; Camk2aE183V/E183V, t(6) = 0.7844, p = 0.4626, n = 7; S2: Camk2aWT/E183V, t(6) = 0.5782, p = 0.5842, n = 7; Camk2aE183V/E183V, t(6) = 2.926, p = 0.0264, n = 7; P2: Camk2aWT/E183V, t(5) = 0.4385, p = 0.6793, n = 6; Camk2aE183V/E183V, t(6) = 9.399, p < 0.0001, n = 7; one-sample t tests).
We extended these analyses of brain subcellular fractions to CaMKIIβ. The total CaMKIIβ levels in each fraction were not significantly different in Camk2aWT/WT, Camk2aWT/E183V, and Camk2aE183V/E183V mice (Fig. 8D,E). Moreover, the fraction of total CaMKIIβ in the three subcellular fractions was unaffected by genotype (data not shown). However, pT287/total levels were reduced in both S2 and P2 fractions from Camk2aE183V/E183V mice compared with Camk2aWT/WT (S2: 66 ± 12%; P2: 45 ± 6%) (Fig. 8F). Thus, although the CaMKIIβ subcellular distribution is unaffected, these data are consistent with the notion that CaMKIIα-E183V exerts a dominant-negative effect to reduce CaMKIIβ phosphorylation.
E183V-CaMKIIα reduces protein stability and enhances ubiquitylation
Because CaMKIIα-E183V is expressed at lower levels than CaMKIIα-WT in both transfected HEK293T cells and in Camk2aE183V/E183V mice (Figs. 1B, 7C), we tested the hypothesis that E183V mutation increases the turnover of CaMKIIα. Cycloheximide was added to HEK293T cells expressing CaMKIIα-WT or CaMKIIα-E183V to inhibit protein synthesis 48 h after transfection. Levels of CaMKIIα-E183V, but not CaMKIIα-WT, in the soluble fraction decreased significantly after 8 h (two-way repeated-measures ANOVA, transfection, p = 0.0001. 0 h vs 8 h: E183V, 54 ± 16%, p = 0.015. WT, 110 ± 13%, n = 4) (Fig. 9A). Thus, the CaMKIIα-E183V mutation enhances protein degradation/turnover.
Figure 9.
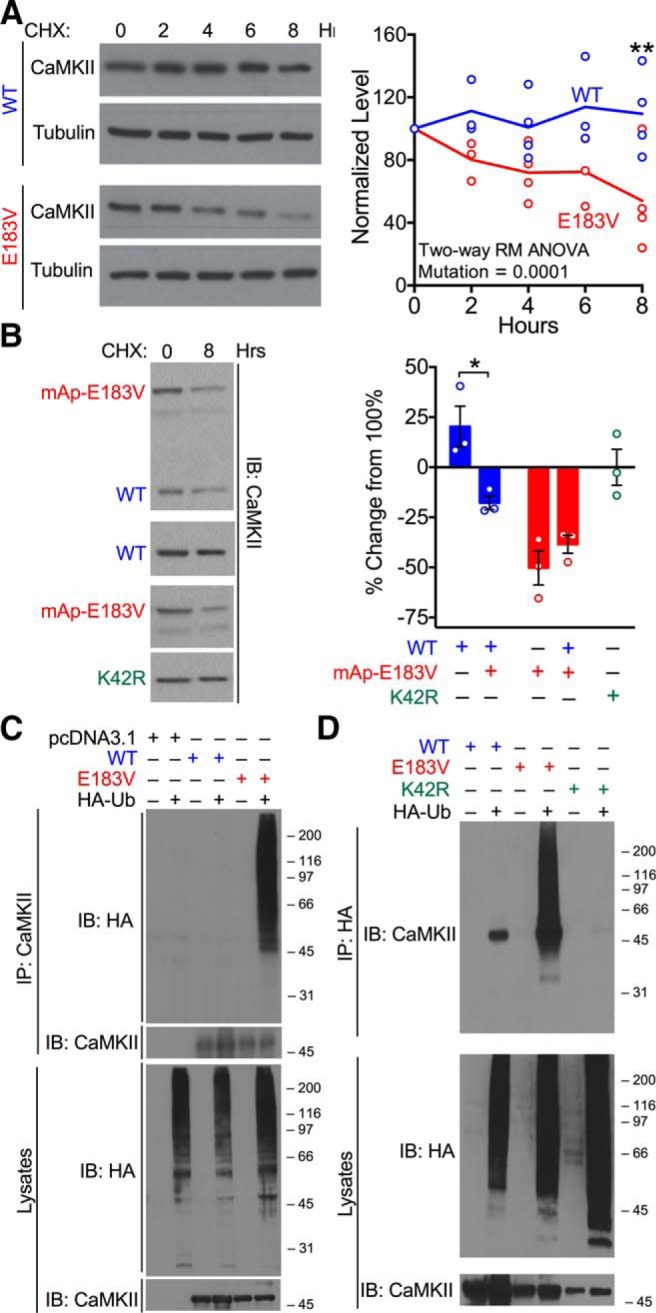
The E183V mutation reduces CaMKIIα stability and enhances ubiquitylation. A, Left, Cycloheximide (50 μg/μl) was added to HEK293T cells expressing CaMKIIα-WT or CaMKIIα-E183V. Cells were lysed at the indicated times and immunoblotted for CaMKIIα and tubulin. Right, Summary graph of CaMKIIα levels normalized to total protein loading (two-way repeated-measures ANOVA with Tukey's post hoc analysis, F(4,26) = 5.447, p = 0.0025, interaction; F(4,26) = 1.458, p = 0.2435, time; F(1,26) = 55.38, p < 0.0001, transfection). Data are mean ± SEM; n = 4. B, Left, Cycloheximide (50 μg/μl) was added to HEK293T cells expressing CaMKIIα-WT, mAp-CaMKIIα-E183V, and/or CaMKIIα-K42R. Cells were lysed after 8 h and immunoblotted for CaMKIIα. Right, Summary of CaMKIIα levels normalized to total protein loading and shown as a percentage change from time 0 (two-way ANOVA with Tukey's post hoc analysis, F(1,8) = 12.01, p = 0.0085, interaction; F(1,8) = 40.29, p = 0.0002, transfection). Data are mean ± SEM; n = 3. C, Soluble fractions of HEK293T cells expressing HA-ubiquitin alone or HA-ubiquitin with either CaMKIIα-WT or CaMKIIα-E183V were immunoprecipitated using a CaMKIIα antibody. Inputs and immune complexes were immunoblotted for HA and CaMKIIα. D, Soluble fractions of HEK293T cells expressing HA-ubiquitin alone or HA-ubiquitin with CaMKIIα-WT, CaMKIIα-E183V, or CaMKIIα-K42R were immunoprecipitated using an HA-antibody. Inputs and immune complexes were immunoblotted for HA and/or CaMKIIα. *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001.
To determine whether CaMKIIα-E183V affects the stability of coexpressed CaMKIIα-WT, HEK293T cells were cotransfected to express untagged CaMKIIα-WT and/or mAp-CaMKIIα-E183V and treated with cycloheximide. The mApple-tagged CaMKIIα-E183V protein is relatively unstable when expressed alone (49 ± 8% residual protein after 8 h), similar to the instability of untagged mutant protein (see above), and this instability was not significantly altered by the coexpression of CaMKIIα-WT (Fig. 9B). However, CaMKIIα-WT levels were significantly lower when coexpressed with CaMKIIα-E183V than when expressed alone (82 ± 3% of time 0 and 120 ± 10%, respectively) (Fig. 9B). Furthermore, levels of a kinase-dead CaMKIIα-K42R mutant protein were stable over an 8 h cycloheximide treatment (99 ± 9%) (Fig. 9B). In combination, these data indicate that the instability of CaMKIIα-E183V is not a direct result of the low levels of kinase activity and that the mutant protein can exert a dominant effect to decrease the stability of coexpressed WT protein.
