

Abstract
Background
The mechanism underlying nonsevere and severe asthma remains unclear, although it is commonly associated with increased airway smooth muscle (ASM) mass. Long noncoding RNAs (lncRNAs) are known to be important in regulating healthy primary airway smooth muscle cells (ASMCs), whereas changed expression has been observed in CD8 T cells from patients with severe asthma.
Methods
Primary ASMCs were isolated from healthy subjects (n = 9) and patients classified as having nonsevere (n = 9) or severe (n = 9) asthma. ASMCs were exposed to dexamethasone and FCS. mRNA and lncRNA expression was measured by using a microarray and quantitative real-time PCR. Bioinformatic analysis was used to examine relevant biological pathways. Finally, the lncRNA plasmacytoma variant translocation 1 (PVT1) was inhibited by transfection of primary ASMCs with small interfering RNAs, and the effect on ASMC phenotype was examined.
Results
The mRNA expression profile was significantly different between patient groups after exposure to dexamethasone and FCS, and these were associated with biological pathways that might be relevant to the pathogenesis of asthma, including cellular proliferation and pathways associated with glucocorticoid activity. We also observed a significant change in lncRNA expression, yet the expression of only one lncRNA (PVT1) is decreased in patients with corticosteroid-sensitive nonsevere asthma and increased in patients with corticosteroid-insensitive severe asthma. Subsequent targeting studies demonstrated the importance of this lncRNA in controlling both proliferation and IL-6 release in ASMCs from patients with severe asthma.
Conclusions
lncRNAs are associated with the aberrant phenotype observed in ASMCs from asthmatic patients. Targeting PVT1 might be effective in reducing airway remodeling in asthmatic patients.
Key words: Asthma, airway smooth muscle, proliferation, IL-6, transcriptome, long noncoding RNA, PVT1
Abbreviations used: ASM, Airway smooth muscle; ASMC, Airway smooth muscle cell; BrdU, Bromodeoxyuridine; cRNA, Coding RNA; lncRNA, Long noncoding RNA; miRNA, MicroRNA; ncRNA, Noncoding RNA; PVT1, Plasmacytoma variant translocation; siRNA, Small interfering RNA
Asthma is characterized by airflow obstruction and chronic airway inflammation and remodeling.1 Airway smooth muscle (ASM) hyperplasia and hypertrophy lead to increased airway wall thickening and airway narrowing, contributing to airway obstruction and inflammation. Epigenetic mechanisms are key regulators of ASM function.2 We have reported that the aberrant phenotype of airway smooth muscle cells (ASMCs) from asthmatic patients is under the negative regulation of the cyclin inhibitors p21WAF1 and p27kip1, which are controlled by microRNA-221.3 Furthermore, using a transcriptomics-based approach, we identified potential microRNA (miRNA) targets and pathways in activated human ASMCs and observed changes in expression of long noncoding RNAs (lncRNAs), including natural antisense, pseudogenes, intronic lncRNAs, and intergenic lncRNAs.4
Having reported results with healthy (or nonasthmatic) ASMCs in 2014,4 we now report on the asthmatic ASMCs that were studied at the same time. Specifically, we report on the differential expression of mRNAs and lncRNAs in ASMCs isolated from patients with nonsevere and severe asthma and treated with dexamethasone before subsequent activation with the growth medium FCS. We have then focused on the only lncRNA found to be decreased in expression in patients with corticosteroid-sensitive nonsevere asthma and increased in expression in patients with corticosteroid-insensitive severe asthmatics, plasmacytoma variant translocation 1 (PVT1), and performed targeting studies to examine its role in a corticosteroid insensitivity model in these cells induced by the combination of TGF-β and FCS.
Methods
Full methodology is available in the Methods section in this article's Online Repository at www.jacionline.org.
Subject selection and ASMC culture
Primary ASMCs from healthy subjects, patients with nonsevere asthma, and patients with severe asthma were treated, as described previously,4, 5, 6, 7 and proliferation rates, cellular viability, and IL-6 release (see Fig E1 in this article's Online Repository at www.jacionline.org), were all comparable to our previous studies.1, 2, 3 Patients' characteristics are shown in Table I.
Table I.
Characteristics of subjects
| Nonasthmatic patients | Patients with nonsevere asthma | Patients with severe asthma | |
|---|---|---|---|
| No. | 9 | 9 | 9 |
| Age (y) | 36.4 ± 12.7 | 42.4 ± 16.2 | 48.9 ± 11 |
| Sex (male/female) | 7/2 | 6/4 | 3/6 |
| Asthma duration (y) | NA | 22.2 ± 16.8 | 25.6 ± 13.2 |
| Inhaled corticosteroid dose (μg of BDP equivalent) | NA | 580 ± 576.9 | 1688.9 ± 176.4 |
| Atopy (no.)∗ | 0 | 8 | 8 |
| Receiving oral corticosteroids (no.) | 0 | 0 | 7 |
| FEV1 (L) | 4.02 ± 0.48 | 2.81 ± 0.71 | 2.7 ± 0.82 |
| FEV1 (% predicted) | 104.23 ± 7.28 | 84.68 ± 12.31 | 80.48 ± 12.34 |
| FEV1/FVC ratio (%) | 78.79 ± 5.98 | 69.87 ± 9.27 | 63.98 ± 9.68 |
| β-Agonist reversibility (%)† | NA | 12.3 ± 11.9 | 19.54 ± 14.56 |
| PC20 (mg/mL) | >16 | 0.69 ± 0.64 | 0.2 ± 0.39 |
Data are shown as means ± SEMs.
BDP, Beclomethasone dipropionate; FVC, forced vital capacity; NA, not available.
Defined as positive skin prick test responses to 1 or more common aeroallergens.
Measured as percentage increase in FEV1 after 400 μg of salbutamol.
