

Summary
Objective
To analyze whether KCNQ2 R201C and R201H variants, which show atypical gain-of-function electrophysiological properties in vitro, have a distinct clinical presentation and outcome.
Methods
Ten children with heterozygous, de novo KCNQ2 R201C or R201H variants were identified worldwide, using an IRB-approved KCNQ2 patient registry and database. We reviewed medical records, and where possible, interviewed parents and treating physicians using a structured, detailed phenotype inventory focusing on the neonatal presentation and subsequent course.
Results
Nine patients had encephalopathy from birth and presented with prominent startle-like myoclonus, which could be triggered by sound or touch. In seven patients EEG was performed in the neonatal period and showed a burst-suppression pattern. However, myoclonus did not have an EEG correlate. In many patients the paroxysmal movements were misdiagnosed as seizures. Seven patients developed epileptic spasms in infancy. In all patients, EEG showed a slow background and multi-focal epileptiform discharges later in life. Other prominent features included respiratory dysfunction (perinatal respiratory failure and/or chronic hypoventilation), hypomyelination, reduced brain volume, and profound developmental delay. One patient had a later onset, and sequencing indicated that a low abundance (~15%) R201C variant had arisen by post-zygotic mosaicism.
Significance
Heterozygous KCNQ2 R201C and R201H gain-of-function variants present with profound neonatal encephalopathy in the absence of neonatal seizures. Neonates present with non-epileptic myoclonus that is often misdiagnosed and treated as seizures. Prognosis is poor. This clinical presentation is distinct from the phenotype associated with loss-of-function variants, supporting the value of in vitro functional screening. These findings suggest that gain-of-function and loss-of-function variants need different targeted therapeutic approaches.
Keywords: KCNQ2, neonatal seizures, myoclonus, epileptic encephalopathy, infantile spasms
Introduction
Pathogenic variants in KCNQ2, a gene well-known to be involved in benign familial neonatal epilepsy (BFNE), have emerged as an important cause of neonatal onset epileptic encephalopathy (KCNQ2 encephalopathy).1–5 BFNE is characterized by seizure onset within the first days of life, seizure remission within months, a normal interictal EEG, normal development, but increased post-neonatal seizure risk.2, 6 Patients with KCNQ2 encephalopathy also present with frequent neonatal seizures, but their interictal EEG is severely abnormal and characterized by multifocal epileptiform abnormalities with random attenuations or a burst-suppression pattern, and they have moderate to profound developmental delay.5, 7
BFNE arises from KCNQ2 alleles causing haploinsufficiency (including deletion of the entire gene) and from missense variants that exhibit partial loss-of-function in vitro.8–10 Studies of several KCNQ2 variants that cause KCNQ2 encephalopathy reveal more extensive loss-of-function or a dominant negative effect.11, 12 These studies suggest a potential direct relationship between the degree of loss-of-function and clinical severity.11, 12 Recently, the KCNQ2 missense variants R201C and R201H5, 13 were shown to increase channel opening in vitro, a gain-of-function effect opposite to the loss-of-function produced by previous BFNE and KCNQ2 encephalopathy variants.14
Given the distinctive molecular physiology of KCNQ2 R201C and R201H in vitro, we investigated whether these variants result in a distinct phenotype. We report detailed clinical data on the neonatal presentation of 10 children heterozygous for KCNQ2 R201C/H and describe a unique electro-clinical phenotype that includes neonatal encephalopathy without seizures, exaggerated startle response that is often misdiagnosed as seizures, burst-suppression EEG pattern at birth with evolution into multifocal epileptiform discharges, early hypoventilation and supplemental oxygen requirement, hypomyelination, and profound developmental delay with early mortality.
Methods
We studied a cohort of patients with de novo KCNQ2 R201C or R201H variants that were included in the Rational Intervention of KCNQ2 Epileptic Encephalopathy (RIKEE) database (http://www.RIKEE.org), a curated database aggregating published information and unpublished patient data since 2011 provided by physicians or families after parental informed consent. Based on the electroclinical features of a newly diagnosed newborn with a de novo R201C variant (patient A, Table 1), a detailed clinical questionnaire was sent to the referring physician of all identified patients, including the one previously published patient with a R201C variant (patient D in current study and patient B in Weckhuysen et al5). Data requested included information on neonatal neurologic exam, presence of paroxysmal events/seizures or abnormal movements in the neonatal period and later in life, whether seizures were EEG-confirmed, EEG findings in the neonatal period and during follow-up, anti-seizure drugs used, neuroimaging findings, other body systems’ involvement, neurodevelopment, and mortality. The actual video-EEG recordings, when available, were reviewed by a neonatal neurophysiologist (MRC). For some patients, members of the investigative team spoke directly with the parent via conference call to better understand the early presentation and course. The respective KCNQ2 variants were identified in different diagnostic or research laboratories, using targeted gene panels that included KCNQ2 or single gene Sanger sequencing. All variants were proven to occur de novo. Parental consent was obtained to include patient videos.
