

Abstract
BACKGROUND
MET gene amplification and Met protein overexpression may be associated with a poor prognosis. The MET/Met status is typically determined with fluorescence in situ hybridization (FISH) and immunohistochemistry (IHC), respectively. Targeted proteomics uses mass spectrometry–based selected reaction monitoring (SRM) to accurately quantitate Met expression. FISH, IHC, and SRM analyses were compared to characterize the prognostic value of MET/Met in gastroesophageal adenocarcinoma (GEC).
METHODS
Samples from 447 GEC patients were analyzed for MET gene amplification (FISH) and Met protein expression (IHC and SRM). Cox proportional hazards models and Kaplan‐Meier estimates were applied to explore relations between Met, overall survival (OS), and clinical/pathological characteristics. Spearman's rank coefficient was used to assess the correlation between parameters.
RESULTS
Patients with MET‐amplified tumors had worse OS when: the MET/centromere enumeration probe for chromosome 7 FISH ratio was ≥ 2 (hazard ratio [HR], 3.13; 95% confidence interval [CI], 1.84‐5.33), the MET gene copy number was ≥5 (HR, 2.51; 95% CI, 1.45‐4.34), or ≥ 10% of the cells had ≥15 copies (HR, 4.28; 95% CI, 2.18‐8.39). Similar observations were made with Met protein overexpression by IHC (≥1 + intensity in ≥ 25% of the tumor cell membrane: HR, 1.39; 95% CI, 1.04‐1.86) or SRM (≥400 amol/μg: HR, 1.76; 95% CI, 1.06‐2.90). A significant correlation was observed between MET FISH/Met IHC, MET FISH/Met SRM, and Met IHC/Met SRM; only MET FISH and Met SRM were independent negative prognostic biomarkers in multivariate analyses.
CONCLUSIONS
MET amplification and overexpression, assessed by multiple methods, were associated with a worse prognosis in univariate analyses. However, only MET amplification by FISH and Met expression by SRM were independent prognostic biomarkers. Compared with IHC, SRM may provide an added benefit for informed decisions about Met‐targeted therapy. Cancer 2017;123:1061–70. © 2016 American Cancer Society.
Keywords: fluorescence in situ hybridization, gastroesophageal adenocarcinoma, gene amplification, immunohistochemistry, MET tyrosine kinase, negative prognostic biomarker, protein overexpression, selected reaction monitoring mass spectrometry
Short abstract
In a large study, MET gene amplification, Met protein overexpression, or both, as assessed by various assays, are associated with a poor prognosis in univariate analyses. However, only MET amplification by fluorescence in situ hybridization and Met expression by selected reaction monitoring mass spectrometry are independent prognostic biomarkers; compared with immunohistochemistry, selected reaction monitoring may provide an added benefit for informed decisions about Met‐targeted therapy.
INTRODUCTION
The Met receptor tyrosine kinase (c‐Met or hepatocyte growth factor [HGF] receptor) is a single‐pass transmembrane receptor that undergoes homodimerization and activation upon the binding of HGF, its only known ligand.1 Numerous signaling pathways are activated by the interaction of HGF and Met2; thus, Met plays a critical role in many biological functions ranging from embryogenesis to wound healing. Mechanisms for Met‐induced oncogenesis include constitutive activation of the kinase domain and/or dysregulated paracrine and/or autocrine signaling.1, 3, 4 The underlying activating mechanism typically involves MET gene amplification, Met and/or HGF protein overexpression, or, rarely, domain‐specific sequence mutations/translocations,2, 3 including the recently observed Met exon 14–skipping mutations in non–small cell lung cancer.5, 6
Aberrant Met activity has been ubiquitously reported across cancers.1, 2, 7, 8 For gastroesophageal adenocarcinoma (GEC), MET amplification and Met protein overexpression are found in 4% to 10% and 25% to 70% of GEC cases, respectively; the rates depend on the definition of positive, and the cohort studied.9, 10, 11, 12, 13, 14, 15, 16 As with other malignancy types,17, 18 patients with Met‐positive GEC (as defined by various criteria) reportedly have a worse prognoses.10, 12, 15, 16, 19, 20, 21 Because of the clinical impact and relevance of Met, the diagnostic determination of its expression and/or mutational/amplification status can provide physicians with critical information for understanding a patient's prognosis, identifying clinical trials for which the patient may be a candidate, and, importantly, making a determination about whether the patient may benefit from Met‐targeted therapies.
This study sought to further evaluate Met's prognostic potential with a large, multi‐institutional, clinically linked cohort of GEC patients and to compare various diagnostic platforms for identifying Met‐positive tumors. Fluorescence in situ hybridization (FISH) and immunohistochemistry (IHC) were used to examine the frequency of MET gene amplification and Met protein overexpression, respectively. Because of the inherent limitations of FISH (time‐consuming, labor‐intensive, subjective scoring) and IHC (variability in tissue staining, lack of intrasample reproducibility over time, and subjective scoring), mass spectrometry–based selected reaction monitoring (SRM)11, 22 was also used to objectively quantitate Met protein expression.11 The relation between the MET/Met status and overall survival (OS) was assessed with all 3 approaches in both univariate and multivariate analyses, with adjustments made for known prognostic covariates of GEC. In addition, the data from MET FISH, Met IHC, and Met SRM were compared for intermethod agreement.