The ubiquitin-proteasome system is a major pathway for cellular protein degradation/turnover. To investigate potential ubiquitylation of CaMKIIα, we expressed CaMKIIα-WT or CaMKIIα-E183V in HEK293T cells with/without HA-tagged ubiquitin. Cell-soluble fractions and immunoprecipitated CaMKIIα complexes were then isolated and immunoblotted for CaMKIIα and HA-ubiquitin. As expected, cell-soluble fractions contained numerous HA-ubiquitylated species that form an immunoreactive smear. Moreover, HA-ubiquitylated species coimmunoprecipitated with CaMKIIα-E183V but could not be detected in CaMKIIα-WT immune complexes (Fig. 9C). Furthermore, samples immunoprecipitated from soluble fractions of cells expressing CaMKIIα-E183V using an antibody to the HA tag on ubiquitin contained a robust smear of CaMKII-immnunoreactive high molecular weight proteins, but HA immunoprecipitates from cells expressing CaMKIIα-WT or CaMKIIα-K42R contain unmodified CaMKIIα (Fig. 9D). Together, these data indicate that CaMKIIα-WT and CaMKIIα-K42R associate directly or indirectly with HA-ubiquitylated proteins and are also consistent with the hypothesis that CaMKIIα-E183V is uniquely polyubiquitylated to enhance CaMKIIα degradation and reduce total expression levels.
CaMKIIα-E183V knock-in mice are hyperactive and display repetitive behaviors
We assessed the behavioral impact of the ASD-associated E183V mutation using a cohort of 3 litters, containing 10 Camk2aWT/WT, 23 Camk2aWT/E183V, and 13 Camk2aE183V/E183V mice. Initial testing in a novel open field arena revealed that Camk2aE183V/E183V mice are hyperactive throughout the 30 min test period compared with Camk2aWT/WT and Camk2aWT/E183V littermates (two-way repeated-measures ANOVA, genotype, p < 0.0001) (Fig. 10A). There was also a significant genotype effect on time spent in the center of the open field arena (two-way repeated-measures ANOVA, genotype, p = 0.0032), with Camk2aE183V/E183V and Camk2aWT/E183V mice spending significantly less time in the center compared with the Camk2aWT/WT controls (Fig. 10B), consistent with an allele-dose-dependent increase of anxiety. Camk2aE183V/E183V mice also exhibited significantly increased rearing and jumping (two-way repeated-measures ANOVA, genotype, p < 0.0001 for both rearing and jumping) compared with Camk2aWT/WT and Camk2aWT/E183V, (Fig. 10C,D). Interestingly, changes in center time, rearing, and jumping behaviors were persistent across the entire 30 min time period, in contrast to the normal habituation of horizontal locomotor activity. The enhanced vertical activities of Camk2aE183V/E183V mice in the open field can be interpreted as an increased repetitive behavior, mimicking a core ASD phenotype. The overall hyperactivity and heightened anxiety of Camk2aE183V/E183V mice also may mimic commonly comorbid symptoms of ASD.
Figure 10.

Behavioral comparisons of CaMKIIα-WT and E183V KI mice. A–D, Measures of activity in a novel open field arena, binned by time in the arena. A, Horizontal locomotion (two-way repeated-measures ANOVA with Tukey's post hoc analysis, F(10,230) = 5.581, p < 0.0001, interaction; F(5,230) = 55.52, p < 0.0001, time; F(2,46) = 14.36, p < 0.0001, genotype; F(46,230) = 11.99, p < 0.0001, subjects). B, Time in center (two-way repeated-measures ANOVA with Tukey's post hoc analysis, F(10,230) = 0.6233, p = 0.7933, interaction; F(5,230) = 1.466, p = 0.2017, time; F(2,46) = 6.545, p = 0.0032, genotype; F(46,230) = 5.835, p < 0.0001, subjects). Asterisks indicate significant difference from control. C, Rearing counts (two-way repeated-measures ANOVA with Tukey's post hoc analysis, F(10,230) = 1.017, p = 0.4296, interaction; F(5,230) = 2.848, p = 0.0162, time; F(2,46) = 13.10, p < 0.0001, genotype; F(46,230) = 8.887, p < 0.0001, subjects). D, Jumping counts (two-way repeated-measures ANOVA with Tukey's post hoc analysis, F(10,230) = 0.6907, p = 7.372, interaction; F(5,230) = 3.63, p = 0.0035, time; F(2,46) = 16.18, p < 0.0001, genotype; F(46,230) = 10.84, p < 0.0001, subjects). E, Three-chamber test for social interactions. Comparisons of time spent interacting with an empty (E) versus novel mouse (M1) pencil cups (Phase 1; left) and familiar mouse (M1) versus a novel mouse (M2) (Phase 2; right) for each genotype (paired t test, Phase 1, E/E: t(9) = 2.434, p = 0.038; Phase 2, E/E: t(9) = 3.362, p = 0.008; Phase 1, E/V: t(24) = 4.117, p = 0.0004; Phase 2, E/V: t(25) = 0.097, p = 0.914; Phase 1, V/V: t(13) = 1.857, p = 0.088; Phase 2, V/V: t(13) = 2.453, p = 0.030). F, Rotational behavior measured in the 3-chamber test (one-way ANOVA with Tukey's post hoc analysis, F(2,46) = 17.33, p < 0.0001). G, Marble burying test: number of marbles (of 12 total) buried in 30 min (one-way ANOVA with Tukey's post hoc analysis, F(2,46) = 17.33, p < 0.0001). For all behavioral analyses: E/E, n = 10; E/V, n = 25; V/V, n = 14. *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001. Data are mean ± SEM.
CaMKIIα-E183V knock-in mice display social deficits and decreased exploratory behavior
To determine the effects of E183V mutation on social interactions, test mice were first habituated to a 3-chamber apparatus containing empty (E) inverted wire pencil cups in the two outer chambers. A novel mouse was then placed under one of the cups (M1), and times spent by the test mice interacting with the two cups were compared. As expected, Camk2aWT/WT mice spent significantly more time with the wire cup containing the novel mouse (E = 88 ± 10 s; M1 = 119 ± 10 s; paired t test, p = 0.038). Camk2aWT/E183V also displayed a preference for the novel mouse (E = 89 ± 8 s; M1 = 143 ± 10 s; paired t test, p = 0.0004), but there was no significant difference between the times that Camk2aE183V/E183V mice spent interacting with the two wire cups (E = 109 ± 12 s; M1 = 141 ± 14 s; paired t test, p = 0.088). In the next phase of this assay, a second novel mouse is placed under the remaining empty cup (M2). As expected, Camk2aWT/WT mice spent more time with the wire cup containing the novel mouse versus the familiar mouse (M1 = 71 ± 8 s; M2 = 100 ± 11 s; paired t test, p = 0.008). In contrast, Camk2aWT/E183V mice displayed no preference for the novel mouse versus the familiar mouse (M1 = 102 ± 10 s; M2 = 100 ± 5 s; paired t test, p = 0.923) (Fig. 10E). However, Camk2aE183V/E183V (M1 = 83 ± 10 s; M2 = 119 ± 15 s; paired t test, p = 0.0304) mice showed a significant preference for a novel mouse. In addition, Camk2aE183V/E183V mice displayed increased repetitive rotational behavior in the 3-chamber assay compared with other groups (Camk2aWT/WT, 98 ± 5, p = 0.0315; Camk2aWT/E183V, 97 ± 4, p = 0.0035; Camk2aE183V/E183V, 119 ± 5; one-way ANOVA with Tukey's post hoc) (Fig. 10F).