Table 1.
| A. Clinical features of R201C/H associated KCNQ2 encephalopathy. | |||||
|---|---|---|---|---|---|
| Patient A | Patient B | Patient C | Patient D* | Patient E | |
| Mutation | c.601C>T; p.R201C |
c.601C>T; p.R201C |
c.601C>T; p.R201C |
c.601C>T; p.R201C |
c.601C>T; p.R201C |
| Age at inclusion | 13m
(deceased- Cardiopulmonary arrest) |
2 yr | 4 mo (deceased) | 2 yr 5 mo (deceased- SUDEP) |
6 yr 4 mo (deceased) |
|
Encephalopathy at birth |
Yes | Yes | Yes | Yes | Yes (day 5) |
|
Presenting paroxysmal events and EEG correlate |
1st day of life- exaggerated and sustained startle reaction to touch (no EEG correlate) |
1st day of life- exaggerated and sustained startle reaction to noise/touch, apnea |
1st day of
life- Exaggerated startle response, apnea |
2nd day of life
– Myoclonic Movements intermixed with tonic component and apnea. (no EEG correlate) |
2nd day of life- myoclonic jerky spontaneous movements, pronounced startle responses |
|
Early neurologic exam |
Axial
hypotonia, Increased peripheral tone, hyperreflexia |
Severe
diffuse hypotonia, hyperreflexiatremo |
Mixed tone, hyperreflexia |
Axial hypotonia | Unknown |
| Neonatal EEG | BS (Figure 1A) | BS | BS | BS | BS |
| Follow-up EEG | Multi-focal Epileptiform discharges with random attenuation (Figure 1B) |
Multi-focal Epileptiform discharges |
Multi-focal Epileptiform discharges |
Multi-focal Epileptiform discharges |
Multi-focal Epileptiform discharges (2 yrs) |
| Infantile spasms | None at 13 mo of age |
Early tonic seizures, IS at 4 mo |
IS after few months |
||
|
Retigabine/ezo- gabine (Yes/No), effect |
Yes (17 days), no clinical response on startle/body movements |
Yes (7 wks), decrease in apneas, no clinical response on startle/body movements |
Yes (3 mo), no clinical response |
No | Yes, (4 yrs) more wakefulness |
|
Vigabatrin (Yes/No), effect |
Yes,
reduced Startle movements |
Yes, increased hypotonia |
Yes,
reduced Startle movements |
Yes, no effect | Yes, no effect |
|
Anti-epileptic medications |
CLZ, P5P, B6, FA |
PB, VPA, PHT, B6, P5P, FA, TPM, LEV, ZNS, LOC, CBZ, STM, KD, CBD enriched cannabis |
CBZ, CLZ, PB, P5P, FA, PHT |
PB, LEV, TPM, VPA, CLZ, KD |
PB, LEV, VPA, LTG, TPM, RUF, CLB |
| Brain MRI | Mild cerebral atrophy (1 wk), hypomyelination, diffuse brain volume loss, subependymal heterotopias (3 mo) (Figure 2) |
Mild brain volume loss (1 wk) |
Severe Hypomyelination (2 and 4 mo) |
Mild frontal lobe volume loss, hypomyelination of the posterior limb of internal capsule, prominent ventricles (1 wk) |
Hypomyelination, reduced brain volume (8 mo) |
| Development | Profound delay | Profound delay | Profound delay | Profound delay | Profound delay |
|
Respiratory involvement |
Chronic hypoventilation, ventilator first 2 wks, non- invasive PPV until 2 mo, caffeine, theophylline, Aminophylline with limited effect, irregular breathing pattern (Figure 3), remains on supplemental O2 |
Ventilator required at birth, apneas stopped at 3 mo, recurrent aspiration pneumonias |
Respiratory failure, rare short periods off the ventilator, caffeine tried with mild effect |
Respiratory impairment with need for supplemental O2 recurrent respiratory infections |
History of
frequent Aspiration pneumonias, O2 dependent at 23 mo, parents decided not to ventilate mechanically |
| B. Clinical features of R201C/H associated KCNQ2 encephalopathy. | |||||
|---|---|---|---|---|---|
| Patient F | Patient G | Patient H | Patient I | Patient J | |
| Mutation | c.601C>T; p.R201C |
c.601C>T; p.R201C |
c.601C>T; p.R201C (mosaic) |
c.602G>A; p.R201Hs |
c.602G>A; p.R201H |
| Age at inclusion | 3 yr | 19 mo | 5 yr | 4 yr 6 mo | 4 yr 10 mo |
|
Encephalopathy at birth |
Yes | Yes | No | Yes (mild) | Yes (mild) |
|
Presenting Paroxysmal events and EEG correlate |
2nd day of
life- Stiffening events (not captured on EEG) |
1st day of life- multi-focal erratic myoclonic movements exaggerated with stimuli (No EEG correlate) |
None reported during infancy |
1st day of
life- Myoclonic Spontaneous movements, exaggerated startle to noise/touch |
1st day of life- early neonatal myoclonic events |
|
Early
neurologic exam |
Severe hypotonia, hyperreflexia |
Severe hypotonia | Mild axial hypotonia |
Axial
hypotonia, Increased peripheral tone, hyperreflexia |
Unknown |
| Neonatal EEG | BS | BS | N/A | Unknown | None |
| Follow-up EEG | Multi-focal Epileptiform discharges, slow background |