MATERIALS AND METHODS
Patients and Samples
Formalin‐fixed, paraffin‐embedded GEC samples were obtained from the University of Chicago (Chicago, Ill) or the University of Urbino (Urbino, Italy) from 2000 to 2015. Samples were collected/annotated under institutional review board–approved protocols. The median follow‐up time was 113.0 months (80% confidence interval [CI], 99.3‐126.3 months); the median follow‐up times for patients with and without curative‐intent surgery were 115.3 months (80% CI, 99.3‐129.0 months) and 31.6 months (80% CI, 14.5‐31.6 months), respectively. The clinical/pathological characteristics of the samples are included in Supporting Tables 1 and 2 (see online supporting information).
MET Analysis
A dual‐color MET FISH assay was conducted with the MET IQFISH probe with centromere enumeration probe for chromosome 7 (CEP7; Agilent Technologies, Santa Clara, Calif) and a histology FISH accessory kit (Dako North America, Carpinteria, Calif); scoring was performed as previously described.11, 23, 24 An average MET gene copy number (GCN) ≥ 5, a MET/CEP7 ratio ≥ 2, and ≥10% of counted tumor nuclei with ≥15 gene copies were the 3 amplification scoring criteria considered positive, and each assessed independently.24, 25
Met IHC was performed with a horseradish peroxidase–labeled, dextrose‐based polymer complex bound to a secondary antibody (Dako North America) as previously described.21, 26 The intensity of cell membrane staining, scored by an experienced pathologist (0, none; 1, low; 2, intermediate; or 3, high), along with the percentage of tumor cells (extensity) for each sample was documented.11, 21 The membrane max intensity parameter of a given tumor was obtained from the highest IHC score that had a nonzero percentage of staining. For example, if a tumor had a score breakdown of 10%, 65%, 25%, and 0% (for scores of 0, 1, 2, and 3, respectively), the membrane max intensity was 2. The predominant score parameter was obtained from the highest percentage‐positive score; in the provided example, this was 1. IHC was defined as positive if ≥25% of the tumor cell membranes were stained with an intensity ≥ 1+; a second analysis was also performed at a higher extensity (≥50%) of cells with an intensity ≥ 1+.21, 26 The pathologists performing FISH and IHC were blinded to the results of previously performed assays and clinical outcomes.
For Met protein quantification by mass spectrometry–based SRM, tissue sections (10 μm) were prepared as previously described.11, 22, 27 This tumor tissue was solubilized with Liquid Tissue, and the resulting tryptic peptides were analyzed with a TSQ Vantage triple‐quadrupole mass spectrometer (Thermo Fisher Scientific, San Jose, Calif) equipped with a nanoACQUITY LC system (Waters, Milford, Mass). The SRM conditions have been described previously.11, 22
Statistical Methods
Spearman's rank correlation coefficient was used to assess correlations between parameters. Cox proportional hazards models and Kaplan‐Meier estimates were applied to explore relations between Met, OS, and other clinical and pathologic characteristics. A multivariate analysis was performed for the Met biomarker status (amplification by FISH, expression by IHC, and expression by Met SRM) and outcomes after adjustments for baseline covariates, including age, sex, race, histology, biopsy, diagnosis, stage, lesion status, curative‐intent surgery, and perioperative therapy.
RESULTS
There were 447 samples available: 344 (77.0%) were evaluable by MET FISH, 332 (74.3%) were evaluable by Met IHC, and 282 (63.1%) were evaluable by Met SRM (Table 1). Intermethod correlations were performed with tumors evaluable by more than 1 approach (discussed in further detail later). A comparison of single‐method subsets by patient demographic and clinical characteristics indicated a balanced representation of each subset (Supporting Tables 1 and 2 [see online supporting information]).
Table 1.
Number of Gastroesophageal Adenocarcinoma Tumor Samples Evaluated for MET/MET by the Method of Assessment
| Method | Samples | Samples With Outcome Data |
|---|---|---|
| FISH | 344 | 338 |
| FISH + IHC | 304 | |
| FISH + SRM | 255 | |
| IHC | 332 | 324 |
| IHC + SRM | 229 | |
| SRM | 282 | 281 |
Abbreviations: FISH, fluorescence in situ hybridization; IHC, immunohistochemistry; SRM, selected reaction monitoring.
Samples were analyzed for MET amplification by FISH and/or for Met protein expression by IHC or SRM; the quantities for each individual subgroup as well as paired evaluation subgroups are presented in the middle column. The quantity of each individual subgroup with corresponding outcome data is presented in the far right column.
When MET gene amplification was defined as a MET/CEP7 ratio ≥ 2 per cell,11 16 of 344 tumors (4.7%) were MET‐amplified (Supporting Table 3 [see online supporting information]). When it was scored as the percentage of tumor cells with a mean MET GCN ≥ 5,12 18 tumors (5.2%) were amplified. When it was defined as ≥10% of tumor cells having at least 15 copies of MET, 9 tumors (2.6%) were amplified. These results were similar to previously published rates of MET gene amplification in GEC.9, 10, 11, 13, 26
Regardless of the definition of amplification, MET FISH was an indicator of a poor prognosis in univariate analyses. Amplification‐positive patients exhibited an increased risk of death (Fig. 1A), and the median OS of unamplified patients was much longer than the median OS of patients who had a MET/CEP7 ratio ≥ 2 (17.8 vs 68.7 months; P < .0001; Fig. 1B), patients who had a mean MET GCN ≥ 5 (28.4 vs 68.7 months; P < .0007; Fig. 1C), or patients for whom ≥10% of tumor cells had ≥15 copies of MET (17.8 vs 59.4 months; P < .0001; Fig. 1D). Using the MET/CEP7 ratio and adjusting for numerous baseline covariates (age, sex, race, histology, biopsy, diagnosis, stage, lesion status, curative‐intent surgery, and perioperative therapy), we found that MET amplification (FISH ratio ≥ 2) was independently prognostic of OS (hazard ratio [HR], 2.55; 95% CI, 1.27‐5.09; P < .008) in comparison with a ratio < 2 (Fig. 2).
