

Abstract
Posttranslational modification—oligomerization, phosphorylation, and proteolytic cleavage—of the human papillomavirus (HPV) E4 protein occurs as the infected keratinocytes migrate up through the suprabasal wart layers. It has been postulated that these events modify E4 function during the virus life cycle. In HPV type 1 (HPV1)-induced warts, N-terminal sequences are progressively cleaved from the full-length E4 protein (E1∧E4) of 17 kDa to produce a series of polypeptides of 16, 11 and 10 kDa. Here, we have shown that in human keratinocytes, a truncated protein (E4-16K), equivalent to the 16-kDa species, mediated a G2 arrest in the cell cycle that was dependent on a threonine amino acid in a proline-rich domain of the protein. Reconstitution of cyclin B1 expression in E4-16K cells reversed the G2 arrest. Expression of E4-16K also induced chromosomal rereplication, and this was associated with aberrant nuclear morphology. Perturbation of the mitotic cell cycle was a biological activity specific to the truncated protein. However, coexpression of the full-length E1∧E4 protein and the truncated E4-16K protein inhibited normal cellular proliferation and cellular DNA rereplication but did not prevent cells from arresting in G2. Our findings provide the first evidence to support the hypothesis that proteolytic cleavage of the E1∧E4 protein modifies its function. Also, different forms of the HPV1 E4 protein cooperate to negatively influence keratinocyte proliferation. We predict that these distinct biological activities of E4 act to support efficient amplification of the viral genome in suprabasal keratinocytes.
Human papillomaviruses (HPVs) are a large group (>200 types) of small DNA viruses that infect cutaneous and mucosal squamous epithelium at a variety of anatomical sites. While the majority of HPV types induce benign hyperproliferative lesions that eventually regress, a small number of virus types can induce the malignant transformation of infected tissue (for a review, see reference 14).
The complete replicative cycle of HPVs is tightly linked to the terminal differentiation program of the keratinocyte. Viral DNA maintenance and replication occur along with cellular chromosomal replication in proliferating basal cells, ensuring that both parent and daughter cells maintain a constant number of viral genomes. As cells migrate up through the epithelium, they undergo a complex pattern of differentiation. Concomitant with cellular differentiation in virally infected cells are the vegetative amplification of the viral genome, the expression of virus capsid proteins (L1 and L2), and finally the assembly of progeny virus. The HPV E4 protein is detected in productively infected cells. The E4 protein is translated from a spliced E1∧E4 transcript to form an E1∧E4 fusion protein that contains the first five amino acids from the E1 protein and almost the entire product of the E4 open reading frame (ORF). Several viral polycistronic mRNA transcripts contain the E1∧E4 gene. The most abundant class (E1∧E4 and E5) has been described for cutaneous and muscosal HPV types and is derived from a differentiation-inducible promoter in the E7 ORF (8, 16, 17, 24, 25, 39). The other major E1∧E4-containing messages derived from this promoter have the potential to also express the L1 and L2 capsid proteins (5, 18, 25).
Induction of E4 protein expression coincides with HPV DNA amplification, and the viral protein persists in cells expressing the capsid proteins and those producing newly synthesized virions (4, 10, 28). The pattern of E4 distribution suggests that E4 function might be required at all stages of the productive cycle. Indeed, cottontail rabbit papillomavirus (CRPV) induced by CRPV mutant genomes that cannot express E4 fail to show evidence of productive viral events such as viral genome amplification or expression of the major capsid protein (27).
In human warts and in in vitro cell culture systems that recapitulate the HPV life cycle, E4 exists as multiple species that are formed through a combination of proteolysis, oligomerization, and phosphorylation (for a review of E4 protein modification, see reference 32 and references therein). Studies, largely of the HPV type 1 (HPV1) E4 protein, have shown that the nature of these modifications is progressively altered as the infected cells migrate upwards within the infected tissue (4, 11, 15). Therefore, although the role of E4 in the virus life cycle has not been elucidated, such posttranslational events might modify E4 function during the different stages of the productive phase of the life cycle.
Proteolysis of the mature HPV1 E1∧E4 molecule involves the progressive cleavage of N-terminal residues (11). Limited proteolysis of the E1∧E4 17-kDa polypeptide removes the N-terminal 15 amino acids to tyrosine 16 to form a 16-kDa species (33). Further proteolysis generates the 10- and 11-kDa proteins, with alanine 59 as the N-terminal residue (33). In HPV1 warts, the 17-kDa polypeptide appears in the parabasal cells coincident with the onset of viral genome amplification (Fig. 1). The 16-, 11-, and 10-kDa polypeptides accumulate in a progressive manner, with the more processed forms becoming abundant in superficial keratinocytes, where capsid protein expression and the assembly of virus particles occur (Fig. 1).
FIG. 1.

Schematic representation of the distribution of E4 proteins in HPV1 infections and their relationship to productive viral events. The full-length E1∧E4 protein (17-kDa [17K]) is proteolytically cleaved to produce smaller polypeptides of 16, 11, and 10 kDa (10). The timing of the appearance of the processed polypeptides in relation to the stage of keratinocyte differentiation and virus events is only an approximation (references 1, 4, 12, 28, and our unpublished data).