Multi-focal Epileptiform discharges, slow background |
Multifocal Epileptiform abnormalities, slow background. |
Multi-focal Epileptiform Discharges (3mo), Hypsarrythmia (3mo) |
Multi-focal Epileptiform Discharges (3mo), Hypsarrythmia (5mo), multi- Focal Epileptiform discharges, slow background (4 yrs) |
| Infantile spasms | IS at 7mo | IS | IS (age 2yr) | IS at 2 mo | IS at 5 mo |
|
Retigabine/ezo gabine (Yes/No) effect |
No | Yes (6
mo), Worsening seizures, prolonged apneas, severe sedation |
No | No | No |
|
Vigabatrin (Yes/No), effect |
Yes, reduced spasms |
Yes, reduced spasms |
Yes, controlled spasms, caused sedation |
Yes, no effect | Yes, limited effect on spasms |
|
Anti-epileptic medications |
PB, TPM, ACTH, KD, LEV, CLZ, FA, B6, P5P |
LEV, OXC, PB, STM, TPM, PHT VPA |
ACTH, LEV | CBZ, ZNS, PB, P5P, FA |
ACTH, PHT, VPA, TPM, LEV |
| Brain MRI | Normal (1 wk), Increased T2 signal in the basal ganglia (1 yr) |
Normal (1.5 wks), hypo myelination (1 mo) |
Hypomyelination (10 mo), marked Hypomyelination and enlarged subarachnoid spaces (4 yr) |
Hypo myelination, diffuse brain volume loss (3 mo) |
Persistent
cavum Septum pellucidum (5 wks), abnormal (2 yr) |
| Development | Profound motor delay |
Profound delay | Age 1 yr-
mild developmental delay Age 5 yr- severe delay |
Profound delay | Profound motor delay, rare words, responsive smile/laugh |
|
Respiratory involvement |
Ventilator required at birth for 4 days, discharged from NICU on supplemental O2, has Occasional apneas |
Supplemental O2 until age 6 mo due to chronic compensated respiratory acidosis (venous pCO2 ≥75 mmHg), irregular breathing pattern, STM had a good effect on O2 need |
Frequent Respiratoryillnesses requiring home nebulizer |
Recurrent aspiration pneumonias, manyadmissions to the pediatric intensive care unit for ventilation |
Frequent respiratory illnesses, aspiration pneumonia, clubbing of finger nails |
Abbreviations: PB-phenobarbital; CLZ-clonazepam; PHT-phenytoin; VPA-valproic acid; LEV-levetiracetam; TPM-topiramate; ZNS-zonisamide; LOC-lacosamide; CBZ-carbamazepine; OXC-oxcarbazepine; STM-sulthiame; LTG-lamotrigine; CLB-clobazam; RUF-rufinamide; CBD-cannabidiol; FA-folinic acid; B6-pyridoxine; P5P–pyridoxal-5-phosphate; KD-ketogenic diet; PPV- positive pressure ventilation; IS-infantile spasms
Results
Neurologic Presentation
Eight patients with heterozygous de novo R201C variants and two patients with heterozygous de novo R201H variants were identified (Table 1). The majority of infants were born at term after uncomplicated gestations and deliveries. Patient C had bradycardia prior to delivery via caesarian section. One child (patient H) was found to carry a post-zygotic mosaic variant; this patient had later symptom onset and is discussed below.
The remaining nine patients presented with encephalopathy at birth (milder in the R201H cases) and developed prominent, abnormal body movements within the first 2 days of age. The movements were characterized by high amplitude spontaneous myoclonic jerks that were triggered by touch and sound, suggesting an exaggerated startle response (video 1). These paroxysmal movements were clinically considered as epileptic seizures in eight patients and treated with anti-seizure medications without success. However, where available for review (patients A, D, G), neonatal video-EEGs demonstrated that the abnormal paroxysmal movements lacked EEG correlate and were consistent non-epileptic myoclonus.
EEG was performed in the neonatal period in seven infants, and in all of them it demonstrated an invariant burst-suppression pattern (Figure 1A). Neurologic exam was notable for diffuse hypotonia and marked hyperreflexia in eight patients. Brain MRI in five patients was obtained at ≤ 10 days of age and either was reported as normal (n = two) or showed mild reduced brain volume (n = three).
Figure 1. Evolution of EEG during Early Infancy (Patient A).

A. EEG (7µV/mm) at two days of age showing a burst-suppression pattern. B. EEG (7µV/mm) at 47 days of age showing a pattern of multi-focal epileptiform discharges with random areas of attenuation while on ezogabine (level 1600ng/mL). C. EEG (7µV/mm), just prior to discharge from the neonatal intensive care unit at 106 days of age showing a pattern of multifocal epileptiform discharges on vigabatrin 50mg/kg/day.