Figure 1.
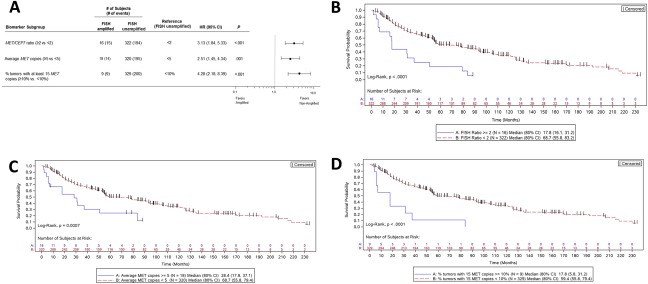
Patient risk and overall survival as assessed by the MET FISH status. (A) Cox proportional hazards model evaluation of FISH classification with respect to overall survival. HRs along with 95% CIs are depicted for MET‐amplified tumors versus unamplified tumors as characterized by 3 separate criteria. (B‐D) Kaplan‐Meier curves depicting the overall survival of subjects with MET‐amplified tumors versus unamplified tumors as characterized by (B) the MET/CEP7 ratio, (C) the average gene copy number, and (D) the percentage of tumor cells containing at least 15 copies of MET. CEP7 indicates centromere enumeration probe for chromosome 7; CI, confidence interval; FISH, fluorescence in situ hybridization; HR, hazard ratio.
Figure 2.
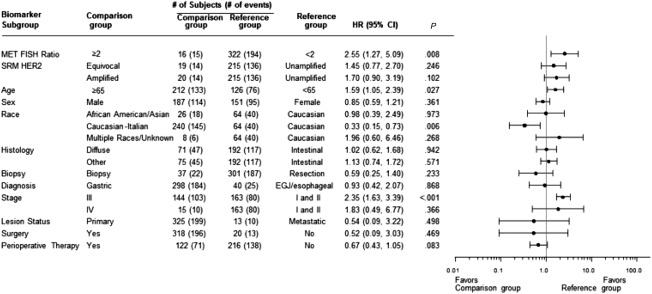
METamplification is an independent indicator of overall survival: Cox proportional hazards model evaluation of the FISH classification with respect to overall survival. A multivariate analysis was performed, and the MET/CEP7 ratio was used as the approach to FISH characterization. HRs along with 95% CIs for each baseline covariate are depicted. CEP7 indicates centromere enumeration probe for chromosome 7; CI, confidence interval; EGJ, esophagogastric junction; FISH, fluorescence in situ hybridization; HER2, human epidermal growth factor receptor 2; HR, hazard ratio; SRM, selected reaction monitoring.
According to IHC, 273 of 332 samples (82.2%) exhibited Met expression at any intensity (intensity score ≥ 1+) and any extensity level (percentage of tumor cells positive; Supporting Table 4 [see online supporting information]). When Met IHC positivity (combined percentages of cells that were positive for Met expression at 1+, 2+, and 3+) was slightly more restrictively defined as any staining intensity ≥ 1 + in either ≥ 25% or ≥ 50% of tumor cells,11, 21, 26 117 samples (35.2%) and 43 samples (13.0%), respectively, were scored as positive.
Increased expression of Met protein according to IHC was also indicative of a poor prognosis for GEC in a univariate analysis. Met IHC–positive patients exhibited a worse prognosis, and there was a general upward trend in the HR with increasing IHC extensity staining (Fig. 3A). Patients whose tumors had ≥25% positive membrane staining at an intensity ≥ 1 + (predefined cutoffs) experienced shorter OS than patients with lower staining levels (54.2 vs 78.4 months; HR, 1.39; 95% CI, 1.04‐1.86, P = .025). The survival difference between patients at the ≥50% positive membrane staining extensity criterion was slightly greater (43.2 vs 78.2 months; HR, 1.62; 95% CI, 1.10‐2.28, P = .013; Fig. 3B,C). However, a multivariate analysis adjusted for known prognostic covariates indicated that Met IHC, according to these 2 definitions of positivity, was not an independent marker of prognosis (P = .36; Fig. 3D).
Figure 3.
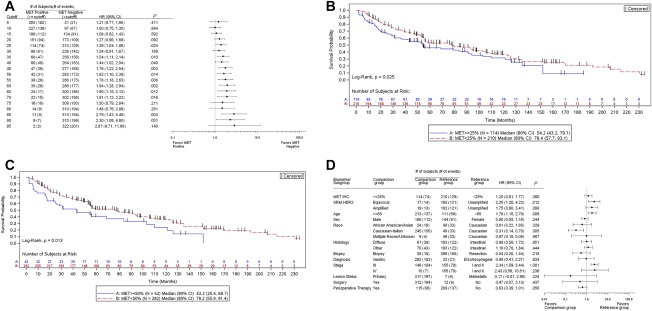
Patient risk and overall survival as assessed by Met IHC expression. (A) Cox proportional hazards model evaluation of IHC classification with respect to overall survival. HRs along with 95% CIs comparing Met‐positive and Met‐negative patients at each cutoff value are depicted. (B,C) Kaplan‐Meier curves depicting the overall survival of the subjects by the Met IHC status with cutoffs of (B) 25% and (C) 50%. (D) Cox proportional hazards model evaluation of the IHC status with respect to overall survival. A multivariate analysis was performed, and 25% staining was used as the Met‐positive cutoff. HRs along with 95% CIs for each baseline covariate are depicted. CI indicates confidence interval; EGJ, esophagogastric junction; HER2, human epidermal growth factor receptor 2; HR, hazard ratio; IHC, immunohistochemistry; SRM, selected reaction monitoring.