RNA extraction
RNA was extracted with the mirVana miRNA isolation kit, as previously described.3, 4, 5, 6, 7
Microarray analysis
lncRNA and mRNA expression was determined by using Agilent SurePrint G3 Human GE microarrays (Agilent Technologies, Santa Clara, Calif), according to the manufacturer's instructions, and as previously described.4 Total RNA samples (50 ng) used in lncRNA and mRNA microarrays were initially labeled with Spike-In control A (for cyanine 3–cytidine triphosphate) or B (for cyanine 5–CTP). Labeled samples were then used for cDNA synthesis by using the cDNA Master Mix (Agilent Technologies) and incubated for 2 hours at 40°C, followed by 15 minutes at 70°C to inactivate the Affinity-Script enzyme. The synthesized cDNA was then used for coding RNA (cRNA) synthesis and amplification by using the Transcription Master Mix with either cyanine 3 or cyanine 5 and incubated at 40°C for 2 hours. Labeled and amplified RNA was then purified with the RNeasy Mini Kit (Qiagen, Hilden, Germany) and quantified with the NanoDrop Spectrophotometer's Microarray Measurement function (NanoDrop, Wilmington, Del). The cyanine 3 or cyanine 5 concentrations and the cRNA concentration were used to calculate the yield (in micrograms) and specific activity (in picomoles of cyanine 3 or cyanine 5 per microgram of cRNA) of each sample. For each microarray reaction, 300 ng of cyanine 3–labeled and 300 ng of cyanine 5–labeled linearly amplified cRNA samples were mixed and incubated together with fragmentation buffer for 30 minutes, followed by addition of hybridization buffer. Samples were loaded onto SurePrint G3 (8 × 60 K) microarray slides (Agilent Technologies) and hybridized at 65°C for 17 hours at 10 rpm by using Agilent's Hybridization oven and SureHyb chamber. The microarrays were then disassembled and washed in GE Wash Buffer 1 (Agilent Technologies) for 60 seconds at room temperature, followed by GE Wash Buffer 2 for 60 seconds at 37°C, followed by an acetonitrile wash (10 seconds at room temperature) and a final wash in stabilization and drying solution for 30 seconds at room temperature, to improve microarray results by preventing ozone-mediated fluorescent signal degradation. The microarrays were scanned with the Agilent Microarray Scanner G2565BA by using the profile for 2-color microarrays (AgilentG3_GX_2Color) at 5-μm resolution (dye channel, Red&Green; scan area, 61 × 21.6 mm).
After normalization against internal controls provided within the labelling kits, probes with background expression (signal value of mRNA < 4.5) were removed. The threshold of background expression was determined by using samples that were processed but contained no RNA. Because initial analysis of mRNA and lncRNA between baseline and FCS-treated cells or dexamethasone plus FCS–treated cells showed none that produced a false discovery rate of less than 0.1, differential expression (P value) was determined by using 3-way ANOVA with the Partek Genomics Suite. We report changes in expression at a P value of less than .05.
Pathway analysis
Differentially expressed mRNAs from each data set were further analyzed by using the bioinformatics software Ingenuity Pathway Analysis application (www.ingenuity.com). A core functional analysis was performed to identify canonical pathways, predicted upstream regulators, and gene networks most associated significantly with the differentially expressed mRNAs.
The significance of the association of a given canonical pathway with the differentially expressed mRNAs was measured in 2 ways: (1) based on the ratio of the number of differentially expressed mRNAs in the data set that mapped to the canonical pathway divided by the total number of genes that map to the canonical pathway and (2) by using the Fisher exact test to calculate a P value for the association between the mRNA and network/canonical pathway.
Quantitative PCR measurement of miRNA and mRNA expression
lncRNA and mRNA expression was measured, as previously described.4
Transfection with small interfering RNAs that target PVT1
ASMCs were transfected, as previously described.3, 5, 8 Small interfering RNAs (siRNAs) designed to target PVT1 and IL6 were purchased from Ambion/Applied Biosystems, Waltham, Mass. ASMCs were transfected with PVT1 inhibitor (30, 100, or 300 nmol/L), IL6 inhibitor (100 nmol/L), or Silencer Negative Control #1 (100 nmol/L) and no siRNA (mock transfection).
Data and statistical analysis
Data were analyzed with GraphPad Prism software, version 5.03 (GraphPad Software, La Jolla, Calif). Data were not normally distributed (as assessed by using the Kolmogorov-Smirnov test), and therefore groups were compared with the Dunn nonparametric test. All data are expressed as means ± SEMs.
Results
mRNAs are differentially expressed between ASMCs from patients with nonsevere and those with severe asthma and are responsible for different pathway activation
Comparison of mRNA expression in ASMCs from healthy subjects and patients with nonsevere or severe asthma showed differential expression (P < .05) of different gene sets depending on the status of the cells being at a “baseline” state after stimulation with FCS and those pretreated with dexamethasone before stimulation with FCS. Full gene lists are shown in Tables E1-E9 in this article's Online Repository at www.jacionline.org.
To identify the pathways in which these mRNAs are involved, we analyzed each data set by using the bioinformatics software application Ingenuity Pathway Analysis. At baseline, those mRNAs that were increased in expression compared with ASMCs from healthy subjects in the nonsevere asthma cohort are proposed to be important in hydrolase activity. Those decreased in expression are involved in the extracellular matrix, and the whole profile is important for cellular assembly (Fig 1, A). Interestingly, hydrolase activity is known to be important for calcium signaling in ASM,9 and the extracellular matrix is involved in asthmatic airway remodeling.10 In the ASMCs from the patients with severe asthma, these profiles changed to an increase in genes necessary for arachidonic acid metabolism and a decrease in those required for multicellular organismal process, and the whole profile is involved in gene expression and organ morphology. Once again, this follows our current knowledge of ASM function and the fact that arachidonic acid induces calcium influx in human airway smooth muscle, which has been proposed to contribute to increased influx in asthmatic patients,11 and previous methylation profiling of the bronchial mucosa of asthmatic patients has demonstrated a positive relationship to atopy for the multicellular organismal process.12 After stimulation with FCS, those mRNAs increased in expression in the patients with nonsevere ASM are associated with the intracellular organelle lumen, those decreased are involved in single-organism development, and together the profile is important in the inflammatory response that is well documented in human ASM (Fig 1, B).13 In ASMCs from patients with severe asthma, those with increased expression are involved in respiratory tube development, and those with decreased expression are involved in signaling. However, together, the mRNAs are proposed to be important in patients with immunologic disease, which helps explain how ASM is important for immunomodulation in acute exacerbations of airway disease, including asthma.14 When these same ASM cohorts were pretreated with dexamethasone, before stimulation with FCS (Fig 1, C), those pathways activated in ASMCs from patients with nonsevere asthma were associated with glucocorticoid activity, such as macrophage activation, the inflammatory response,15 and phospholipase activity.16 However, ASMCs from patients with severe asthma responded by activating genes associated with cellular growth and proliferation, a phenotype well documented as being aberrant.3, 4, 5, 6
Fig 1.