After one month of age, the EEG showed a pattern of multi-focal epileptiform discharges in all 10 infants (Figure 1B). Seven infants developed epileptic spasms at a median age of five months (range three to 24 months). All four infants who had neonatal EEG and developed epileptic spasms showed a burst-suppression pattern in the neonatal period. Brain MRI was obtained in eight infants between one and 24 months of age and revealed hypomyelination and reduced brain volume in six patients (Figure 2) and abnormal signal in the basal ganglia in one patient (F). Reduced brain volume became more evident on follow-up brain MRI. Patient A also had subependymal heterotopias (Figure 2). The patients had profound developmental delay without the achievement of discernable developmental motor milestones and were non-verbal. One patient (patient I) later in life (video 2) developed severe choreoathetosis; however it is possible that similar movement disorders, including choreoathetosis and myoclonus-dystonia, occurred in some of the other patients. Four patients had early mortality ranging from 4.5 months to 6.3 years. The other six children are alive and currently between 19 months and five years of age.
Figure 2. Brain MRI at Three months of age in Infant with KCNQ2 R201C Variant.

A. Axial T2 weighted image shows diffuse reduced brain volume with ex vacuo enlargement of the lateral ventricles, also notice subependymal heterotopias in the right ventricular atrium (white arrow). B. Sagittal T1 weighted image shows diffuse thinning of the corpus callosum (white arrow) that reflects bilateral volume loss in the cortex. C. Axial T1 weighted inversion recovery image shows no myelination in the anterior limb of internal capsule (should be present at three months of age), suggesting hypomyelination (patient A).
One patient (patient H) did not have encephalopathy nor paroxysmal events as a newborn. She was noted to have only mild axial hypotonia, and by age one year could sit, crawl in a supine position and roll over independently, and, although showing some developmental delay, she could reach milestones not attained by the other nine patients. However, in the second year of life, developmental regression occurred coinciding with the onset of epileptic spasms despite aggressive physical therapy efforts. At age five years, she was non-verbal, non-ambulatory, but could smile and communicate pleasure to her family. Whole exome sequencing revealed a low abundance KCNQ2 c.601C>T variant in 16 of 81 reads, or 19.8% (95% confidence interval, 11.7% – 30.1%). The difference between the observed frequency and 50% (expected for heterozygosity) was highly significant (P = 3.6 × 10−8, Binomial exact test). 15 Consistent with whole exome sequencing results, when bidirectional Sanger seqeuncing was performed in parallel on the trio, the variant peak was estimated as 15–25% of the total at base 601.
Treatment
All patients were trialed on multiple medications (Table 1). Given the genetic mutation in KCNQ2, five patients were trialed on retigabine/ezogabine (European name/United States name) between 17 days and four years of age with little or no response. Patient A was slowly escalated to a dose of 7mg/kg every eight hours over a period of three weeks to a level of 1,600ng/mL. While on ezogabine, the EEG continued to have a pattern of severe encephalopathy and there was no clinical improvement in respiratory or neurological status. GGT became elevated to 404 U/L, although AST and ALT remained normal. Given the lack of response, ezogabine was discontinued. Patient G had clinical worsening on retigabine at six months of age with increased paroxysmal events, prolonged apneas, and severe sedation. Patient E had subjective improvement with increased wakefulness when on retigabine at four years of age. Three patients with epileptic spasms were treated with hormonal therapy (adrenocorticotropic hormone) with poor response (patients F, H, and J). In two of these patients, spasms reduced, although did not resolve, after starting vigabatrin. All 10 infants were treated with vigabatrin early in life. In five patients, a noticeable reduction in either the exaggerated startle/myoclonic jerks (patients A and C) or a reduction in spasms (patients F, G, and H) was noted. Interestingly, patient A was treated with vigabatrin for control of exaggerated startle and jerking at four months of age and did not develop epileptic spasms.
Other System Involvement
Respiratory problems, including respiratory failure and recurrent aspiration pneumonia, were common among these patients (Table 1). In six patients (A-D, F, and G) there was respiratory failure and hypoventilation at birth and mechanical ventilation or oxygen supplementation was required. Patient A required invasive mechanical ventilation until two weeks of age and then required non-invasive positive pressure ventilation until two months of age due to hypoventilation and chronically elevated pCO2, unresponsive to caffeine, theophylline, and aminophylline treatments. After discontinuation of mechanical ventilation, patient A exhibited excessive sighing and atypical periodic breathing associated with intermittent hypoxia (Figure 3). Congenital central hypoventilation syndrome (CCHS) was considered, although patients with CCHS usually have a normal EEG. However, Sanger sequencing revealed no variants in PHOX2b nor did whole exome sequencing reveal a mutation in other genes linked to CCHS.16 Chronic compensated respiratory acidosis with an irregular breathing pattern was seen in patient G who received supplemental oxygen until six months of age. Patient D also had chronically elevated pCO2, received supplemental oxygen, and died suddenly, overnight, without preceding infection. Patients E, I, and J had recurrent aspiration pneumonias and often required admission to the intensive care unit for respiratory insufficiency. Patient J had clubbing of the finger nails at five years of age.
Figure 3. Respiratory and SpO2 Recording in Infant with KCNQ2 R201C Variant.