According to mass spectrometry–based SRM, 231 of 282 samples (81.9%) had Met levels below the lower limit of quantitation of 200 amol/μg (Supporting Table 5 [see online supporting information]). Protein concentrations in the remaining 51 samples (18.1%) ranged from 200 to 3245.5 amol/μg.11, 27 Using expression levels previously established in an independent cohort11 and minimum P value and hazard modeling, we tested and established 400 amol/μg as the cutoff for Met‐positive expression because this value was determined to have the most significant effect on OS (Fig. 4A). As such, 22 tumors (7.8%) were classified as Met‐positive in this study. The median OS for patients above this cutoff was significantly shorter than the median OS for their Met‐negative counterparts (22.6 vs 59.4 months; HR 1.76 (95% CI 1.06‐2.9); p = 0.0258; P = .0258; Fig. 4B). After adjustments for covariates, patients with Met expression levels ≥ 400 amol/μg exhibited a significantly higher risk of death (HR, 1.85; 95% CI, 1.04‐3.30; Fig. 4C).
Figure 4.
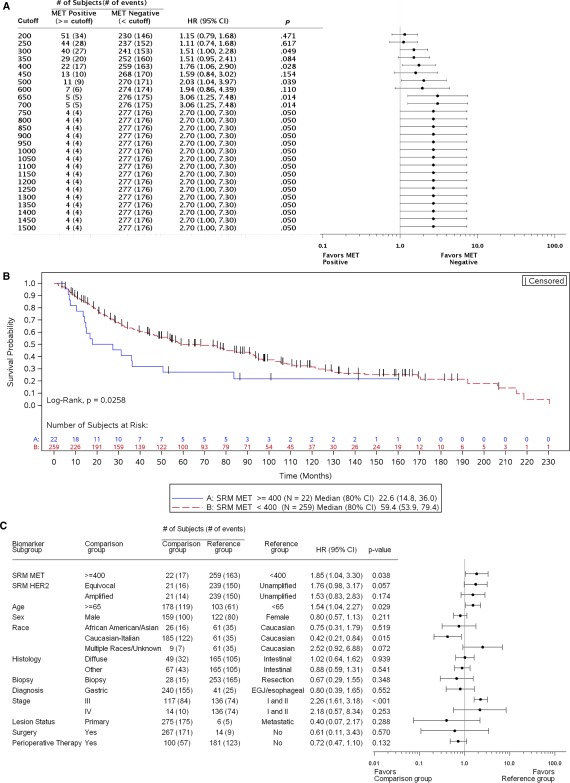
Patient risk and overall survival as assessed by Met SRM expression. (A) Cox proportional hazards model evaluation of Met SRM quantitation with respect to overall survival. HRs along with 95% CIs comparing Met‐positive and Met‐negative patients at each cutoff value are depicted. The number of MET‐amplified tumors present within each Met‐positive cutoff group is presented. (B) Kaplan‐Meier curves depicting the overall survival of subjects by Met SRM expression with 400 amol/μg as the Met‐positive cutoff. (C) Cox proportional hazards model evaluation of Met SRM expression with respect to overall survival. A multivariate analysis was performed, and 400 amol/μg was used as the Met‐positive cutoff. HRs along with 95% CIs for each baseline covariate are depicted. CI indicates confidence interval; HER2, human epidermal growth factor receptor 2; HR, hazard ratio; SRM, selected reaction monitoring
FISH and SRM results were assessed for correlations with the clinicopathological variables listed in Supporting Table 1 (see online supporting information). FISH amplification was associated with an esophageal primary location (16.3%) versus a distal gastric location (3.7%), and a Met SRM level ≥ 400 amol/μg was associated with a higher tumor stage (III/IV; see online supporting information). There were no other statistically significant correlations observed.
FISH, IHC, and SRM results were also assessed for cross‐method sensitivity and specificity; we used subsets of tumor samples that were analyzed by multiple methods (Table 1). With a MET/CEP7 ratio ≥ 2 as the reference, Met IHC at the ≥25% staining cutoff was 80% sensitive (12 of 15 FISH‐positive samples were identified) and 69.2% specific (200 of 289 negative samples were identified) for MET amplification by FISH (Fig. 5A), and there was a significant correlation (P < .0001). With SRM at ≥ 400 amol/μg as the reference, Met IHC exhibited 68.8% sensitivity (11 of 16 Met SRM–positive samples were identified) with 68.5% specificity (146 of 213 Met SRM–negative samples; P = .002; Fig. 5B). Finally, in comparison with a MET/CEP7 ratio ≥ 2, SRM (≥400 amol/μg) identified 5 of 13 MET‐amplified tumors (38.5% sensitivity) and 230 of 242 unamplified tumors (95.0% specificity) with a significant correlation between the 2 methods (P < .001; Fig. 5C). Thus, although both Met IHC and Met SRM correlated with MET FISH, they were relatively insensitive for discerning the MET FISH amplification status in this study. In addition, although a very limited subset was used, applying a previously defined cutoff of ≥1500 amol/μg11 resulted in a Met SRM specificity of 100% for identifying MET amplification (no negative FISH samples were identified within this high Met‐positive SRM cutoff).
Figure 5.