Venn diagrams showing pathway analysis of differentially expressed mRNAs in ASM from asthmatic patients. A-C, Pathway analysis showing intergroup comparisons at baseline (Fig 1, A), after stimulation with FCS (2.5%; Fig 1, B), and after stimulation with FCS (2.5%) plus TGF-β (1 ng/mL; Fig 1, C). Data represent 9 members of each patient type. D, Expression of 9 mRNAs was confirmed by using TaqMan RT-PCR to validate the array data. Bars represent means ± SEMs from 9 primary ASMC donors. *P < .05, **P < .01, and ***P < .001.
Levels of NAV2, NFIB, PTGIS, CHI3L1, NOVA1, PTPRD, IL6, PGM5, and NNAT were selected to be verified by means of quantitative RT-PCR (Fig 1, D). For each of these, FCS caused a significant increase in expression in ASMCs from both patients with nonsevere and those with severe asthma (P < .05). Pretreatment of ASMCs from patients with nonsevere asthma with dexamethasone inhibited this increase in expression in all mRNAs, apart from NAV2. In ASMCs from patients with severe asthma, dexamethasone caused a further increase in NAV2 and PTGIS expression (P < .01); in all other instances dexamethasone only slightly inhibited the FCS-induced mRNA expression to levels that were not comparable with ASMCs from patients with nonsevere asthma.
lncRNAs are differentially expressed between ASMCs from patients with nonsevere and those with severe asthma
We have previously shown that more than 30 lncRNAs are increased in expression in healthy primary ASMCs after treatment with dexamethasone and stimulation with FCS.4 Hence to identify novel lncRNAs in ASMCs from asthmatic patients, we used ENSEMBLE (www.ensembl.org/index.html) to determine the genomic position of those probe sets from the microarray that did not match known protein-coding genes.
At baseline, 21 lncRNAs were differentially expressed in ASMCs from patients with nonsevere asthma (15 increased and 6 decreased) when compared with healthy subjects (see Table E10 in this article's Online Repository at www.jacionline.org). ASMCs from patients with severe asthma differentially expressed 19 lncRNAs (13 increased and 6 decreased) when compared with healthy subjects (see Table E11 in this article's Online Repository at www.jacionline.org).4 Interestingly, 4 lncRNAs were altered in expression in both disease phenotypes (RP5-1158E12.3, FKBP1A-SDCBP2, LINC00472, and PVT1), and PVT1 was also differentially expressed in ASMCs from healthy subjects.4 After stimulation with FCS, ASMCs from patients with nonsevere asthma expressed a completely different set of lncRNAs (15 increased and 16 decreased), with the exception of PVT1 and RP11-141M1 (see Table E12 in this article's Online Repository at www.jacionline.org), and treatment of the same cells with dexamethasone, before said stimulation resulted in an increase in expression of 60 lncRNAs, and a decrease in 19 (see Table E13 in this article's Online Repository at www.jacionline.org). A similar pattern was observed in FCS-stimulated ASMCs from patients with severe asthma; of the 32 lncRNAs that changed in expression, only 2 (LINC00940 and RP11-120D5.1) were the same as seen in the baseline ASMCs from patients with nonsevere asthma (see Table E14 in this article's Online Repository at www.jacionline.org). Furthermore, when ASMCs from patients with severe asthma were treated with dexamethasone before subsequent stimulation with FCS, the number of differentially expressed lncRNAs doubled (36 increased and 38 decreased in expression, see Table E15 in this article's Online Repository at www.jacionline.org).
Of those lncRNAs that were expressed in ASMCs from asthmatic patients, only PVT1 was found to be decreased in expression in the patients with corticosteroid-sensitive nonsevere asthma and increased in expression in patients with corticosteroid-insensitive severe asthma (see Tables E10 and E11). Therefore we further examined the function of this lncRNA.
Effect of FCS and TGF-β on PVT1 lncRNA expression in ASMCs from asthmatic patients
We have previously demonstrated that combined stimulation with FCS (2.5%) and TGF-β (1 ng/mL) is required to induce a differential response in both cellular proliferation and cytokine release in ASMCs from asthmatic patients.3, 5 To determine the potential role of PVT1 in this proliferative and inflammatory response, we examined the time course of its expression in the presence of FCS and TGF-β. FCS plus TGF-β did not change the expression of PVT1 up to 24 hours in ASMCs from healthy subjects (Fig 2, A), whereas at 24 hours, there was a significant reduction in the expression of PVT1 in ASMCs from patients with nonsevere asthma (P < .01 vs unstimulated control ASMCs). Furthermore, FCS plus TGF-β led to an approximately 3-fold increase (P < .01 vs unstimulated control ASMCs) in PVT1 expression, which reached a plateau at 3 hours in ASMCs from patients with severe asthma and remained increased at 24 hours (Fig 2, A).
Fig 2.