In combination, data from the 3-chamber test indicate that E183V mutation disrupt normal social behavior. Camk2aWT/E183V mice exhibited deficits in social memory or preference for social novelty, whereas Camk2aE183V/E183V mice exhibited a deficit in social motivation, as well as increased repetitive circling behavior, reminiscent of core repetitive behavior symptoms of ASD. Notably in this context, the ASD proband harboring the E183V de novo mutation showed typical impairments in core phenotypic domains of social reciprocity, language/communication, repetitive behaviors, and restricted interests (Table 1). Moreover, this proband does not carry any other obvious pathogenic mutations (Iossifov et al., 2014), including de novo CNVs or SNVs in genes implicated as high confidence or probable ASD candidate genes based on large-scale genetic studies (i.e., CNV and whole exome sequencing) (Sanders et al., 2015). The only other de novo nonsynonymous mutations identified in this proband were a nonsense mutation in a tumor suppressor gene, TUSC1, and a splice site disruption in TERF2, which encodes a component of the telomere nucleoprotein complex.
Table 1.
SSC proband 14620 phenotypic measuresa
| Diagnostic instrument | Scale | SSC14620.p1 (male) | SSC version 15 (all records, n = 2759) |
||
|---|---|---|---|---|---|
| N | Mean (SD) | Range | |||
| Chronological age (yr) | 5.25 | 2759 | 9.03 (3.57) | 4–18 | |
| Verbal IQ | 81 | 2757 | 78.04 (31.27) | 5–167 | |
| Nonverbal IQ | 60 | 2757 | 84.54 (26.16) | 9–161 | |
| ADI-R | Social interaction | 10 | 2757 | 20.34 (5.71) | 8–30 |
| Communication, verbal | 14 | 2424 | 16.50 (4.26) | 6–26 | |
| Communication, nonverbal | 3 | 2757 | 9.26 (3.44) | 0–14 | |
| Restricted/repetitive behaviors | 3 | 2757 | 6.52 (2.50) | 0–12 | |
| ADOS | Calibrated severity scores | 8 | 2684 | 7.44 (1.68) | 4–10 |
| Social + communication | 14 | 2758 | 13.34 (4.16) | 4–24 | |
| Social affect | 10 | 2684 | 11.17 (4.01) | 2–20 | |
| Restricted/repetitive behaviors | 4 | 2758 | 3.97 (2.06) | 0–8 | |
| SRS | SRS parent, raw | 91 | 2747 | 98.01 (27.00) | 11–177 |
| SRS parent, t score | 77 | 2747 | 79.55 (10.46) | 39–91 | |
| ABC | ABC total | 32 | 2754 | 46.40 (25.71) | 0–138 |
| I, Irritability | 11 | 2755 | 11.49 (8.73) | 0–45 | |
| II, Lethargy | 1 | 2755 | 9.79 (7.17) | 0–40 | |
| III, Stereotypy | 5 | 2755 | 4.90 (4.31) | 0–21 | |
| IV, Hyperactivity | 15 | 2754 | 16.60 (10.46) | 0–48 | |
| V, Inappropriate Speech | 0 | 2754 | 3.63 (2.96) | 0–12 | |
| RBS | RBS-R overall | 9 | 2756 | 27.13 (17.39) | 0–105 |
| I, Stereotyped Behavior | 3 | 2756 | 4.46 (3.36) | 0–18 | |
| II, Self-Injurious | 1 | 2756 | 2.09 (2.87) | 0–21 | |
| III, Compulsive Behavior | 0 | 2757 | 4.11 (3.93) | 0–24 | |
| IV, Ritualistic Behavior | 2 | 2756 | 5.04 (3.96) | 0–18 | |
| V, Sameness Behavior | 3 | 2755 | 7.75 (5.99) | 0–33 | |
| VI, Restricted Behavior | 0 | 2755 | 3.67 (2.83) | 0–12 | |
aVerbal/nonverbal I. Values are from the following diagnostic instruments: Autism Diagnostic Interview-Revised (ADI-R) (Lord et al., 1994), Autism Diagnostic Observation Schedule (ADOS) (Lord et al., 2000b), Social Responsiveness Scale (SRS) (Constantino et al., 2003), Aberrant Behavior Checklist (ABC) (Aman et al., 1985), and Repetitive Behavior Scales-Revised (RBS-R) (Wolff et al., 2012), shown for the male ASD proband from SSC family 14620, the overall SSC cohort (both males and females; version 15, https://sfari.org/resources/sfari-base), males only, and females only. ADI-R/ADOS domain and ABC/RBS-R subscale scores are provided to illustrate degree of severity across different dimensions of the phenotype. Ages are expressed in months.
We also tested mice using a marble-burying task, which has been used to test for repetitive/perseverative behaviors, as well as for exploratory behavior in a novel environment (Moy et al., 2014). Interestingly, there were significant decreases in the number of marbles buried by both Camk2aWT/E183V and Camk2aE183V/E183V mice compared with Camk2aWT/WT (Camk2aWT/WT, 8.9 ± 0.6; Camk2aWT/E183V, 5.3 ± 0.8, p = 0.0069; Camk2aE183V/E183V, 1.6 ± 0.5, p <0.0001; one-way ANOVA with Tukey's post hoc) (Fig. 10G). These data indicate that there is an E183V allele dose-dependent decrease in exploratory behavior in this novel environment.
Discussion
CaMKII is among the most intensively studied neuronal proteins, with well-defined roles at excitatory synapses linked to LTP and spatial learning and memory (see Introduction). Studies described herein characterize at multiple levels the functional impact of the first human CAMK2A mutation identified in an individual diagnosed with any neuropsychiatric disorder, explicitly linking CaMKII to ASD-related behaviors, such as social interaction deficits and increased repetitive behaviors.
The de novo missense E183V CAMK2A mutation was recently detected in a proband with a typical ASD diagnosis (Iossifov et al., 2014); social interaction deficits and repetitive/stereotyped behavior, as well as hyperactivity and anxiety (Table 1). This mutation likely contributes to ASD risk in this proband because of: (1) prior knowledge of the neuronal roles of CaMKIIα, particularly binding to and regulating proteins strongly implicated in ASD, such as SHANK3 and GRIN2B; (2) lack of other known significant genetic risk factors in this proband; (3) clear biochemical deficits caused by E183V mutation; (4) effects of CaMKIIα-E183V on dendritic morphology and synaptic function; and (5) ASD-related behavioral phenotypes of E183V knock-in mice.
Mice with a knock-in CaMKIIα-E183V mutation had novel behavioral deficits. Camk2aE183V/E183V mice displayed enhanced repetitive behaviors and deficits in social interactions (Fig. 10), which are generally believed to mimic core ASD symptoms and are similar to other mouse ASD models (Silverman et al., 2010). Homozygous mice were also hyperactive in an open field test, with reduced center time suggesting increased anxiety; hyperactivity and anxiety are frequent comorbid symptoms in ASD patients. Heterozygous mice had less severe phenotypes, but a striking deficit in the novelty phase of the social testing paradigm, which contains components of social memory and recognition (Yang et al., 2011), and more modestly increased anxiety. Camk2aE183V/E183V mice also had a robust increase of repetitive vertical behaviors. Thus, the reduced marble-burying behavior of both Camk2aE183V/E183V and Camk2aWT/E183V mice was initially surprising. However, reduced marble burying was observed in other ASD-mouse models, perhaps indicating reduced exploration in a complex environment (Balemans et al., 2010; Feyder et al., 2010; DeLorey et al., 2011; Moy et al., 2014). It is possible that the hyperactivity and/or increased anxiety of CaMKII-E183V KI mice contributes to social interaction deficits and reduced marble burying. Despite extensive studies of other CaMKII mutant mice, this is the first study showing that CaMKII mutation causes core ASD-related phenotypes.