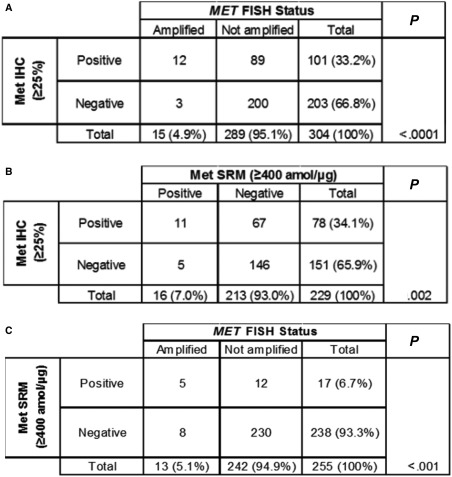
Correlation of MET/MET analytical approaches. MET gene amplification results were compared with (A) Met IHC and (C) Met SRM cutoff–derived protein expression; (B) the correlation between Met IHC and Met SRM was also examined. FISH indicates fluorescence in situ hybridization; IHC, immunohistochemistry; SRM, selected reaction monitoring.
DISCUSSION
This study evaluated Met as a prognostic biomarker for GEC by contrasting the technical methods of FISH, IHC, and SRM and by assessing various criteria for defining genomic or proteomic positivity. Moreover, an association of each of these methods/definitions with clinical outcomes was evaluated via univariate and covariate adjusted analyses. The findings are consistent with some smaller studies demonstrating similar concordance patterns between Met SRM and IHC and/or FISH11 and comparable adverse prognoses in univariate analyses when the Met biomarker (FISH, IHC, or SRM) was positive.10, 12, 15, 21 Notably, the findings are unique in demonstrating an independently worse prognosis for amplification as determined by the FISH ratio and overexpression by SRM according to an adjusted multivariate analysis; in contrast, IHC with the currently applied positivity criteria was not an independent prognostic biomarker.26
Numerous complexities contribute to determining the strength of Met as a predictive and/or prognostic biomarker; these can often be associated with a lack of consensus on a diagnostic assay (FISH, next‐generation sequencing, IHC, and SRM) and varying reported incidence rates of (and thresholds for) Met positivity (Supporting Table 6 [see online supporting information]). In this study, we assessed the prognostic value of MET amplification by FISH and Met expression by IHC and SRM without consideration of their predictive value for the benefit of anti‐Met therapy or of other emerging prognostic/predictive Met biomarkers, including MET mutations and/or exon 14 skipping, HGF aberrations, and Met aberrations within circulating tumor cells or circulating tumor DNA.5, 6, 12, 28, 29, 30, 31, 32, 33, 41, 42, 43, 44
Generally, MET amplification denotes an increased GCN, is considered a genomic driver of the tumor, and results in extreme overexpression of the Met protein.11, 14 However, with respect to amplification and consequent overexpression, the degree of amplification is important to consider: low‐level amplification (eg, MET/CEP7 ratio = 3:1.5 = 2) generally does not correlate with extreme overexpression of the protein (Supporting Table 6 [see online supporting information]). This was previously demonstrated with the FISH ratio and SRM expression both evaluated as linear variables in comparison with a binary FISH ratio ≥ 2.11 This is also quite analogous to human epidermal growth factor receptor 2 (HER2) amplification, as recently described.27 Therefore, the ease of categorizing into binary subsets (ratio ≥ or < 2) should be weighed against the weaker observed correlation with expression when higher and lower FISH ratios are combined within 1 category. This may have implications for the strength of FISH as a prognostic and predictive biomarker. Ultimately, targeted therapies inhibit proteins, so excluding patients with gene amplification (low‐level) who lack resulting overexpression may enrich the cohort most likely to benefit. Regardless, the rates of MET amplification in this study with a FISH ratio ≥ 2 (4.7%), a FISH GCN ≥ 5 (5.2%), and a FISH GCN ≥ 15 in > 10% of cells (2.6%) were consistent with rates previously reported,9, 10, 11, 13, 15 and they were the poorest prognostic factors among the MET biomarkers evaluated. This is intuitive because when it is amplified, MET is the driver oncogene portending significant metastatic and aggressive tumor behavior. In this setting, Met is exceptionally overexpressed (several‐fold higher than Met‐expressing tumors in the absence of gene amplification).11 Although a lower SRM cutoff of ≥400 amol/μg (set to define overexpression and not amplification) had low accuracy in discerning MET amplification by FISH, the predefined level of ≥1500 amol/μg did show 100% specificity (in a small cohort analyzed with both FISH and SRM analysis). This level of ≥ 1500 amol/μg has consistently demonstrated excellent accuracy in identifying truly cluster MET–amplified tumors.11 However, when heterogeneous MET amplification was present within a sample, FISH identified the amplified areas, whereas SRM represented the aggregate Met expression level of the entire tumor; this resulted in lower than expected expression levels of a homogeneously amplified tumor (Supporting Fig. 1 [see online supporting information]).11
In contrast to amplification, Met protein overexpression determined by IHC or SRM includes the small subset with overexpression due to gene amplification but consists mainly of expression without amplification. The incidence of Met overexpression depends on the lower limit set for positivity. Indeed, the rate reportedly varies widely from 24% to 70%, in part because of the variability of this lower limit along with other analytic variables associated with IHC, the assay typically used for Met expression. As previously described, Met SRM is more objective than IHC and correlates better with FISH ratio and GCN values.11 This was confirmed in this study: Met overexpression, as determined by SRM, accounted for 18.1% of the samples at any level above the lower limit of detection (≥200 amol/μg) and for 7.8% of the samples at the predefined threshold of ≥400 amol/μg. In contrast, 82.2% of the samples in this study demonstrated any positivity by IHC, and 35.2% and 13% demonstrated any positivity by any staining intensity (intensity ≥ 1 + in ≥ 25% and ≥50% of tumor cells, respectively). Interestingly, a recent phase 3 trial, RILOMET‐1, reported a Met positivity rate of 81% versus a phase 2 rate of 64% with the Dako IHC criteria (intensity ≥ 1 + with extensity ≥ 25%); this suggests interstudy variability even with the same IHC antibody and scoring.21, 26 In addition to observed variability between the 2 rilotumumab trials using the Dako antibody, it is also possible that the rate of Met positivity differs between locally advanced disease and metastatic disease and between primary tumor and metastatic biopsies.