Effect of dexamethasone and FCS plus TGF-β on PVT1 lncRNA expression in ASMCs from asthmatic patients. A, FCS plus TGF-β–induced PVT1 lncRNA expression was measured by using quantitative RT-PCR over 1, 3, 6, and 24 hours. B, Dexamethasone (Dex) and FCS plus TGF-β–induced PVT1 expression was measured by using quantitative RT-PCR at 24 hours. Points/bars represent means ± SEMs of 9 ASMC donors. **P < .01 and ***P < .001. ##P < .01.
At 24 hours, dexamethasone had no effect on PVT1 expression in ASMCs from healthy subjects (Fig 2, B). However, the FCS plus TGF-β–induced reduction of PVT1 expression in ASMCs from patients with nonsevere asthma returned to basal levels in the presence of dexamethasone (P < .01). Furthermore, dexamethasone alone increased PVT1 expression in ASMCs from patients with severe asthma (approximately 4-fold, P < .001), and when these cells were subsequently stimulated with FCS plus TGF-β, an even greater increase in expression was observed (approximately 8-fold, P < .001; Fig 2, B).
Inhibition of PVT1 with siRNAs on ASMC proliferation and IL-6 release
To elucidate the role of PVT1, we examined the action of siRNA-mediated inhibition of PVT1 on dexamethasone-exposed, FCS plus TGF-β–induced bromodeoxyuridine (BrdU) incorporation and IL-6 release. As previously reported,3, 5 a significant increase in BrdU incorporation and IL-6 release was observed in both healthy subjects and patients with severe asthma after stimulation with FCS plus TGF-β (P < .001 vs unstimulated control ASMCs; Fig 3, C-F). Dexamethasone inhibited BrdU incorporation and IL-6 release in the healthy ASMCs but had no effect on BrdU incorporation in ASMCs from patients with severe asthma and a limited effect on IL-6 release (Fig 3, C-F). These results also demonstrate the relative corticosteroid insensitivity of the cells from patients with severe asthma compared with those from healthy subjects with respect to BrdU incorporation and IL-6 release.
Fig 3.

Effect of targeting PVT1 with siRNAs on IL-6 release and BrdU incorporation in ASMCs from healthy subjects and patients with severe asthma. A and B,PVT1 lncRNA expression was measured by using RT-PCR after ASM from healthy subjects and patients with severe asthma was transfected with siRNA (300 nmol/L), which was designed to target PVT1. C-F, BrdU incorporation (Fig 3, C and D) and IL-6 release (Fig 3, E and F) were measured by using the BrdU ELISA and DuoSet ELISA, respectively, at 8 days. Bars/points represent means ± SEMs of 9 ASMC donors. */#P < .05, ##/++P < .01, and ***/###P < .001. Dex, Dexamethasone.
Transfection with Amaxa electroporation with siRNAs designed to target PVT1 (300 nmol/L) knocked down expression of PVT1 in ASMCs from healthy subjects both at baseline and after stimulation with FCS plus TGF-β (P < .01; Fig 3, A). When the same cells were exposed to dexamethasone before stimulation with FCS plus TGF-β, a reduction was still observed, although not to the same level as without dexamethasone (P < .05). When PVT1 was targeted with siRNAs in ASMCs from patients with severe asthma, the FCS plus TGF-β– and dexamethasone plus FCS plus TGF-β–induced PVT1 expression was returned to basal levels (Fig 3, B).
Knocking down PVT1 had no effect on BrdU incorporation induced by FCS plus TGF-β in either cohort of ASMCs (Fig 3, C and D). However, PVT1 knockdown increased BrdU incorporation in ASMCs from patients with severe asthma when they were exposed to dexamethasone before stimulation with FCS plus TGF-β (P < .01 vs dexamethasone plus FCS plus TGF-β; Fig 3, D). Furthermore, siRNA-targeted inhibition of PVT1 both resulted in an increase in FCS plus TGF-β–induced IL-6 release (P < .01 vs FCS plus TGF-β) and reversed the inhibitory action of dexamethasone in ASMCs from healthy subjects (P < .01 vs dexamethasone plus FCS plus TGF-β; Fig 3, E). This suggests that inhibiting expression of PVT1 increases corticosteroid insensitivity in ASMCs; however, there was no effect observed on IL-6 release in ASMCs from patients with severe asthma when PVT1 was targeted, although this might be because of IL-6 release already being at maximal levels (Fig 3, F).
A nontargeting negative control (30-300 nmol/L) demonstrated no effect on either BrdU incorporation or IL-6 release in either cohort, as previously shown (data not shown).3, 5, 8 To demonstrate evidence of efficient transfection, concurrent studies were performed to examine the effect of siRNAs (100 nmol/L) targeted to IL6 mRNA. A reduction in IL-6 release induced by FCS plus TGF-β stimulation in ASMCs from healthy subjects (P < .001) and those from patients with severe asthma (P < .01) was observed, as previously described (see Fig E2, A and B, in this article's Online Repository at www.jacionline.org).3, 8 Furthermore, to demonstrate that electroporation had no adverse effect on cellular viability, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays were performed both before (see Fig E2, C and D) and after (see Fig E2, E and F) stimulation with FCS plus TGF-β. In either situation, transfection by using Amaxa electroporation had no effect on cellular viability.
Effect of PVT1 inhibition on IL6 mRNA expression in ASMCs from asthmatic patients
In a similar pattern to that seen in the increasing IL-6 protein release induced by FCS plus TGF-β in these ASMCs,3 IL6 mRNA expression is increased in healthy cells (approximately 15-fold), greater still in cells from patients with nonsevere asthma (approximately 20-fold), and increased to the highest degree in cells from patients with severe ASMCs (approximately 45-fold) when these cells are stimulated with FCS plus TGF-β (P < .01 vs unstimulated control; Fig 4, A). Incubation with dexamethasone before stimulation with FCS plus TGF-β resulted in inhibition of IL6 mRNA expression in ASMCs from healthy subjects and patients with nonsevere asthma (P < .001 and P < .01, respectively), with no significant effect in ASMCs from patients with severe asthma (Fig 4, A).