The biochemical properties of CaMKIIα are profoundly affected by E183V mutation in the catalytic domain. First, catalytic activity is dramatically reduced. The CaMKIIα-E183V mutant retained some activity toward GluA1 AMPA receptor subunits (∼30% of WT) but had negligible activity toward a peptide substrate or by Thr286 autophosphorylation. This difference may relate to prior observations that substrates can interact with CaMKIIα in multiple ways (e.g., Coultrap et al., 2010) and that some CaMKII activators and inhibitors exhibit substrate-selective effects (Jiao et al., 2011; Jalan-Sakrikar et al., 2012). Notably, CaMKIIα-E183V forms holoenzymes with CaMKIIα-WT, presumably accounting for the dominant-negative activity of CaMKIIα-E183V to reduce Thr286 autophosphorylation of CaMKIIα-WT. Second, CaMKIIα-E183V mutation disrupted binding to several CaMKAPs. Approximately 10% of the >100 CaMKAPs specifically associated with synaptic CaMKII holoenzymes (Baucum et al., 2015) have been linked to ASD, including GluN2B; Shank-1, -2, and -3; SAPAP-1, -2, -3, -4; and Homer-1 (Durand et al., 2007; Berkel et al., 2010; Pinto et al., 2010; O'Roak et al., 2012; Sato et al., 2012). Notably, Shank3 was one of the most abundant components of synaptic CaMKII holoenzyme complexes (Baucum et al., 2015) and SHANK3 is among the most commonly mutated genes in ASD (Moessner et al., 2007; Jiang and Ehlers, 2013). Moreover, E183V mutation disrupts Shank3 coimmunoprecipitation with CaMKIIα from transfected cells. CaMKIIα also interacts directly with and/or regulates other ASD-linked synaptic proteins, such as voltage-gated calcium channels and neuroligins (Leonard et al., 1999; Abiria and Colbran, 2010; Jenkins et al., 2010; Bemben et al., 2014; Baucum et al., 2015). In combination, these data indicate that E183V mutation disrupts CaMKII association with an ASD-linked synaptic protein network. Further characterization of biochemical consequences of CaMKIIα-E183V mutation in vitro and in neurons seems likely to provide novel insights into molecular mechanisms underlying ASD.
The expression of CaMKIIα-E183V in neurons that endogenously express CaMKIIα-WT might mimic a heterozygous genotype and had a significant impact on dendritic morphology and synaptic transmission. CaMKIIα-WT was targeted to dendritic spines, as expected, but E183V mutation interfered with synaptic targeting, presumably reflecting reduced interactions with GluN2B, Shank3, and other CaMKAPs. Reduced Thr286 autophosphorylation may also contribute to the disrupted subcellular targeting (McNeill and Colbran, 1995; Strack and Colbran, 1998; Shen and Meyer, 1999; Robison et al., 2005a; Bayer et al., 2006; Bingol et al., 2010; Jiao et al., 2011). CaMKIIα-E183V expression strikingly decreased dendritic spine density and enhanced dendritic arborization relative to neurons expressing CaMKIIα-WT, reminiscent of prior observations that alterations in synaptic maturation are observed with ASD-linked mutations in other synaptic proteins in cultured neurons and in mice (e.g., Shank3) (Peça et al., 2011; Durand et al., 2012). It is perhaps surprising that we did not detect changes in the distribution of different spine types following expression of CaMKIIα-E183V because persistent CaMKIIα translocation to spines has been linked to increased spine size (Zhang et al., 2008; Pi et al., 2010). Moreover, increased dendritic arborization following CaMKIIα-E183V expression in neurons contrasts with the decreased arborization in striatal MSNs following transgenic overexpression of a CaMKII inhibitor (Klug et al., 2012). CaMKII can regulate dendritic morphology by interacting with and/or phosphorylating numerous proteins, including kalirin-7 (Xie et al., 2007), F-actin (Kim et al., 2015), and Rem2, a small Ras-like GTPase (Ghiretti et al., 2013). Therefore, the inhibitory actions of CaMKIIα-E183V on endogenous CaMKIIα and/or CaMKIIβ may disrupt one or more of these pathways to affect dendritic morphology. Moreover, the effects of CaMKIIα-E183V expression on neuronal morphology may be cell-type specific.
Expression of CaMKIIα-E183V in cultured neurons also reduced mEPSC amplitude and frequency compared with neurons expressing CaMKIIα-WT. Impaired basal synaptic transmission and/or synaptic plasticity have been observed with ASD-linked mutations of other synaptic proteins. For example, expression of Shank3 mutants reduces mEPSC amplitude and frequency in dissociated hippocampal neurons (Durand et al., 2012), and basal synaptic transmission is altered in Shank3 heterozygous mice (Peça et al., 2011). Mutations in the neuroligin family of postsynaptic density proteins have also been linked to ASD (Ylisaukko-oja et al., 2005), and CaMKIIα phosphorylates neuroligin-1, regulating its surface expression and excitatory synaptic transmission (Bemben et al., 2014). Overexpression of an ASD-associated neuroligin-3 mutant also reduces basal synaptic transmission in cultured neurons compared with WT neuroligin-3 (Gutierrez et al., 2009). It will be important to investigate the impact of CaMKIIα-E183V mutation on synaptic transmission and plasticity in multiple brain regions in future studies.
Biochemical analyses indicate that total CaMKIIα protein levels are reduced in Camk2aE183V/E183V and Camk2aWT/E183V mice, perhaps due to increased polyubiquitylation and turnover of CaMKIIα-E183V. Furthermore, CaMKIIα-E183V can act in a dominant manner to enhance CaMKIIα-WT degradation when the two proteins are coexpressed in heterologous cells, in addition to its dominant-negative actions to reduce autophosphorylation (see above). CaMKIIα synaptic targeting and autophosphorylation are disrupted in both Camk2aE183V/E183V and Camk2aWT/E183V mice, and CaMKIIβ autophosphorylation is reduced in Camk2aE183V/E183V mice, providing evidence for a dominant-negative impact of CaMKIIα-E183V on both isoforms. Further studies of the cellular mechanisms controlling CaMKIIα turnover are clearly warranted.