In this study, although IHC was associated with a poor prognosis in the univariate analysis, this did not persist after adjustments for other prognostic covariates. The low‐level expression of Met may be associated with these poor prognostic covariates yet not be independently associated with outcomes upon adjustments. In contrast, extreme overexpression as a result of MET amplification or by SRM (determined here to be ≥400 amol/μg) in the absence of MET amplification remained prognostic after adjustments for other variables.
Finally, Met may be a negative predictive biomarker for standard cytotoxic therapies15, 34 and a positive predictive biomarker for Met‐directed therapies, particularly when one is selecting for MET amplification.13, 14, 15, 35, 36 However, all trials to date selecting for MET amplification have been open‐label, single‐arm trials, undoubtedly because of the low incidence of MET amplification, aggressive tumors with a quickly progressing clinical course, and difficulty in accruing cases for proper randomization. Because of the putative negative prognosis rendered by MET amplification, single‐arm trials may underestimate the benefit of anti‐Met therapies if this overall poor baseline prognosis is not considered. On the other hand, despite early evidence for the predictive value of anti‐Met therapy for tumors that overexpress Met (regardless of the GCN/amplification status),21, 23 2 recent phase 3 trials, RILOMET‐1 (with the anti‐HGF antibody rilotumumab) and METGastric (with the anti‐Met antibody onartuzumab), both failed to meet their primary endpoints in Met‐positive patients as determined by 2 different IHC methods.26, 37 Although it is quite plausible that Met inhibition, in the setting of Met overexpression lacking amplification, is not sufficient to improve outcomes (ie, a negative prognostic biomarker is not by default also a positive predictive biomarker for a targeted therapy), it is also possible that the positivity criteria were too lenient. The phase 1 patient who responded to onartuzumab monotherapy23 was later found to have an expression level of 526.93 amol/μg.11 Indeed, the positivity rate in RILOMET‐1 (≥1 + intensity, ≥ 25% extensity) was 81%, with only 21% of the patients (71 patients on rilotumumab vs 57 patients on a placebo) having higher level expression (≥2 + intensity, ≥ 50% extensity).26 In fact, in the METGastric trial, a predefined subset analysis (IHC: ≥ 2 + intensity, ≥ 50% extensity) suggested improved OS with an HR of 0.64 (P = .06) with 105 and 109 patients in the onartuzumab and control arms, respectively, or 38% of all enrolled patients. Because the trial closed early with only 70% of the intended accrual (562 of 800), the intended power to detect an HR of 0.49 in the higher expressing subset was compromised; moreover, if the more likely true HR is 0.6 to 0.8, the trial was significantly underpowered to detect this true benefit. Unfortunately, because METGastric and even more so RILOMET‐1 did not have sufficient power to evaluate the benefit in those samples/patients with higher expression cutoffs, they do not provide guidance for this select patient population. Coupling these results with the inherent limitations associated with IHC (antigen dependency, variability in tissue staining, lack of temporal reproducibility, and subjective scoring of positive staining) suggests that the technique may not be a definitive method for identifying patients likely to respond to Met‐targeted therapy. Regardless, it is clear that low‐level Met expression or, worse, treatment of all‐comers with no selection does not predict a benefit from anti‐Met agents.26, 37, 38
In summary, MET FISH and Met SRM were independently associated with a poor prognosis in this large cohort of GEC patients, whereas Met IHC at the currently applied positivity cutoffs was not independently associated with outcomes. SRM is a novel technology demonstrating clinical utility with Met and Her211, 27, 39, 45 and additionally has the advantage of multiplex peptide analysis,22 which is analogous to next‐generation sequencing for genomic aberrations. The results of this study suggest that amplification by FISH and/or overexpression by SRM should be incorporated into multivariate models when one is assessing the prognostic significance of other novel covariates. Moreover, amplification and overexpression, as determined by FISH and SRM, may better direct treatment and, in particular, Met‐targeted therapies for GEC principally in the setting of next‐generation clinical trial designs.40
FUNDING SUPPORT
This work was supported by the National Institutes of Health (K12 award CA139160‐01A and K23 award CA178203‐01A1), the University of Chicago Comprehensive Cancer Center Award in Precision Oncology (Cancer Center Support Grant P30 CA014599), the Cancer Research Foundation Young Investigator Award, the Alliance for Clinical Trials in Oncology Foundation Young Investigator Award, an Amgen collaborative research agreement, an OncoPlexDx collaborative research agreement, the Live Like Katie Foundation Award, and the Sal Ferrara II Fund for Pangea (to Daniel V. T. Catenacci).
CONFLICT OF INTEREST DISCLOSURES
Daniel V. T. Catenacci received honoraria for advisory boards/consulting from Amgen, Genentech/Roche, Lilly Oncology, and OncoPlexDx/NantOmics and research funding from Amgen, Genentech, and OncoPlexDx/NantOmics. Agnes Ang, Jing Shen, and Robert D. Loberg are Amgen employees. Wei‐Li Liao, Fabiola Cecchi, and Todd Hembrough are OncoPlexDx/NantOmics employees. Todd Hembrough also has a patent pending for MET selected reaction monitoring.