Fig 4.

Effect of targeting PVT1 with siRNAs on IL6 and c-MYC mRNA in ASMCs from healthy subjects and patients with severe asthma. A and B,IL6 and c-MYC mRNA expression was measured by using RT-PCR after exposure to dexamethasone (Dex; 10−7 mol/L) and stimulation with FCS (2.5%) plus TGF-β (1 ng/mL) for 24 hours. C-F, ASMCs from healthy subjects and patients with severe asthma were transfected with siRNAs designed to target PVT1. Also, the expression of IL6 (Fig 4, C and E) and c-MYC (Fig 4, D and F) mRNA was measured by using RT-PCR. Bars represent means ± SEMs of 9 ASMC donors. #P < .05, **/##/++P < .01, and ***/###/+++P < .001.
The effect of PVT1 targeting by siRNAs on IL6 mRNA was then examined. In ASMCs from healthy subjects, the FCS plus TGF-β–induced expression of IL6 was similar to that observed in Fig 4, A, when the cells were either mock transfected or transfected with 100 nmol/L of negative control siRNA (Fig 4, C). When the healthy ASMCs were transfected with an siRNA designed to target IL6 mRNA, the expression of this mRNA was almost completely attenuated (P < .001; Fig 4, C). However, when the basal expression of PVT1 (as described in Fig 2, B), was inhibited in these ASMCs with 300 nmol/L siRNA, a significant increase in IL6 mRNA expression was observed when the cells were stimulated with FCS plus TGF-β (P < .05; Fig 4, C). Addition of dexamethasone before FCS plus TGF-β resulted in a decrease in IL6 mRNA levels in the mock and negative siRNA-transfected cells, and IL-6 siRNA transfection resulted in inhibition of IL6 mRNA expression (Fig 4, C).
IL6 mRNA expression was similarly affected by the different transfection variables in ASMCs from patients with severe asthma (Fig 4, E). Either mock transfection or transfection with a negative control siRNA had no effect on IL6 mRNA expression. Transfecting with an siRNA designed to target IL6 inhibited IL6 mRNA expression (Fig 4, E). However, inhibiting the expression of PVT1 with siRNAs reduced IL6 mRNA expression after stimulation with FCS plus TGF-β, both with and without exposure to dexamethasone (P < .001; Fig 4, E).
Effect of PVT1 inhibition on c-MYC mRNA in ASMCs from asthmatic patients
PVT1 is transcriptionally activated by the oncogene c-MYC,4, 17 and we have previously reported that c-MYC is important in controlling ASMC proliferation but not cytokine release.5 Therefore we examined the effect of targeting PVT1 with siRNAs on c-MYC mRNA expression. Stimulation with FCS plus TGF-β induced an increase in c-MYC mRNA expression in ASMCs from patients with severe asthma (P < .01), with no effect seen in ASMCs from healthy subjects or patients with nonsevere asthma (Fig 4, B). Exposure of ASMCs from patients with severe asthma to dexamethasone, either on its own or before stimulation with FCS plus TGF-β, resulted in a larger increase in c-MYC mRNA expression (both P < .001; Fig 4, B).
Transfecting siRNAs designed to target PVT1 (300 nmol/L) knocked down expression of c-MYC in healthy ASMCs both at baseline and after stimulation with FCS plus TGF-β (P < .01; Fig 4, D). When PVT1 was targeted with siRNAs in ASMCs from patients with severe asthma, the FCS plus TGF-β– and dexamethasone plus FCS plus TGF-β–induced c-MYC expression was returned to basal levels (Fig 4, F).
In summary, these results show that in primary ASMCs from nonasthmatic subjects, inhibiting the endogenous expression of PVT1 results in reversal of the inhibitory action of dexamethasone on FCS plus TGF-β–induced IL6 mRNA expression and protein release. Furthermore, in ASMCs from patients with severe asthma, dexamethasone increases PVT1 expression, and inhibition of this with siRNAs results in an increase in FCS plus TGF-β–induced cellular proliferation through targeting of the transcription factor c-MYC.
Discussion
Using a transcriptomics-based approach, we examined the expression of RNAs (mRNA, miRNA, and lncRNA) in primary ASMCs isolated from nonasthmatic subjects, patients with nonsevere asthma, and patients with severe asthma. We published the results of the nonasthmatic subjects in 2014,4 and now, after some additional functional studies, report on the patients with nonsevere and severe asthma.