Mice overexpressing or underexpressing CaMKIIα or with mutated Camk2a alleles that disrupt CaMKIIα signaling have revealed diverse cellular and behavioral roles relevant to psychiatric disorders (Elgersma et al., 2004; Robison, 2014). Hyperactivity was observed in heterozygous CaMKIIα null mice with ∼50% lower CaMKIIα levels (Yamasaki et al., 2008) and in CaMKIIα-T286A KI mice with reduced kinase activity, but no change in expression levels (Easton et al., 2011; Shonesy et al., 2013), similar to Camk2aE183V/E183V mice. However, CaMKIIα-T286A KI mice displayed reduced anxiety in a light/dark box test and elevated plus maze (Easton et al., 2011; Gustin et al., 2011), but not in an open-field test (Easton et al., 2011), in contrast to Camk2aE183V/E183V mice. In addition, dendritic outgrowth of hippocampal neurons is reduced in heterozygous CaMKIIα null mice (Yamasaki et al., 2008). Moreover, mice with an inactivating K42R knock-in mutation in CaMKIIα have normal CaMKIIα expression and synaptic targeting (Yamagata et al., 2009). In contrast, neuronal expression of CaMKIIα-E183V disrupts synaptic targeting and enhances dendritic arborization while reducing spine density (Fig. 5). Total levels of CaMKIIα were reduced in Camk2aE183V/E183V and Camk2aWT/E183V mice, with decreased relative synaptic targeting Camk2aE183V/E183V mice. Thus, our initial studies indicate that the E183V mutation has relatively unique effects compared with other CaMKIIα mutations. However, elucidating relationships between the molecular, cellular, and behavioral consequences of E183V mutation will require further investigations.
Some studies indicate that CaMKII has indirect roles in developmental disorders with symptoms that overlap those of ASD. For example, the Fragile X mental retardation protein directly regulates CaMKIIα mRNA translation (Darnell et al., 2011). Moreover, CaMKII hyper-phosphorylates Homer proteins in a Fragile X mouse model, and reducing CaMKIIα levels or activity rescues behavioral phenotypes in these mice (Guo et al., 2015). Similarly, CaMKII is hyper-autophosphorylated in an Angelman syndrome mouse model, and preventing CaMKIIα Thr306 phosphorylation rescues multiple behavioral phenotypes of these mice (van Woerden et al., 2007). However, the current findings provide strong support for a more direct role for CaMKIIα in ASD-related synaptic and behavioral phenotypes.
In conclusion, the E183V mutation of CaMKIIα profoundly affects multiple biochemical activities, resulting in dysregulation of dendritic morphology and synaptic transmission, and ultimately causes robust ASD-like behavioral phenotypes in mice. Thus, our studies identified a novel role for CaMKII in a synaptic signaling protein network that modulates ASD-related behaviors. Ultimately, it will be important to integrate this novel perspective with other information about this network to develop a more comprehensive understanding of the molecular disruptions that cause ASD at the synaptic and circuit levels.
Footnotes
This work was supported by the National Institutes of Health Grants T32-MH065215 to J.R.S., K01-MH107765 to B.C.S., F31-MH109196 to C.R.M., and R01-MH063232 and R01-NS078291 to R.J.C.; AHA Grants 14PRE18420020 to X.W. and 15PRE25110020 to C.R.M.; and Vanderbilt Kennedy Center Intramural Hobbs Discovery Grant. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Confocal Imaging and Imaris analyses were performed in the Vanderbilt University School of Medicine Cell Imaging Shared Resource (supported by National Institutes of Health Grants CA68485, DK20593, DK58404, DK59637 and EY08126). CRISPR/Cas9 pronuclear injections were performed by the Vanderbilt University School of Medicine Transgenic Mouse/ES Cell Shared Resource (supported through the Cancer Center Support Grant CA68485, the Vanderbilt Diabetes Research and Training Center DK020593, and the Center for Stem Cell Biology). Behavioral studies were performed in the Murine Neurobehavior Core laboratory at Vanderbilt University (supported through the Eunice Kennedy Shriver Intellectual and Developmental Disabilities Research Center 1U54HD083211-01A1). Molecular graphics were performed using the UCSF Chimera package, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by National Institute of General Medical Sciences P41-GM103311). We thank Dr. Brian Wadzinski for providing HA-ubiquitin expression constructs; Dr. Craig Garner for providing the GFP-Shank3 expression construct; Dr. P. Jeffrey Conn for providing the mGlu5 expression construct; and Dr. Robert Macdonald for providing the px330 plasmid.
The authors declare no competing financial interests.
References
- Abiria SA, Colbran RJ (2010) CaMKII associates with CaV1.2 l-type calcium channels via selected beta subunits to enhance regulatory phosphorylation. J Neurochem 112:150–161. 10.1111/j.1471-4159.2009.06436.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aman MG, Singh NN, Stewart AW, Field CJ (1985) Psychometric characteristics of the aberrant behavior checklist. Am J Ment Defic 89:492–502. [PubMed] [Google Scholar]
- Arons MH, Thynne CJ, Grabrucker AM, Li D, Schoen M, Cheyne JE, Boeckers TM, Montgomery JM, Garner CC (2012) Autism-associated mutations in ProSAP2/Shank3 impair synaptic transmission and neurexin-neuroligin-mediated transsynaptic signaling. J Neurosci 32:14966–14978. 10.1523/JNEUROSCI.2215-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balemans MC, Huibers MM, Eikelenboom NW, Kuipers AJ, van Summeren RC, Pijpers MM, Tachibana M, Shinkai Y, van Bokhoven H, Van der Zee CE (2010) Reduced exploration, increased anxiety, and altered social behavior: autistic-like features of euchromatin histone methyltransferase 1 heterozygous knockout mice. Behav Brain Res 208:47–55. 10.1016/j.bbr.2009.11.008 [DOI] [PubMed] [Google Scholar]
- Barria A, Derkach V, Soderling T (1997a) Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-type glutamate receptor. J Biol Chem 272:32727–32730. 10.1074/jbc.272.52.32727 [DOI] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR (1997b) Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science 276:2042–2045. 10.1126/science.276.5321.2042 [DOI] [PubMed] [Google Scholar]
- Baucum AJ 2nd, Strack S, Colbran RJ (2012) Age-dependent targeting of protein phosphatase 1 to Ca2+/calmodulin-dependent protein kinase II by spinophilin in mouse striatum. PLoS One 7:e31554. 10.1371/journal.pone.0031554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baucum AJ 2nd, Brown AM, Colbran RJ (2013) Differential association of postsynaptic signaling protein complexes in striatum and hippocampus. J Neurochem 124:490–501. 10.1111/jnc.12101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baucum AJ 2nd, Shonesy BC, Rose KL, Colbran RJ (2015) Quantitative proteomics analysis of CaMKII phosphorylation and the CaMKII interactome in the mouse forebrain. ACS Chem Neurosci 6:615–631. 10.1021/cn500337u [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, LeBel E, McDonald GL, O'Leary H, Schulman H, De Koninck P (2006) Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J Neurosci 26:1164–1174. 10.1523/JNEUROSCI.3116-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemben MA, Shipman SL, Hirai T, Herring BE, Li Y, Badger JD 2nd, Nicoll RA, Diamond JS, Roche KW (2014) CaMKII phosphorylation of neuroligin-1 regulates excitatory synapses. Nat Neurosci 17:56–64. 10.1038/nn.3601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkel S, Marshall CR, Weiss B, Howe J, Roeth R, Moog U, Endris V, Roberts W, Szatmari P, Pinto D, Bonin M, Riess A, Engels H, Sprengel R, Scherer SW, Rappold GA (2010) Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat Genet 42:489–491. 10.1038/ng.589 [DOI] [PubMed] [Google Scholar]