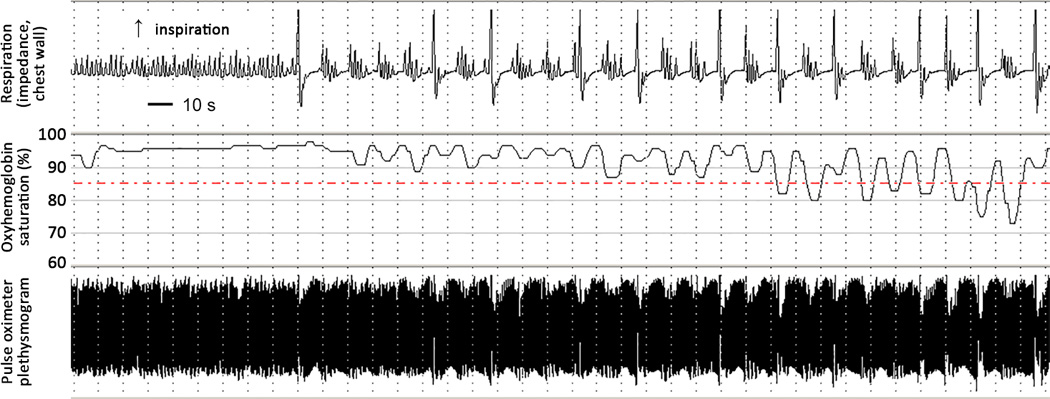
Respiratory and pulse oximetry recording at three months of age showing excessive sighing and unusual periodic breathing associated with intermittent hypoxia (patient A).
Three patients developed diabetes insipidus (A, C, and F) between one to two months of age that in patient A required prolonged DDAVP and did not self-resolve. The only dysmorphic feature documented in the infants was an abnormal palate; patient D had a cleft soft palate, patient G had a cleft palate, and patient C had a high-arched palate. All patients were fed by gastrostomy tube.
Discussion
Patients with the gain-of-function KCNQ2 R201C and R201H variants have a severe form of KCNQ2 encephalopathy with a phenotype that is novel and distinct from what has been reported for other KCNQ2 encephalopathy variants. Previously undescribed features include neonatal encephalopathy without seizures, exaggerated and prolonged startle response with bilateral high amplitude myoclonic jerks without EEG correlate, hypoventilation with periodic breathing, and profound developmental impairment with early mortality. Our electro-clinical data support the hypothesis that in vitro gain-of function correlate in vivo with clinical features that are different from the ones observed in neonates with variants causing loss-of-function.
The position of the variant likely has an impact on the clinical expression. While BFNE-associated variants are randomly distributed throughout the channel protein and include missense, splice, stop, and frameshift variants as well as exon and whole-gene deletions, currently known KCNQ2 encephalopathy variants are missense or single codon deletions, clustering in four functionally important protein domains: the voltage sensor domain, the pore, the C-terminus proximal region, and the calmodulin-binding B helix region.17 Within the voltage sensor domain, most KCNQ2 variants causing encephalopathy reduce channel function (often with dominant-negative effects).11, 12 However, a few affecting residues involved in voltage sensor domain resting state stabilization (i.e. R201C, R201H, and R198Q) produce gain-of-function effects by shifting activation to more hyperpolarized potentials and/or increasing maximal current density.14, 18, 19
Both de novo loss-of-function and the R201C/H gain-of-function variants lead to a neonatal encephalopathy characterized by a burst-suppression EEG pattern. However, even though 70% of the patients in our series developed epileptic spasms in infancy, the absence of seizures in the neonatal period seems to be a peculiarity of the gain-of-function variants. In fact, no neonatal seizures or encephalopathy were reported in four patients carrying a milder KCNQ2 R198Q gain-of-function variant who later developed epileptic spasms at 4–6 months of age.19
Excessive startle response as a prominent neonatal feature was observed for the R201C/H gain-of-function variants. The paroxysmal movements were described as myoclonic in many of the cases. Myoclonic jerks associated with encephalopathy and a burst-suppression EEG pattern are seen in neonates with Early Myoclonic Encephalopathy (EME). However, in EME, myoclonus is typically fragmentary, erratic, of low amplitude, tends to shift from one body part to another in a random asynchronous fashion, and is not induced by a stimulus.20 In our patients, the myoclonic movements were massive, elicited by tactile or acoustic stimuli, tended to persist after the end of the stimulus, and were difficult to distinguish from epileptic movements without simultaneous video-EEG recording. They were indeed clinically diagnosed as seizures and treated as such in most cases of our series. Such a misdiagnosis is not surprising, since it is often impossible to accurately differentiate between seizures and non-epileptic paroxysmal movements in neonates using clinical evaluation alone.21
Semiologically, the exaggerated startle response is reminiscent of hyperekplexia, a genetic condition due to mutations affecting spinal glycine neurotransmission that is characterized by an exaggerated startle response to stimuli, with a brief period of stiffness following the startle reflex.22 However, EEG in hyperekplexia is normal and there is no encephalopathy.22, 23 The movements of R201C/H neonates are different from the myokymia that affects older patients with some specific loss-of-function KCNQ2 variants.24, 25 Such myokymia is rhythmic, and is a sign of lower motor neuron hyperexcitability.