Of the mRNAs we examined, we found that the mRNA profile differentially regulated in ASMCs from patients with nonsevere asthma is important in ASMC calcium signaling,9 airway remodeling,10 the inflammatory response,13 and glucocorticoid activity.15, 16 Interestingly, the mRNA profile in ASMCs from patients with severe asthma appears to be important in increased calcium influx in the smooth muscle,11 atopy,12 and possible immunomodulation in patients with acute exacerbations of airway disease, including asthma.14 Furthermore, when confirming our array results using RT-PCR, we described mRNAs with expression that has previously been shown to be increased in response to dexamethasone (NAV2)18 to regulate cell viability, cell growth, cellular proliferation, and/or airway remodeling (NFIB, NOVA1, PTPRD, and PGM5)19, 20, 21, 22 and those acting as potential tumor suppressors in patients with lung carcinoma (PTPRD and NNAT).23, 24 Furthermore, we describe a large increase in CHI3L1 expression, which has not only been reported in a number of patients with inflammatory diseases and cancer but also has been implicated in asthmatic patients, with the discovery of an allele that doubles the risk.25 However, PTGIS is the only one of these mRNA targets to have been associated with ASM and can induce both bronchodilation and reverse ASMC remodeling in a mouse model of asthma.26
We also measured the expression of lncRNAs in our patient cohorts. A large number of lncRNAs were differentially expressed under all situations (baseline, corticosteroid treatment, and mitogen stimulation). Interestingly, only PVT1 was found to be decreased in ASMCs from patients with nonsevere asthma and increased in ASMCs from patients with severe asthma, and therefore we performed functional studies on this lncRNA in our model of asthmatic hyperproliferation and corticosteroid insensitivity.3, 5 The action of PVT1 in our ASMCs from asthmatic patients was complex. As such, we have proposed a figure that shows potential mechanisms for PVT1 in ASMC proliferation and IL-6 release (Fig 5). In ASMCs from nonasthmatic healthy subjects, TGF-β plus FCS induces IL6 mRNA expression, which translates into IL-6 protein release. Interestingly, the addition of dexamethasone results in an increase in expression of PVT1, which, when inhibited by siRNAs (Fig 5, A), induces greater IL6 mRNA expression and more IL-6 protein release. In ASMCs from patients with severe asthma, TGF-β plus FCS not only induces IL6 mRNA expression and protein release but also PVT1 expression. The effect of dexamethasone was the same, and when we inhibited PVT1 in these ASMCs with siRNAs (Fig 5, B), a decrease in IL6 mRNA and IL-6 protein release was observed, along with a concurrent increase in cellular proliferation. The mechanism of action of PVT1 in human primary ASM is clearly complex. Very little is known about the role of PVT1 in human pathologies. PVT1 is frequently expressed in patients with numerous cancers (including lung)27, 28, 29, 30, 31 and diabetes.32, 33, 34 Furthermore, targeting of PVT1 in these pathologies has been shown to have a range of effects, including decreasing proliferation and increasing apoptosis,28, 35 decreasing resistance to gemcitabine (a chemotherapy drug),36 and regulation of genes and proteins involved in ECM deposition.32 For the first time, we have demonstrated the novel way in which targeting PVT1 in human primary ASMCs can affect phenotype.
Fig 5.

A and B, Potential mechanisms for PVT1 contribution to ASMC proliferation and IL-6 release in asthmatic patients. Dex, Dexamethasone; FCS, fetal calf serum; TGF-β, transforming growth factor-beta.
The variable mechanism of action of PVT1 could simply be a consequence of different cells, tissues, and pathologies examined; however, it might also be due to the action and configuration of PVT1 itself. PVT1 is a downstream target of c-MYC that targets and binds PVT1, driving its transcription.17 We show that in our severe asthmatic ASMC cohorts, dexamethasone increases PVT1 expression, and inhibition of this results in an increase in FCS plus TGF-β–induced cellular proliferation, possibly by targeting of c-MYC.
Finally, PVT1 is unlikely to be the only noncoding RNA (ncRNA) acting in our ASMC model. We have previously reported on the role of miRNAs in these cells after stimulation with TGF-β plus FCS,3 and now it is known that PVT1 can express a cluster of 6 miRNAs, such as miR-1207-5p (which we discussed previously4). Although these ncRNAs are coexpressed, regulation of miR-1207-5p expression by TGF-β is independent of PVT1.33
In conclusion, we have demonstrated a large difference in ncRNA expression profiles (including miRNAs and lncRNAs) in primary human ASMCs from patients with nonsevere and severe asthma and the differential effects of adding corticosteroids and mitogen. Furthermore, we have found that PVT1 regulates both IL-6 release and proliferation in ASMCs from patients with severe asthma.
Key messages.
-
•
Severe asthma is a worldwide health issue that is mostly unresponsive to existing therapy.
-
•
lncRNAs are differentially expressed between ASMCs from patients with nonsevere and severe asthma.
-
•
Targeting of the lncRNA PVT1 can reduce increased cellular proliferation and IL-6 release from ASMCs in patients with severe asthma.
Footnotes
Supported by a fellowship from Imperial College London (to M.M.P.), grants from Asthma UK (08/041) and the Wellcome Trust (085935; to K.F.C.). This project was supported by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London. The views expressed in this publication are those of the authors(s) and not necessarily those of the National Health Service, National Institute for Health Research (NIHR), or Department of Health. K.F.C. is a Senior Investigator of the NIHR. M.M.P. was an Imperial College Research Fellow. M.M.P., I.M.A., and K.F.C. are members of the Interuniversity Attraction Poles Program-Belgian State-Belgian Science Policy- project P7/30.
Disclosure of potential conflict of interest: The authors declare that they have no relevant conflicts of interest.
Supplementary data
Fig E1.
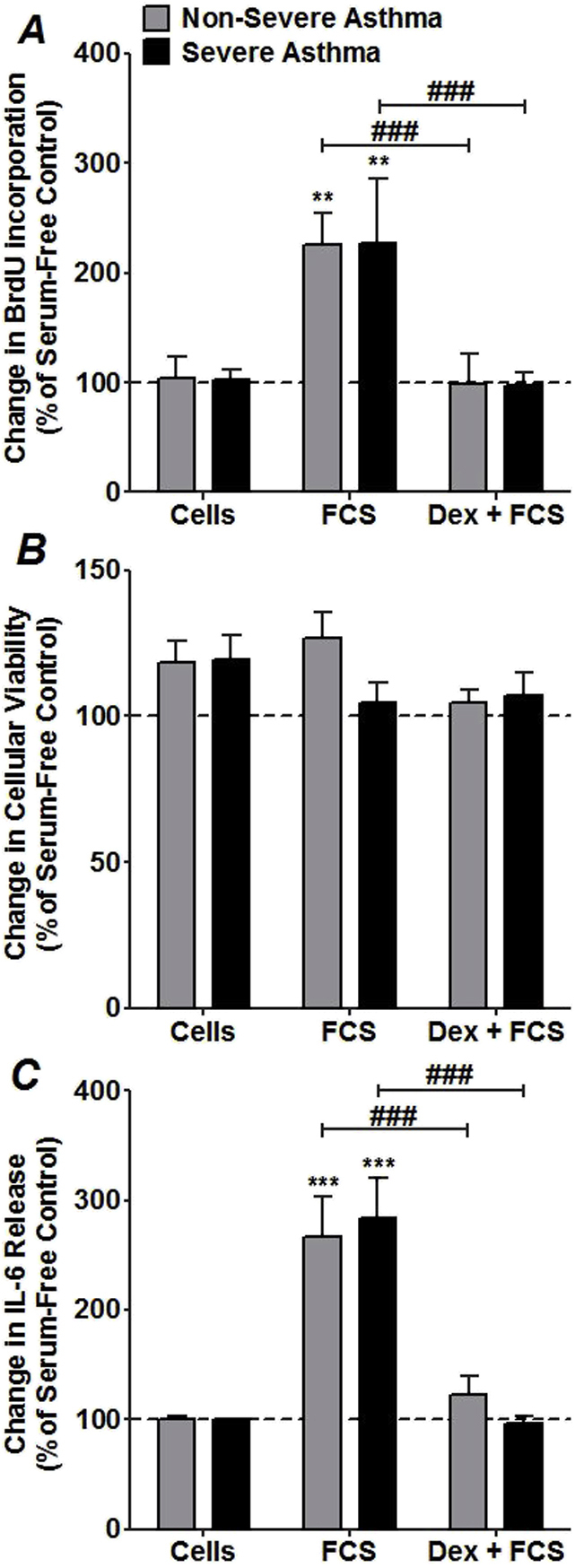
Fig E2.