- Bingol B, Wang CF, Arnott D, Cheng D, Peng J, Sheng M (2010) Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell 140:567–578. 10.1016/j.cell.2010.01.024 [DOI] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW (1996) CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell 87:1203–1214. 10.1016/S0092-8674(00)81816-4 [DOI] [PubMed] [Google Scholar]
- Bourgeron T. (2015) From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci 16:551–563. 10.1038/nrn3992 [DOI] [PubMed] [Google Scholar]
- Chao LH, Stratton MM, Lee IH, Rosenberg OS, Levitz J, Mandell DJ, Kortemme T, Groves JT, Schulman H, Kuriyan J (2011) A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin-dependent kinase II holoenzyme. Cell 146:732–745. 10.1016/j.cell.2011.07.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Rainnie DG, Greene RW, Tonegawa S (1994) Abnormal fear response and aggressive behavior in mutant mice deficient for alpha-calcium-calmodulin kinase II. Science 266:291–294. 10.1126/science.7939668 [DOI] [PubMed] [Google Scholar]
- Constantino JN, Davis SA, Todd RD, Schindler MK, Gross MM, Brophy SL, Metzger LM, Shoushtari CS, Splinter R, Reich W (2003) Validation of a brief quantitative measure of autistic traits: comparison of the social responsiveness scale with the autism diagnostic interview-revised. J Autism Dev Disord 33:427–433. 10.1023/A:1025014929212 [DOI] [PubMed] [Google Scholar]
- Coultrap SJ, Buard I, Kulbe JR, Dell'Acqua ML, Bayer KU (2010) CaMKII autonomy is substrate-dependent and further stimulated by Ca2+/calmodulin. J Biol Chem 285:17930–17937. 10.1074/jbc.M109.069351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Freund RK, O'Leary H, Sanderson JL, Roche KW, Dell'Acqua ML, Bayer KU (2014) Autonomous CaMKII mediates both LTP and LTD using a mechanism for differential substrate site selection. Cell Rep 6:431–437. 10.1016/j.celrep.2014.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB (2011) FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146:247–261. 10.1016/j.cell.2011.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLorey TM, Sahbaie P, Hashemi E, Li WW, Salehi A, Clark DJ (2011) Somatosensory and sensorimotor consequences associated with the heterozygous disruption of the autism candidate gene, Gabrb3. Behav Brain Res 216:36–45. 10.1016/j.bbr.2010.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsäter H, Sponheim E, Goubran-Botros H, Delorme R, Chabane N, Mouren-Simeoni MC, de Mas P, Bieth E, Rogé B, Héron D, Burglen L, et al. (2007) Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet 39:25–27. 10.1038/ng1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Perroy J, Loll F, Perrais D, Fagni L, Bourgeron T, Montcouquiol M, Sans N (2012) SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol Psychiatry 17:71–84. 10.1038/mp.2011.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton AC, Lucchesi W, Schumann G, Giese KP, Müller CP, Fernandes C (2011) αCaMKII autophosphorylation controls exploratory activity to threatening novel stimuli. Neuropharmacology 61:1424–1431. 10.1016/j.neuropharm.2011.08.036 [DOI] [PubMed] [Google Scholar]
- Easton AC, Lucchesi W, Lourdusamy A, Lenz B, Solati J, Golub Y, Lewczuk P, Fernandes C, Desrivieres S, Dawirs RR, Moll GH, Kornhuber J, Frank J, Hoffmann P, Soyka M, Kiefer F, Kiefer F, Schumann G, Peter Giese K, Müller CP, et al. (2013) αCaMKII autophosphorylation controls the establishment of alcohol drinking behavior. Neuropsychopharmacology 38:1636–1647. 10.1038/npp.2013.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgersma Y, Fedorov NB, Ikonen S, Choi ES, Elgersma M, Carvalho OM, Giese KP, Silva AJ (2002) Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity, and learning. Neuron 36:493–505. 10.1016/S0896-6273(02)01007-3 [DOI] [PubMed] [Google Scholar]
- Elgersma Y, Sweatt JD, Giese KP (2004) Mouse genetic approaches to investigating calcium/calmodulin-dependent protein kinase II function in plasticity and cognition. J Neurosci 24:8410–8415. 10.1523/JNEUROSCI.3622-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyder M, Karlsson RM, Mathur P, Lyman M, Bock R, Momenan R, Munasinghe J, Scattoni ML, Ihne J, Camp M, Graybeal C, Strathdee D, Begg A, Alvarez VA, Kirsch P, Rietschel M, Cichon S, Walter H, Meyer-Lindenberg A, Grant SG, et al. (2010) Association of mouse Dlg4 (PSD-95) gene deletion and human DLG4 gene variation with phenotypes relevant to autism spectrum disorders and Williams' syndrome. Am J Psychiatry 167:1508–1517. 10.1176/appi.ajp.2010.10040484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink CC, Bayer KU, Myers JW, Ferrell JE Jr, Schulman H, Meyer T (2003) Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron 39:283–297. 10.1016/S0896-6273(03)00428-8 [DOI] [PubMed] [Google Scholar]
- Fischbach GD, Lord C (2010) The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron 68:192–195. 10.1016/j.neuron.2010.10.006 [DOI] [PubMed] [Google Scholar]
- Ghiretti AE, Kenny K, Marr MT 2nd, Paradis S (2013) CaMKII-dependent phosphorylation of the GTPase Rem2 is required to restrict dendritic complexity. J Neurosci 33:6504–6515. 10.1523/JNEUROSCI.3861-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, Silva AJ (1998) Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 279:870–873. 10.1126/science.279.5352.870 [DOI] [PubMed] [Google Scholar]
- Guo W, Ceolin L, Collins KA, Perroy J, Huber KM (2015) Elevated CaMKIIα and hyperphosphorylation of Homer mediate circuit dysfunction in a fragile X syndrome mouse model. Cell Rep 13:2297–2311. 10.1016/j.celrep.2015.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin RM, Bichell TJ, Bubser M, Daily J, Filonova I, Mrelashvili D, Deutch AY, Colbran RJ, Weeber EJ, Haas KF (2010) Tissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndrome. Neurobiol Dis 39:283–291. 10.1016/j.nbd.2010.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin RM, Shonesy BC, Robinson SL, Rentz TJ, Baucum AJ 2nd, Jalan-Sakrikar N, Winder DG, Stanwood GD, Colbran RJ (2011) Loss of Thr286 phosphorylation disrupts synaptic CaMKIIalpha targeting, NMDAR activity and behavior in pre-adolescent mice. Mol Cell Neurosci 47:286–292. 10.1016/j.mcn.2011.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez RC, Hung J, Zhang Y, Kertesz AC, Espina FJ, Colicos MA (2009) Altered synchrony and connectivity in neuronal networks expressing an autism-related mutation of neuroligin 3. Neuroscience 162:208–221. 10.1016/j.neuroscience.2009.04.062 [DOI] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, Hell JW (2012) CaMKII binding to GluN2B is critical during memory consolidation. EMBO J 31:1203–1216. 10.1038/emboj.2011.482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW. (2014) CaMKII: claiming center stage in postsynaptic function and organization. Neuron 81:249–265. 10.1016/j.neuron.2013.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS (2005) CaMKII tethers to l-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol 171:537–547. 10.1083/jcb.200505155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, Smith JD, Paeper B, Nickerson DA, Dea J, Dong S, Gonzalez LE, Mandell JD, Mane SM, Murtha MT, Sullivan CA, et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature 515:216–221. 