One patient harboring the R201C variant had a less severe phenotype and lacked the early neonatal features that were consistently seen among the other nine patients. Her genetic testing indicated post-zygotic mosaicism, which was previously shown to underlie milder presentation of KCNQ2 encephalopathy loss-of-function variants.4, 26
Synchronized activity during prenatal and postnatal periods is essential for the development of cortical neuronal circuits.27 Our findings suggest that the R201C gain-of-function may disrupts network activity in the neonatal period, but (unlike loss-of-function variants) somehow prevents some aspects of synchronization and/or excitability required for expression of seizures. KCNQ2 channels are also found in neurons of the dorsal root ganglia and nociceptive afferent terminals, where they play a role in pain sensation.28–30 Since the paroxysmal movements in our patients were triggered by tactile stimulation, it could be hypothesized that the exaggerated startle response may be secondary to an abnormal sensation to touch.
Early central hypoventilation and abnormal breathing patterns are additional prominent clinical features observed in patients with KCNQ2 R201C/H variants. Apneic episodes and respiratory failure along with neonatal encephalopathy and severe hypotonia have been described in males with methyl-CpG-binding protein 2 (MECP2) mutations,31 but these infants lack both the distinct paroxysmal movements and severe EEG findings observed in infants with KCNQ2 gain-of-function variants. Apneas are described in loss-of-function variants of KCNQ2 encephalopathy and typically occur as part of a tonic seizure.5, 32 In our R201C variant patients however, hypoventilation and apnea observed in the neonatal period were demonstrated to be associated with central hypoventilation rather than seizure activity. Interestingly, the KCNQ2 channel has key functions in respiratory drive.33 Chemosensitive neurons in the retrotrapezoid nucleus of the brainstem are regulated through a pathway which involves serotonergic inhibition of KCNQ2 channels to increase respiratory drive in response to a change in CO2/H+.34 A gain-of-function channel variant by causing excessive channel opening may result in a reduction of respiratory drive due to a decreased sensitivity to inhibition by serotonin despite chronic elevated CO2 levels in infants with a gain-of-function variant. Sulthiame, a carbonic anhydrase inhibitor, given to patient G had a beneficial effect on breathing. KCNQ2 potassium channel subunits have also been detected in branches of the carotid sinus nerve, which provides sensory innervation to the carotid body chemoreceptors, as well as in carotid body glomus cells, which are the main O2-sensing cell in the carotid bodies.35 Little is known about the role of KCNQ2 in the carotid body glomus cells or carotid sinus nerve fibers. Of note, mice homozygous for a KCNQ2 mutation die of respiratory failure after birth.36 Respiratory problems may contribute to early mortality in KCNQ2 encephalopathy due to R201C/H variants. Animal model and human studies are needed to understand the effect of gain-of-function variants on central and peripheral chemoreflex respiratory control.
In the majority of our patients, MRI showed progressive diffuse brain volume reduction, thin corpus callosum and hypomyelination. The thinning of the corpus callosum could reflect the bilateral volume loss in the cortex as seen in many other encephalopathies. Brain MRI at three months of age in one infant (patient A) showed an additional finding of altered neuronal migration with gray matter heterotopias. Interestingly, a neuropathological report of a neonate with KCNQ2 encephalopathy found mild malformation of cortical development and heterotopic gray matter,37 and heterotopias were also found in a KCNQ2 transgenic mouse model.38 The cellular and circuit mechanisms through which altered KCNQ2 channel activity results in such neurodevelopmental structural abnormalities are currently unknown.
Recognition of genotype-phenotype correlations in patients with KCNQ2 variants can have important treatment implications. Retigabine/ezogabine, a KCNQ-channel opener and therefore a presumed targeted therapy for epilepsy involving KCNQ2 loss-of-function,5, 39 did not impact the myoclonic jerks and/or the encephalopathy in patients with R201C/H variants and even seemed to lead to a worsening of the symptoms in at least one patient (patient G). Given this observation, it is possible that treatment with retigabine/ezogabine in patients with a gain-of-function variant may result in a detrimental clinical effect. KCNQ2-related epilepsies are therefore a prime example of the need for functional classification of allele subgroups, as gain-of-function and loss-of function require different targeted therapeutic approaches.
The patients with KCNQ2 R201C and R201H variants described in this report define a distinct and very severe form of neonatal encephalopathy and provide a clinical translation of in vitro functional studies. We believe that gain-of-function variants of KCNQ2 should be considered in newborns with unexplained severe encephalopathy, burst-suppression EEG pattern, and stimulus sensitive myoclonic jerks, in the absence of epileptic seizures, as well as in newborns with central hypoventilation. In our series, prognosis was poor. Future investigations should aim at identifying drugs that counteract excessive channel opening as an early targeted therapy that may improve outcome.
Supplementary Material
Video 1 Paroxysmal myoclonic movements, without EEG correlate, that are stimulus sensitive in an infant with the R201C variant at 2 months of age.
Video 2. Chorea is shown in a 4 year old girl with the R201H variant. The movements wax and wane with episodes that last for a few days.
Key Point Box.
-
-
The KCNQ2 gain-of-function variants R201C and R201H lead to a phenotype clearly distinct from the one associated with KCNQ2 loss-of-function variants
-
-
Children have a neonatal encephalopathy with non-epileptic myoclonic jerks, central hypoventilation, and profound developmental delay with early mortality
-
-
In vitro functional studies in KCNQ2 translate to in vivo clinical entities, supporting the use of distinct targeted treatments for specific patient subgroups
Acknowledgments
The authors thank the wonderful families and the children for teaching us about KCNQ2. We also thank Dr. David Goldstein (Columbia University) and Dr. Slave Petrovski (University of Melbourne) for performing and interpreting next generation and Sanger sequencing data (Patient H) and Dr. E.J. Kamsteeg (Radboud University Medical Center) for next generation and Sanger sequence data (Patient I).