References
- 1.Bousquet J., Jeffery P.K., Busse W.W., Johnson M., Vignola A.M. Asthma. From bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med. 2000;161:1720–1745. doi: 10.1164/ajrccm.161.5.9903102. [DOI] [PubMed] [Google Scholar]
- 2.Clifford R.L., Singer C.A., John A.E. Epigenetics and miRNA emerge as key regulators of smooth muscle cell phenotype and function. Pulm Pharmacol Ther. 2013;26:75–85. doi: 10.1016/j.pupt.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perry M.M., Baker J.E., Gibeon D.S., Adcock I.M., Chung K.F. Airway smooth muscle hyperproliferation is regulated by MicroRNA-221 in severe asthma. Am J Respir Cell Mol Biol. 2013;50:7–17. doi: 10.1165/rcmb.2013-0067OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perry M., Tsitsiou E., Austin P., Lindsay M., Gibeon D., Adcock I. Role of non-coding RNAs in maintaining primary airway smooth muscle cells. Respir Res. 2014;15:58. doi: 10.1186/1465-9921-15-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perry M.M., Durham A.L., Austin P.J., Adcock I.M., Chung K.F. BET bromodomains regulate TGF-beta-induced proliferation and cytokine release in asthmatic airway smooth muscle. J Biol Chem. 2015;290:9111–9121. doi: 10.1074/jbc.M114.612671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perry M.M., Hui C.K., Whiteman M., Wood M.E., Adcock I., Kirkham P. Hydrogen sulfide inhibits proliferation and release of IL-8 from human airway smooth muscle cells. Am J Respir Cell Mol Biol. 2011;45:746–752. doi: 10.1165/rcmb.2010-0304OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Leary L., Sevinc K., Papazoglou I.M., Tildy B., Detillieux K., Halayko A.J. Airway smooth muscle inflammation is regulated by microRNA-145 in COPD. FEBS Lett. 2016;590:1324–1334. doi: 10.1002/1873-3468.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larner-Svensson H.M., Williams A.E., Tsitsiou E., Perry M.M., Jiang X., Chung K.F. Pharmacological studies of the mechanism and function of interleukin-1beta-induced miRNA-146a expression in primary human airway smooth muscle. Respir Res. 2010;11:68. doi: 10.1186/1465-9921-11-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jude J.A., Wylam M.E., Walseth T.F., Kannan M.S. Calcium signaling in airway smooth muscle. Proc Am Thorac Soc. 2008;5:15–22. doi: 10.1513/pats.200704-047VS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumawat K., Koopmans T., Gosens R. beta-catenin as a regulator and therapeutic target for asthmatic airway remodeling. Expert Opin Ther Targets. 2014;18:1023–1034. doi: 10.1517/14728222.2014.934813. [DOI] [PubMed] [Google Scholar]
- 11.Thompson M.A., Prakash Y.S., Pabelick C.M. Arachidonate-regulated Ca(2+) influx in human airway smooth muscle. Am J Respir Cell Mol Biol. 2014;51:68–76. doi: 10.1165/rcmb.2013-0144OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim Y.J., Park S.W., Kim T.H., Park J.S., Cheong H.S., Shin H.D. Genome-wide methylation profiling of the bronchial mucosa of asthmatics: relationship to atopy. BMC Med Genet. 2013;14:39. doi: 10.1186/1471-2350-14-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung K.F. Airway smooth muscle cells: contributing to and regulating airway mucosal inflammation? Eur Respir J. 2000;15:961–968. doi: 10.1034/j.1399-3003.2000.15e26.x. [DOI] [PubMed] [Google Scholar]
- 14.Koziol-White C.J., Panettieri R.A., Jr. Airway smooth muscle and immunomodulation in acute exacerbations of airway disease. Immunol Rev. 2011;242:178–185. doi: 10.1111/j.1600-065X.2011.01022.x. [DOI] [PubMed] [Google Scholar]
- 15.Boardman C., Chachi L., Gavrila A., Keenan C.R., Perry M.M., Xia Y.C. Mechanisms of glucocorticoid action and insensitivity in airways disease. Pulm Pharmacol Ther. 2014;29:129–143. doi: 10.1016/j.pupt.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 16.Perretti M., Ahluwalia A. The microcirculation and inflammation: site of action for glucocorticoids. Microcirculation. 2000;7:147–161. [PubMed] [Google Scholar]
- 17.Carramusa L., Contino F., Ferro A., Minafra L., Perconti G., Giallongo A. The PVT-1 oncogene is a Myc protein target that is overexpressed in transformed cells. J Cell Physiol. 2007;213:511–518. doi: 10.1002/jcp.21133. [DOI] [PubMed] [Google Scholar]
- 18.Liu L., Walker E.A., Kissane S., Khan I., Murray P.I., Rauz S. Gene expression and miR profiles of human corneal fibroblasts in response to dexamethasone. Invest Ophthalmol Vis Sci. 2011;52:7282–7288. doi: 10.1167/iovs.11-7463. [DOI] [PubMed] [Google Scholar]