10.1038/nature13908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalan-Sakrikar N, Bartlett RK, Baucum AJ 2nd, Colbran RJ (2012) Substrate-selective and calcium-independent activation of CaMKII by alpha-actinin. J Biol Chem 287:15275–15283. 10.1074/jbc.M112.351817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MA, Christel CJ, Jiao Y, Abiria S, Kim KY, Usachev YM, Obermair GJ, Colbran RJ, Lee A (2010) Ca2+-dependent facilitation of Cav1.3 Ca2+ channels by densin and Ca2+/calmodulin-dependent protein kinase II. J Neurosci 30:5125–5135. 10.1523/JNEUROSCI.4367-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH, Ehlers MD (2013) Modeling autism by SHANK gene mutations in mice. Neuron 78:8–27. 10.1016/j.neuron.2013.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Robison AJ, Bass MA, Colbran RJ (2008) Developmentally regulated alternative splicing of densin modulates protein-protein interaction and subcellular localization. J Neurochem 105:1746–1760. 10.1111/j.1471-4159.2008.05280.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Jalan-Sakrikar N, Robison AJ, Baucum AJ 2nd, Bass MA, Colbran RJ (2011) Characterization of a central Ca2+/calmodulin-dependent protein kinase IIalpha/beta binding domain in densin that selectively modulates glutamate receptor subunit phosphorylation. J Biol Chem 286:24806–24818. 10.1074/jbc.M110.216010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Lakhanpal G, Lu HE, Khan M, Suzuki A, Hayashi MK, Narayanan R, Luyben TT, Matsuda T, Nagai T, Blanpied TA, Hayashi Y, Okamoto K (2015) A temporary gating of actin remodeling during synaptic plasticity consists of the interplay between the kinase and structural functions of CaMKII. Neuron 87:813–826. 10.1016/j.neuron.2015.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug JR, Mathur BN, Kash TL, Wang HD, Matthews RT, Robison AJ, Anderson ME, Deutch AY, Lovinger DM, Colbran RJ, Winder DG (2012) Genetic inhibition of CaMKII in dorsal striatal medium spiny neurons reduces functional excitatory synapses and enhances intrinsic excitability. PLoS One 7:e45323. 10.1371/journal.pone.0045323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm N, O'Roak BJ, Shendure J, Eichler EE (2014) A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci 37:95–105. 10.1016/j.tins.2013.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TS, Karl R, Moosmang S, Lenhardt P, Klugbauer N, Hofmann F, Kleppisch T, Welling A (2006) Calmodulin kinase II is involved in voltage-dependent facilitation of the l-type Cav1.2 calcium channel: identification of the phosphorylation sites. J Biol Chem 281:25560–25567. 10.1074/jbc.M508661200 [DOI] [PubMed] [Google Scholar]
- Leonard AS, Lim IA, Hemsworth DE, Horne MC, Hell JW (1999) Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-d-aspartate receptor. Proc Natl Acad Sci U S A 96:3239–3244. 10.1073/pnas.96.6.3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AS, Bayer KU, Merrill MA, Lim IA, Shea MA, Schulman H, Hell JW (2002) Regulation of calcium/calmodulin-dependent protein kinase II docking to N-methyl-d-aspartate receptors by calcium/calmodulin and alpha-actinin. J Biol Chem 277:48441–48448. 10.1074/jbc.M205164200 [DOI] [PubMed] [Google Scholar]
- Lisman J, Yasuda R, Raghavachari S (2012) Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci 13:169–182. 10.1038/nrn3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C, Rutter M, Le Couteur A (1994) Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord 24:659–685. 10.1007/BF02172145 [DOI] [PubMed] [Google Scholar]
- Lord C, Cook EH, Leventhal BL, Amaral DG (2000a) Autism spectrum disorders. Neuron 28:355–363. 10.1016/S0896-6273(00)00115-X [DOI] [PubMed] [Google Scholar]
- Lord C, Risi S, Lambrecht L, Cook EH Jr, Leventhal BL, DiLavore PC, Pickles A, Rutter M (2000b) The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord 30:205–223. 10.1023/A:1005592401947 [DOI] [PubMed] [Google Scholar]
- Loweth JA, Singer BF, Baker LK, Wilke G, Inamine H, Bubula N, Alexander JK, Carlezon WA Jr, Neve RL, Vezina P (2010) Transient overexpression of alpha-Ca2+/calmodulin-dependent protein kinase II in the nucleus accumbens shell enhances behavioral responding to amphetamine. J Neurosci 30:939–949. 10.1523/JNEUROSCI.4383-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen AL, Kameyama K, Roche KW, Huganir RL (1997) Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J Biol Chem 272:32528–32533. 10.1074/jbc.272.51.32528 [DOI] [PubMed] [Google Scholar]
- McNeill RB, Colbran RJ (1995) Interaction of autophosphorylated Ca2+/calmodulin-dependent protein kinase II with neuronal cytoskeletal proteins: characterization of binding to a 190-kDa postsynaptic density protein. J Biol Chem 270:10043–10049. 10.1074/jbc.270.17.10043 [DOI] [PubMed] [Google Scholar]
- Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, Zwaigenbaum L, Fernandez B, Roberts W, Szatmari P, Scherer SW (2007) Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet 81:1289–1297. 10.1086/522590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy SS, Riddick NV, Nikolova VD, Teng BL, Agster KL, Nonneman RJ, Young NB, Baker LK, Nadler JJ, Bodfish JW (2014) Repetitive behavior profile and supersensitivity to amphetamine in the C58/J mouse model of autism. Behav Brain Res 259:200–214. 10.1016/j.bbr.2013.10.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Futai K, Lashuel HA, Lo I, Okamoto K, Walz T, Hayashi Y, Sheng M (2004) Quaternary structure, protein dynamics, and synaptic function of SAP97 controlled by L27 domain interactions. Neuron 44:453–467. 10.1016/j.neuron.2004.10.012 [DOI] [PubMed] [Google Scholar]
- Omkumar RV, Kiely MJ, Rosenstein AJ, Min KT, Kennedy MB (1996) Identification of a phosphorylation site for calcium/calmodulin dependent protein kinase II in the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem 271:31670–31678. 10.1074/jbc.271.49.31670 [DOI] [PubMed] [Google Scholar]
- O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, et al. (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485:246–250. 10.1038/nature10989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G (2011) Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472:437–442. 10.1038/nature09965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM (2011) Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 14:285–293. 10.1038/nn.2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- Pi HJ, Otmakhov N, El Gaamouch F, Lemelin D, De Koninck P, Lisman J (2010) CaMKII control of spine size and synaptic strength: role of phosphorylation states and nonenzymatic action. Proc Natl Acad Sci U S A 107:14437–14442. 10.1073/pnas.1009268107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bölte S, Bolton PF, et al. (2010) Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466:368–372. 10.1038/nature09146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison AJ. (2014) Emerging role of CaMKII in neuropsychiatric disease. Trends Neurosci 37:653–662. 10.1016/j.tins.2014.07.001 [DOI] [PubMed] [Google Scholar]
- Robison AJ, Bartlett RK, Bass MA, Colbran RJ (2005a) Differential modulation of Ca2+/calmodulin-dependent protein kinase II activity by regulated interactions with N-methyl-d-aspartate receptor NR2B subunits and alpha-actinin. J Biol Chem 280:39316–39323. 10.1074/jbc.M508189200 [DOI] [PubMed] [Google Scholar]