The RIKEE registry has been supported by a Research Infrastructure grant from the American Epilepsy Society and the Epilepsy Foundation, by the Jack Pribaz Foundation, and by Baylor College of Medicine. Dr. Mulkey receives support from the UAMS Center for Translational Neuroscience (NIH P30 GM110702). Dr. Taglialatela received support from Telethon (GGP15113). Dr. Cooper and Ms. Joshi were supported in part by NINDS R01 NS49119.
Joost Nicolai receives financial support for congress visits from UCB Pharma and Actelion. Edward C. Cooper has Investigator Initiated Research Grants: GlaxoSmithKline, SciFluor Biosciences. Maria Roberta Cilio receives financial support from Insys Therapeutics for Company Sponsored Clinical Trials.
Footnotes
Author disclosures
The remaining authors have no conflicts of interest.
Ethical Publication Statement
The authors confirm that they have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting Information
Supplementary material legend
References
- 1.Biervert C, Schroeder BC, Kubisch C, et al. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- 2.Grinton BE, Heron SE, Pelekanos JT, et al. Familial neonatal seizures in 36 families: Clinical and genetic features correlate with outcome. Epilepsia. 2015;56:1071–1080. doi: 10.1111/epi.13020. [DOI] [PubMed] [Google Scholar]
- 3.Singh NA, Charlier C, Stauffer D, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- 4.Weckhuysen S, Mandelstam S, Suls A, et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol. 2012;71:15–25. doi: 10.1002/ana.22644. [DOI] [PubMed] [Google Scholar]
- 5.Weckhuysen S, Ivanovic V, Hendrickx R, et al. Extending the KCNQ2 encephalopathy spectrum: clinical and neuroimaging findings in 17 patients. Neurology. 2013;81:1697–1703. doi: 10.1212/01.wnl.0000435296.72400.a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ronen GM, Rosales TO, Connolly M, Anderson VE, Leppert M. Seizure characteristics in chromosome 20 benign familial neonatal convulsions. Neurology. 1993;43:1355–1360. doi: 10.1212/wnl.43.7.1355. [DOI] [PubMed] [Google Scholar]
- 7.Kato M, Yamagata T, Kubota M, et al. Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia. 2013;54:1282–1287. doi: 10.1111/epi.12200. [DOI] [PubMed] [Google Scholar]
- 8.Miceli F, Soldovieri MV, Joshi N, Weckhuysen S, Cooper EC, Taglialatela M. KCNQ2-Related Disorders. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. Seattle (WA): University of Washington, Seattle; 2010. Apr 27, [[Updated 2016 Mar 31]]. pp. 1993–2016. GeneReviews [online] Available at: www.ncbi.nlm.nih.gov/books/NBK32534/ [Google Scholar]
- 9.Schroeder BC, Kubisch C, Stein V, Jentsch TJ. Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature. 1998;396:687–690. doi: 10.1038/25367. [DOI] [PubMed] [Google Scholar]
- 10.Singh NA, Westenskow P, Charlier C, et al. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain. 2003;126:2726–2737. doi: 10.1093/brain/awg286. [DOI] [PubMed] [Google Scholar]
- 11.Orhan G, Bock M, Schepers D, et al. Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann Neurol. 2014;75:382–394. doi: 10.1002/ana.24080. [DOI] [PubMed] [Google Scholar]
- 12.Miceli F, Soldovieri MV, Ambrosino P, et al. Genotype-phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of K(v)7.2 potassium channel subunits. Proc Natl Acad Sci U S A. 2013;110:4386–4391. doi: 10.1073/pnas.1216867110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45:825–830. doi: 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miceli F, Soldovieri MV, Ambrosino P, et al. Early-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. J Neurosci. 2015;35:3782–3793. doi: 10.1523/JNEUROSCI.4423-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Halvorsen M, Petrovski S, Shellhaas R, et al. Mosaic mutations in early-onset genetic diseases. Genetics in medicine : official journal of the American College of Medical Genetics. 2016;18:746–749. doi: 10.1038/gim.2015.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carroll JL, Agarwal A. Development of ventilatory control in infants. Paediatr Respir Rev. 2010;11:199–207. doi: 10.1016/j.prrv.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 17.Millichap JJ, Park KL, Tsuchida T, et al. KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients. Neurology Genetics. 2016;2:e96. doi: 10.1212/NXG.0000000000000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Devaux J, Abidi A, Roubertie A, et al. A Kv7.2 mutation associated with early onset epileptic encephalopathy with suppression-burst enhances Kv7/M channel activity. Epilepsia. 2016;57:e87–e93. doi: 10.1111/epi.13366. [DOI] [PubMed] [Google Scholar]