- 19.Buckanovich R.J., Yang Y.Y., Darnell R.B. The onconeural antigen Nova-1 is a neuron-specific RNA-binding protein, the activity of which is inhibited by paraneoplastic antibodies. J Neurosci. 1996;16:1114–1122. doi: 10.1523/JNEUROSCI.16-03-01114.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dooley A.L., Winslow M.M., Chiang D.Y., Banerji S., Stransky N., Dayton T.L. Nuclear factor I/B is an oncogene in small cell lung cancer. Genes Dev. 2011;25:1470–1475. doi: 10.1101/gad.2046711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shyur S.D., Wang J.Y., Lin C.G.-J., Hsiao Y.H., Liou Y.H., Wu Y.J. The polymorphisms of protein-tyrosine phosphatase receptor-type delta gene and its association with pediatric asthma in the Taiwanese population. Eur J Hum Genet. 2008;16:1283–1288. doi: 10.1038/ejhg.2008.79. [DOI] [PubMed] [Google Scholar]
- 22.Moiseeva E.P., Critchley D.R. Characterisation of the promoter which regulates expression of a phosphoglucomutase-related protein, a component of the dystrophidutrophin cytoskeleton predominantly expressed in smooth muscle. Eur J Biochem. 1997;248:634–643. doi: 10.1111/j.1432-1033.1997.00634.x. [DOI] [PubMed] [Google Scholar]
- 23.Kohno T., Otsuka A., Girard L., Sato M., Iwakawa R., Ogiwara H. A catalog of genes homozygously deleted in human lung cancer and the candidacy of PTPRD as a tumor suppressor gene. Genes Chromosom Cancer. 2010;49:342–352. doi: 10.1002/gcc.20746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okubo C., Minami Y., Tanaka R., Uchihara T., Anami Y., Furuya S. Analysis of differentially expressed genes in neuroendocrine carcinomas of the lung. J Thorac Oncol. 2006;1:780–786. [PubMed] [Google Scholar]
- 25.Ober C., Tan Z., Sun Y., Possick J.D., Pan L., Nicolae R. Effect of variation in CHI3L1 on serum YKL-40 level, risk of asthma, and lung function. N Engl J Med. 2008;358:1682–1691. doi: 10.1056/NEJMoa0708801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamabayashi C., Koya T., Kagamu H., Kawakami H., Kimura Y., Furukawa T. A novel prostacyclin agonist protects against airway hyperresponsiveness and remodeling in mice. Am J Respir Cell Mol Biol. 2012;47:170–177. doi: 10.1165/rcmb.2011-0350OC. [DOI] [PubMed] [Google Scholar]
- 27.Iwakawa R., Takenaka M., Kohno T., Shimada Y., Totoki Y., Shibata T. Genome-wide identification of genes with amplification and/or fusion in small cell lung cancer. Genes Chromosom Cancer. 2013;52:802–816. doi: 10.1002/gcc.22076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takahashi Y., Sawada G., Kurashige J., Uchi R., Matsumura T., Ueo H. Amplification of PVT-1 is involved in poor prognosis via apoptosis inhibition in colorectal cancers. Br J Cancer. 2014;110:164–171. doi: 10.1038/bjc.2013.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim H.P., Cho G.A., Han S.W., Shin J.Y., Jeong E.G., Song S.H. Novel fusion transcripts in human gastric cancer revealed by transcriptome analysis. Oncogene. 2014;33:5434–5441. doi: 10.1038/onc.2013.490. [DOI] [PubMed] [Google Scholar]
- 30.Bisio A., De Sanctis V., Del Vescovo V., Denti M., Jegga A., Inga A. Identification of new p53 target microRNAs by bioinformatics and functional analysis. BMC Cancer. 2013;13:552. doi: 10.1186/1471-2407-13-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Z., Zhu Z., Zhang B., Li W., Li X., Wu X. Frequent mutation of rs13281615 and its association with PVT1 expression and cell proliferation in breast cancer. J Genet Genomics. 2014;41:187–195. doi: 10.1016/j.jgg.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 32.Alvarez M.L., DiStefano J.K. Functional characterization of the plasmacytoma variant translocation 1 gene (PVT1) in diabetic nephropathy. PLoS One. 2011;6:e18671. doi: 10.1371/journal.pone.0018671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alvarez M.L., Khosroheidari M., Eddy E., Kiefer J. Role of microRNA 1207-5P and its host gene, the long non-coding RNA Pvt1, as mediators of extracellular matrix accumulation in the kidney: implications for diabetic nephropathy. PLoS One. 2013;8:e77468. doi: 10.1371/journal.pone.0077468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alwohhaib M., Alwaheeb S., Alyatama N., Dashti A.A., Abdelghani A., Hussain N. Single nucleotide polymorphisms at erythropoietin, superoxide dismutase 1, splicing factor, arginine/serin-rich 15 and plasmacytoma variant translocation genes association with diabetic nephropathy. Saudi J Kidney Dis Transpl. 2014;25:577–581. doi: 10.4103/1319-2442.132190. [DOI] [PubMed] [Google Scholar]
- 35.Guan Y., Kuo W.L., Stilwell J.L., Takano H., Lapuk A.V., Fridlyand J. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin Cancer Res. 2007;13:5745–5755. doi: 10.1158/1078-0432.CCR-06-2882. [DOI] [PubMed] [Google Scholar]
- 36.You L., Chang D., Du H.Z., Zhao Y.P. Genome-wide screen identifies PVT1 as a regulator of Gemcitabine sensitivity in human pancreatic cancer cells. Biochem Biophys Res Commun. 2011;407:1–6. doi: 10.1016/j.bbrc.2011.02.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.