- Robison AJ, Bass MA, Jiao Y, MacMillan LB, Carmody LC, Bartlett RK, Colbran RJ (2005b) Multivalent interactions of calcium/calmodulin-dependent protein kinase II with the postsynaptic density proteins NR2B, densin-180, and alpha-actinin-2. J Biol Chem 280:35329–35336. 10.1074/jbc.M502191200 [DOI] [PubMed] [Google Scholar]
- Robison AJ, Vialou V, Mazei-Robison M, Feng J, Kourrich S, Collins M, Wee S, Koob G, Turecki G, Neve R, Thomas M, Nestler EJ (2013) Behavioral and structural responses to chronic cocaine require a feedforward loop involving DeltaFosB and calcium/calmodulin-dependent protein kinase II in the nucleus accumbens shell. J Neurosci 33:4295–4307. 10.1523/JNEUROSCI.5192-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronemus M, Iossifov I, Levy D, Wigler M (2014) The role of de novo mutations in the genetics of autism spectrum disorders. Nat Rev Genet 15:133–141. 10.1038/nrg3585 [DOI] [PubMed] [Google Scholar]
- Sala C, Futai K, Yamamoto K, Worley PF, Hayashi Y, Sheng M (2003) Inhibition of dendritic spine morphogenesis and synaptic transmission by activity-inducible protein Homer1a. J Neurosci 23:6327–6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, Goldberg AP, Jinlu C, Keaney JF 3rd, Klei L, Mandell JD, Moreno-De-Luca D, Poultney CS, Robinson EB, Smith L, Solli-Nowlan T, et al. (2015) Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87:1215–1233. 10.1016/j.neuron.2015.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato D, Lionel AC, Leblond CS, Prasad A, Pinto D, Walker S, O'Connor I, Russell C, Drmic IE, Hamdan FF, Michaud JL, Endris V, Roeth R, Delorme R, Huguet G, Leboyer M, Rastam M, Gillberg C, Lathrop M, Stavropoulos DJ, et al. (2012) SHANK1 deletions in males with autism spectrum disorder. Am J Hum Genet 90:879–887. 10.1016/j.ajhg.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks NF, Maruo T, Farina AN, Ellisman MH, Nakagawa T (2010) Contribution of the global subunit structure and stargazin on the maturation of AMPA receptors. J Neurosci 30:2728–2740. 10.1523/JNEUROSCI.5146-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Meyer T (1999) Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science 284:162–166. 10.1126/science.284.5411.162 [DOI] [PubMed] [Google Scholar]
- Shonesy BC, Wang X, Rose KL, Ramikie TS, Cavener VS, Rentz T, Baucum AJ 2nd, Jalan-Sakrikar N, Mackie K, Winder DG, Patel S, Colbran RJ (2013) CaMKII regulates diacylglycerol lipase-alpha and striatal endocannabinoid signaling. Nat Neurosci 16:456–463. 10.1038/nn.3353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shonesy BC, Jalan-Sakrikar N, Cavener VS, Colbran RJ (2014) CaMKII: a molecular substrate for synaptic plasticity and memory. Prog Mol Biol Transl Sci 122:61–87. 10.1016/B978-0-12-420170-5.00003-9 [DOI] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S (1992) Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science 257:206–211. 10.1126/science.1321493 [DOI] [PubMed] [Google Scholar]
- Silverman JL, Yang M, Lord C, Crawley JN (2010) Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci 11:490–502. 10.1038/nrn2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- State MW, Šestan N (2012) Neuroscience: the emerging biology of autism spectrum disorders. Science 337:1301–1303. 10.1126/science.1224989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S, Colbran RJ (1998) Autophosphorylation-dependent targeting of calcium/calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem 273:20689–20692. 10.1074/jbc.273.33.20689 [DOI] [PubMed] [Google Scholar]
- Strack S, McNeill RB, Colbran RJ (2000) Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem 275:23798–23806. 10.1074/jbc.M001471200 [DOI] [PubMed] [Google Scholar]
- Swanger SA, Yao X, Gross C, Bassell GJ (2011) Automated 4D analysis of dendritic spine morphology: applications to stimulus-induced spine remodeling and pharmacological rescue in a disease model. Mol Brain 4:38. 10.1186/1756-6606-4-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavalin SJ, Colbran RJ (2016) CaMKII-mediated phosphorylation of GluN2B regulates recombinant NMDA receptor currents in a chloride-dependent manner. Mol Cell Neurosci 79:45–52. 10.1016/j.mcn.2016.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Woerden GM, Harris KD, Hojjati MR, Gustin RM, Qiu S, de Avila Freire R, Jiang YH, Elgersma Y, Weeber EJ (2007) Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci 10:280–282. 10.1038/nn1845 [DOI] [PubMed] [Google Scholar]
- Watkins GR, Wang N, Mazalouskas MD, Gomez RJ, Guthrie CR, Kraemer BC, Schweiger S, Spiller BW, Wadzinski BE (2012) Monoubiquitination promotes calpain cleavage of the protein phosphatase 2A (PP2A) regulatory subunit alpha4, altering PP2A stability and microtubule-associated protein phosphorylation. J Biol Chem 287:24207–24215. 10.1074/jbc.M112.368613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsby PJ, Wang H, Wolfe JT, Colbran RJ, Johnson ML, Barrett PQ (2003) A mechanism for the direct regulation of T-type calcium channels by Ca2+/calmodulin-dependent kinase II. J Neurosci 23:10116–10121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff JJ, Bodfish JW, Hazlett HC, Lightbody AA, Reiss AL, Piven J (2012) Evidence of a distinct behavioral phenotype in young boys with fragile X syndrome and autism. J Am Acad Child Adolesc Psychiatry 51:1324–1332. 10.1016/j.jaac.2012.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GY, Cline HT (1998) Stabilization of dendritic arbor structure in vivo by CaMKII. Science 279:222–226. 10.1126/science.279.5348.222 [DOI] [PubMed] [Google Scholar]
- Xie Z, Srivastava DP, Photowala H, Kai L, Cahill ME, Woolfrey KM, Shum CY, Surmeier DJ, Penzes P (2007) Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron 56:640–656. 10.1016/j.neuron.2007.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata Y, Kobayashi S, Umeda T, Inoue A, Sakagami H, Fukaya M, Watanabe M, Hatanaka N, Totsuka M, Yagi T, Obata K, Imoto K, Yanagawa Y, Manabe T, Okabe S (2009) Kinase-dead knock-in mouse reveals an essential role of kinase activity of Ca2+/calmodulin-dependent protein kinase II alpha in dendritic spine enlargement, long-term potentiation, and learning. J Neurosci 29:7607–7618. 10.1523/JNEUROSCI.0707-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki N, Maekawa M, Kobayashi K, Kajii Y, Maeda J, Soma M, Takao K, Tanda K, Ohira K, Toyama K, Kanzaki K, Fukunaga K, Sudo Y, Ichinose H, Ikeda M, Iwata N, Ozaki N, Suzuki H, Higuchi M, Suhara T, et al. (2008) Alpha-CaMKII deficiency causes immature dentate gyrus, a novel candidate endophenotype of psychiatric disorders. Mol Brain 1:6. 10.1186/1756-6606-1-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Silverman JL, Crawley JN (2011) Automated three-chambered social approach task for mice. Curr Protoc Neurosci 8:26. 10.1002/0471142301.ns0826s56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda R, Sabatini BL, Svoboda K (2003) Plasticity of calcium channels in dendritic spines. Nat Neurosci 6:948–955. 10.1038/nn1112 [DOI] [PubMed] [Google Scholar]
- Ylisaukko-oja T, Rehnström K, Auranen M, Vanhala R, Alen R, Kempas E, Ellonen P, Turunen JA, Makkonen I, Riikonen R, Nieminen-von Wendt T, von Wendt L, Peltonen L, Järvelä I (2005) Analysis of four neuroligin genes as candidates for autism. Eur J Hum Genet 13:1285–1292. 10.1038/sj.ejhg.5201474 [DOI] [PubMed] [Google Scholar]
- Zhang YP, Holbro N, Oertner TG (2008) Optical induction of plasticity at single synapses reveals input-specific accumulation of alphaCaMKII. Proc Natl Acad Sci U S A 105:12039–12044. 10.1073/pnas.0802940105 [DOI] [PMC free article] [PubMed] [Google Scholar]