- 19.Millichap JJ, Miceli F, De Maria M, et al. Infantile spasms and encephalopathy without preceding neonatal seizures caused by KCNQ2 R198Q, a gain-of-function variant. Epilepsia. 2016 doi: 10.1111/epi.13601. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beal JC, Cherian K, Moshe SL. Early-onset epileptic encephalopathies: Ohtahara syndrome and early myoclonic encephalopathy. Pediatr Neurol. 2012;47:317–323. doi: 10.1016/j.pediatrneurol.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 21.Malone A, Ryan CA, Fitzgerald A, Burgoyne L, Connolly S, Boylan GB. Interobserver agreement in neonatal seizure identification. Epilepsia. 2009;50:2097–2101. doi: 10.1111/j.1528-1167.2009.02132.x. [DOI] [PubMed] [Google Scholar]
- 22.Bode A, Lynch JW. The impact of human hyperekplexia mutations on glycine receptor structure and function. Mol Brain. 2014;7:2. doi: 10.1186/1756-6606-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bakker MJ, van Dijk JG, van den Maagdenberg AM, Tijssen MA. Startle syndromes. Lancet Neurol. 2006;5:513–524. doi: 10.1016/S1474-4422(06)70470-7. [DOI] [PubMed] [Google Scholar]
- 24.Wuttke TV, Jurkat-Rott K, Paulus W, Garncarek M, Lehmann-Horn F, Lerche H. Peripheral nerve hyperexcitability due to dominant-negative KCNQ2 mutations. Neurology. 2007;69:2045–2053. doi: 10.1212/01.wnl.0000275523.95103.36. [DOI] [PubMed] [Google Scholar]
- 25.Dedek K, Fusco L, Teloy N, Steinlein OK. Neonatal convulsions and epileptic encephalopathy in an Italian family with a missense mutation in the fifth transmembrane region of KCNQ2. Epilepsy Res. 2003;54:21–27. doi: 10.1016/s0920-1211(03)00037-8. [DOI] [PubMed] [Google Scholar]
- 26.Milh M, Lacoste C, Cacciagli P, et al. Variable clinical expression in patients with mosaicism for KCNQ2 mutations. Am J Med Genet A. 2015;167A:2314–2318. doi: 10.1002/ajmg.a.37152. [DOI] [PubMed] [Google Scholar]
- 27.Khazipov R, Luhmann HJ. Early patterns of electrical activity in the developing cerebral cortex of humans and rodents. Trends Neurosci. 2006;29:414–418. doi: 10.1016/j.tins.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 28.Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156:1185–1195. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Passmore GM, Selyanko AA, Mistry M, et al. KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci. 2003;23:7227–7236. doi: 10.1523/JNEUROSCI.23-18-07227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Passmore GM, Reilly JM, Thakur M, et al. Functional significance of M-type potassium channels in nociceptive cutaneous sensory endings. Front Mol Neurosci. 2012;5:63. doi: 10.3389/fnmol.2012.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Falsaperla R, Pavone L, Fichera M, Striano P, Pavone P. Apneic crises: a clue for MECP2 testing in severe neonatal hypotonia-respiratory failure. Eur J Paediatr Neurol. 2012;16:744–748. doi: 10.1016/j.ejpn.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 32.Numis AL, Angriman M, Sullivan JE, et al. KCNQ2 encephalopathy: delineation of the electroclinical phenotype and treatment response. Neurology. 2014;82:368–370. doi: 10.1212/WNL.0000000000000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mulkey DK, Hawkins VE, Hawryluk JM, Takakura AC, Moreira TS, Tzingounis AV. Molecular underpinnings of ventral surface chemoreceptor function: focus on KCNQ channels. J Physiol. 2015;593:1075–1081. doi: 10.1113/jphysiol.2014.286500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hawkins VE, Hawryluk JM, Takakura AC, Tzingounis AV, Moreira TS, Mulkey DK. HCN channels contribute to serotonergic modulation of ventral surface chemosensitive neurons and respiratory activity. J Neurophysiol. 2015;113:1195–1205. doi: 10.1152/jn.00487.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buniel M, Glazebrook PA, Ramirez-Navarro A, Kunze DL. Distribution of voltage-gated potassium and hyperpolarization-activated channels in sensory afferent fibers in the rat carotid body. J Comp Neurol. 2008;510:367–377. doi: 10.1002/cne.21796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe H, Nagata E, Kosakai A, et al. Disruption of the epilepsy KCNQ2 gene results in neural hyperexcitability. J Neurochem. 2000;75:28–33. doi: 10.1046/j.1471-4159.2000.0750028.x. [DOI] [PubMed] [Google Scholar]
- 37.Dalen Meurs-van der Schoor, van WM, van KM, et al. Severe Neonatal Epileptic Encephalopathy and KCNQ2 Mutation: Neuropathological Substrate? Front Pediatr. 2014;2:136. doi: 10.3389/fped.2014.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci. 2005;8:51–60. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- 39.Millichap JJ, Cooper EC. KCNQ2 Potassium Channel Epileptic Encephalopathy Syndrome: Divorce of an Electro-Mechanical Couple? Epilepsy Curr. 2012;12:150–152. doi: 10.5698/1535-7511-12.4.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video 1 Paroxysmal myoclonic movements, without EEG correlate, that are stimulus sensitive in an infant with the R201C variant at 2 months of age.
Video 2. Chorea is shown in a 4 year old girl with the R201H variant. The movements wax and wane with episodes that last for a few days.