AUTHOR CONTRIBUTIONS
Daniel V. T. Catenacci: Study conception and design, clinical trial and translational statistical design, sample collection, assay performance, data management, data interpretation, and manuscript writing and editing. Agnes Ang: Study conception and design, sample collection, assay performance, data management, data interpretation, and manuscript writing and editing. Wei‐Li Liao: Sample collection, assay performance, data management, and manuscript writing and editing. Jing Shen: Clinical trial and translational statistical design and manuscript writing and editing. Emily O'Day: Sample collection, assay performance, data management, and manuscript writing and editing. Robert D. Loberg: Data interpretation and manuscript writing and editing. Fabiola Cecchi: Manuscript writing and editing. Todd Hembrough: Study conception and design, data interpretation, and manuscript writing and editing. Annamaria Ruzzo: sample collection, assay performance, data management, and manuscript writing and editing. Francesco Graziano: Study conception and design, data interpretation, and manuscript writing and editing.
Supporting information
Additional supporting information may be found in the online version of this article.
Supporting Information
Supporting Information
This study was presented in part at poster sessions of the 2015 American Society of Clinical Oncology Annual Meeting (Chicago, IL) and the 2015 European Cancer Congress (Vienna, Austria).
We acknowledge Ellen Wertheimer and Christopher Parsons for their assistance with the preparation and review of this article. Daniel V. T. Catenacci also thanks the Live Like Katie Foundation and the Sal Ferrara II Fund, without which this work would not have been possible.
REFERENCES
- 1. Gherardi E, Birchmeier W, Birchmeier C, et al. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12:89–103. [DOI] [PubMed] [Google Scholar]
- 2. Petrini I. Biology of MET: a double life between normal tissue repair and tumor progression. Ann Transl Med. 2015;3:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cecchi F, Rabe DC, Bottaro DP. Targeting the HGF/Met signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:553–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834–848. [DOI] [PubMed] [Google Scholar]
- 5. Awad MM, Oxnard GR, Jackman DM, et al. MET exon 14 mutations in non–small‐cell lung cancer are associated with advanced age and stage‐dependent MET genomic amplification and c‐Met overexpression. J Clin Oncol. 2016;34:721–730. [DOI] [PubMed] [Google Scholar]
- 6. Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5:842–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Piguet AC, Medova M, Keogh A, et al. Impact of MET targeting on tumor‐associated angiogenesis and growth of MET mutations–driven models of liver cancer. Genes Cancer. 2015;6:317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sacco JJ, Clague MJ. Dysregulation of the Met pathway in non–small cell lung cancer: implications for drug targeting and resistance. Transl Lung Cancer Res. 2015;4:242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bass AJ, Thorsson V, Shmulevich I, et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Catenacci DV, Cervantes G, Yala S, et al. RON (MST1R) is a novel prognostic marker and therapeutic target for gastroesophageal adenocarcinoma. Cancer Biol Ther. 2011;12:9–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Catenacci DV, Liao WL, Thyparambil S, et al. Absolute quantitation of Met using mass spectrometry for clinical application: assay precision, stability, and correlation with MET gene amplification in FFPE tumor tissue. PLoS One. 2014;9:e100586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Graziano F, Galluccio N, Lorenzini P, et al. Genetic activation of the MET pathway and prognosis of patients with high‐risk, radically resected gastric cancer. J Clin Oncol. 2011;29:4789–4795. [DOI] [PubMed] [Google Scholar]
- 13. Jardim DL, Tang C, Gagliato Dde M, et al. Analysis of 1,115 patients tested for MET amplification and therapy response in the MD Anderson Phase I Clinic. Clin Cancer Res. 2014;20:6336–6345. [DOI] [PubMed] [Google Scholar]
- 14. Smolen GA, Sordella R, Muir B, et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA‐665752. Proc Natl Acad Sci U S A. 2006;103:2316–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lennerz JK, Kwak EL, Ackerman A, et al. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J Clin Oncol. 2011;29:4803–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Metzger ML, Behrens HM, Boger C, et al. MET in gastric cancer—discarding a 10% cutoff rule. Histopathology. 2016;68:241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goyal L, Muzumdar MD, Zhu AX. Targeting the HGF/c‐MET pathway in hepatocellular carcinoma. Clin Cancer Res. 2013;19:2310–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Raghav KP, Wang W, Liu S, et al. cMET and phospho‐cMET protein levels in breast cancers and survival outcomes. Clin Cancer Res. 2012;18:2269–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hack SP, Bruey JM, Koeppen H. HGF/MET‐directed therapeutics in gastroesophageal cancer: a review of clinical and biomarker development. Oncotarget. 2014;5:2866–2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu S, Yu Y, Zhao N, et al. C‐Met as a prognostic marker in gastric cancer: a systematic review and meta‐analysis. PLoS One. 2013;8:e79137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Iveson T, Donehower RC, Davidenko I, et al. Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first‐line treatment for gastric or oesophagogastric junction adenocarcinoma: an open‐label, dose de‐escalation phase 1b study and a double‐blind, randomised phase 2 study. Lancet Oncol. 2014;15:1007–1018. [DOI] [PubMed] [Google Scholar]
- 22. Hembrough T, Thyparambil S, Liao WL, et al. Application of selected reaction monitoring for multiplex quantification of clinically validated biomarkers in formalin‐fixed, paraffin‐embedded tumor tissue. J Mol Diagn. 2013;15:454–465. [DOI] [PubMed] [Google Scholar]
- 23. Catenacci DV, Henderson L, Xiao SY, et al. Durable complete response of metastatic gastric cancer with anti‐Met therapy followed by resistance at recurrence. Cancer Discov. 2011;1:573–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Catenacci DVT, Tang R, Oliner KS, et al. MET as a prognostic biomarker of survival in a large cohort of patients with gastroesophageal cancer (GEC) [abstract 4034]. J Clin Oncol. 2015;33(suppl):4034. [Google Scholar]
- 25. Cappuzzo F, Marchetti A, Skokan M, et al. Increased MET gene copy number negatively affects survival of surgically resected non–small‐cell lung cancer patients. J Clin Oncol. 2009;27:1667–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cunningham D, Tebbutt NC, Davidenko I, et al. Phase III, randomized, double‐blind, multicenter, placebo (P)‐controlled trial of rilotumumab (R) plus epirubicin, cisplatin and capecitabine (ECX) as first‐line therapy in patients (pts) with advanced MET‐positive (pos) gastric or gastroesophageal junction (G/GEJ) cancer: RILOMET‐1 study [abstract 4000]. J Clin Oncol. 2015;33(suppl):4000. [Google Scholar]
- 27. Catenacci DV, Liao WL, Zhao L, et al. Mass‐spectrometry–based quantitation of Her2 in gastroesophageal tumor tissue: comparison to IHC and FISH. Gastric Cancer. 2016;19:1066–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Asaoka Y, Tada M, Ikenoue T, et al. Gastric cancer cell line Hs746T harbors a splice site mutation of c‐Met causing juxtamembrane domain deletion. Biochem Biophys Res Commun. 2010;394:1042–1046. [DOI] [PubMed] [Google Scholar]
- 29. Frampton GM, Ali SM, Rosenzweig M, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5:850–859. [DOI] [PubMed] [Google Scholar]
- 30. Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee JH, Han SU, Cho H, et al. A novel germ line juxtamembrane Met mutation in human gastric cancer. Oncogene. 2000;19:4947–4953. [DOI] [PubMed] [Google Scholar]
- 32. Penuel E, Li C, Parab V, et al. HGF as a circulating biomarker of onartuzumab treatment in patients with advanced solid tumors. Mol Cancer Ther. 2013;12:1122–1130. [DOI] [PubMed] [Google Scholar]
- 33. Wallenius V, Hisaoka M, Helou K, et al. Overexpression of the hepatocyte growth factor (HGF) receptor (Met) and presence of a truncated and activated intracellular HGF receptor fragment in locally aggressive/malignant human musculoskeletal tumors. Am J Pathol. 2000;156:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nakajima M, Sawada H, Yamada Y, et al. The prognostic significance of amplification and overexpression of c‐met and c‐erb B‐2 in human gastric carcinomas. Cancer. 1999;85:1894–1902. [DOI] [PubMed] [Google Scholar]
- 35. Kang YK, LoRusso P, Salgia R, et al. Phase I study of ABT‐700, an anti–c‐Met antibody, in patients (pts) with advanced gastric or esophageal cancer (GEC) [abstract 167]. J Clin Oncol. 2015;33(suppl 3):167. [Google Scholar]
- 36. Kwak EL, LoRusso P, Hamid O, et al. Clinical activity of AMG 337, an oral MET kinase inhibitor, in adult patients (pts) with MET‐amplified gastroesophageal junction (GEJ), gastric (G), or esophageal (E) cancer [abstract 1]. J Clin Oncol. 2015;33(suppl 3):1. 25332246 [Google Scholar]
- 37. Shah MA, Bang YJ, Lordick F, et al. METGastric: a phase III study of onartuzumab plus mFOLFOX6 in patients with metastatic HER2‐negative (HER2‐) and MET‐positive (MET+) adenocarcinoma of the stomach or gastroesophageal junction (GEC) [abstract 4012]. J Clin Oncol. 2015;33(suppl):4012. [Google Scholar]
- 38. Shah MA, Wainberg ZA, Catenacci DV, et al. Phase II study evaluating 2 dosing schedules of oral foretinib (GSK1363089), cMET/VEGFR2 inhibitor, in patients with metastatic gastric cancer. PLoS One. 2013;8:e54014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nuciforo P, Thyparambil S, Aura C, et al. High HER2 protein levels correlate with increased survival in breast cancer patients treated with anti‐HER2 therapy. Mol Oncol. 2016;10:138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Catenacci DV. Next‐generation clinical trials: novel strategies to address the challenge of tumor molecular heterogeneity. Mol Oncol. 2015;9:967–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bardelli A, Corso S, Bertotti A, et al. Amplification of the MET receptor drives resistance to anti‐EGFR therapies in colorectal cancer. Cancer Discov. 2013;3:658–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu J, McCleland M, Stawiski EW, et al. Integrated exome and transcriptome sequencing reveals ZAK isoform usage in gastric cancer. Nat Commun. 2014;5:3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Choi J, Lee HE, Kim MA, et al. Analysis of MET mRNA expression in gastric cancers using RNA in situ hybridization assay: its clinical implication and comparison with immunohistochemistry and silver in situ hybridization. PLoS One. 2014;9:e111658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang F, Flanagan J, Su N, et al. RNAscope: a novel in situ RNA analysis platform for formalin‐fixed, paraffin‐embedded tissues. J Mol Diagn. 2012;14:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. An E, Ock CY, Kim TY, Lee KH, Han SW, Im SA, Kim TY, Liao WL, Cecchi F, Blackler A, Thyparambil S, Kim WH, Burrows J, Hembrough T, Catenacci D, Oh DY, Bang YJ, et al. Quantitative proteomic analysis of HER2 expression in the selection of gastric cancer patients for trastuzumab treatment. Ann Oncol. 2016. Sep 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article.
Supporting Information
Supporting Information