Recombinant molecules that closely resemble the 16- and 11-kDa processed forms do not colocalize with keratin intermediate filaments (33), nor do they induce reorganization of nuclear dot 10 (ND10) domains (36). Extreme N-terminal sequences of E1∧E4 appear to be critical for these activities, such as the LLXLL motif (10LLGLL14) that is necessary for coalignment with the keratin cytoskeleton (33). We would, therefore, predict that loss of N-terminal amino acids through proteolytic cleavage during the virus life cycle would eliminate those E4 functions dependent on sequences in the N-terminal domain (33). An important question is whether the smaller forms of the E4 protein are functional proteins, and, if so, what role they play in the virus life cycle. Therefore, in this study we have investigated whether truncated HPV1 E4 proteins that closely resemble the naturally occurring processed proteins encode biological activities. We show that in immortalized and HPV-transformed human keratinocytes, a truncated E4 protein, equivalent to the 16-kDa species, can mediate a G2 arrest in the cell cycle and that this arrest is reversed by exogenous expression of cyclin B1. Cells expressing the truncated protein also undergo rereplication of the cellular genome, and this is associated with aberrant nuclear morphology. Perturbation of cell division is specific to the truncated form of E4 and is not mediated by the full-length E1∧E4 protein. Surprisingly, cells coexpressing the full-length and truncated E4 proteins do not show evidence of the rereplication of cellular DNA or aberrant mitotic division, despite retaining the ability to arrest in G2. Furthermore, coexpression is also associated with a significant inhibition of cellular proliferation. We predict that these biological activities of E4 act to support efficient amplification of the viral genome in differentiating keratinocytes.
MATERIALS AND METHODS
Cell culture.
SCC-12F keratinocytes were grown in a 3:1 mixture of Dulbecco's modified Eagle's medium and Ham's F-12 medium supplemented with 5% fetal calf serum, 2 mM glutamine, hydrocortisone (0.4 μg ml−1), and antibiotics. SV-JD keratinocytes (simian virus 40 [SV40]-immortalized human epidermal keratinocytes) were maintained in culture as described previously (34). The cervical tumor cell line HeLa and the epithelial cell line COS-1 were cultured as described for SV-JD keratinocytes. G418-resistant NIH 3T3 mouse fibroblasts were maintained in Dulbecco's modified Eagle's medium supplemented with 10% bovine serum, 2 mM glutamine, and antibiotics. For preparation of the NIH 3T3 fibroblasts for the feeder layer, cells were treated with 4,000 Rads of γ radiation.
Plasmid constructs and transfections.
The construction of plasmids pcDNA-1E4 (E4-17K), pcDNA-1Δ2-15 (E4-16K), and pcDNA-1Δ2-58 (E4-11K) was described previously (36).Plasmid pcDNA-1Δ2-15/Hyg was constructed by inserting the 1E4Δ2-15 cDNA into the BamHI site of the pcDNA3.1 vector containing the hygromycin B resistance gene (Invitrogen Life Technologies). The panel of E4-16K deletion mutations was PCR amplified from E4-17K mutant plasmids containing the amino acid deletions Δ21-24, Δ24-27, Δ27-30, Δ44-48, and Δ49-53 (38) by using a forward primer (5′-GATGATAAGATCTATGTACACTCCGACTACCCAA-3′) that deletes the DNA sequence coding for N-terminal residues 2 to 15 and a reverse primer (5′-GATATGAGATCTTTACACAGACCACGGGT-3′) that reconstructs the C terminus of the E4 protein. PCR products were cloned by TOPO TA cloning (Invitrogen Life Technologies) into the expression vector pcDNA 3.1. The deleted amino acids correspond to the following HPV1 E4-16K sequences: Δ7-10, Δ10-13, Δ13-16, Δ30-34, and Δ35-39. The QuikChange site-directed mutagenesis kit (Stratagene) was used to generate the alanine substitutions of Thr13, Pro14, Pro15, and Ser16. The primers used were 5′-CTATCCGAGAGTGGCACCGCCCAGCAATCGAC-3′ (T13A), 5′-CTATCCGAGAGTGACAGCGCCCAGCAATCGACGTC-3′ (P14A), 5′-GAGAGTGACACCGGCCAGCAATCGACGTCCATC-3′ (P15A), and 5′-AGTGACACCGCCCGCCAATCGACGTC-3′ (S16A). Bi-directional sequencing with an ABI Prism 3100 genetic analyzer was used to verify the sequences of all mutated plasmids. The cyclin B1-green fluorescent protein (GFP) expression plasmid, pcDNA3/cyclin B1-GFP was a generous gift from Jonathan Pines (Wellcome/Cancer Research UK Institute, Cambridge, United Kingdom).
For colony formation assays, subconfluent cultures of SCC-12F and HeLa keratinocytes were harvested by trypsinization, and 2 × 106 cells were electroporated in the presence of 10 μg of plasmid DNA by using 120 μF/0.45 kV (Gene Pulser II; Bio-Rad). After 10 min on ice, the cells were transferred to 10 ml of normal growth medium and plated out into 10 6-cm dishes. To each dish of electroporated SCC-12F cells, 2 ×105 irradiated NIH 3T3 fibroblasts were added as a feeder layer. Transfected cells were selected by the addition of 400 μg of G418 sulfate (Invitrogen Life Technologies) per ml and/or 200 μg of hygromycin B (Sigma Chemicals) per ml; after 4 to 5 weeks of selection, they were fixed with 70% ethanol, and colonies were stained with 5% (wt/vol) Giemsa stain. Control dishes of mock-transfected cells were selected by using the same conditions; complete cell death was noted within the first week of selection, and no drug-resistant colonies were formed.
To establish transient expression, 5 × 105 cells were seeded into 6-cm dishes. The following day, cells were transfected with between 4 and 5 μg of plasmid DNA by using the Lipofectamine Plus or Lipofectamine 2000 reagent (Invitrogen Life Technologies). Cells were harvested at various times posttransfection either for Western blot analysis or for cell cycle analysis.
Construction and infection of rAd.
Recombinant adenoviruses (rAds) expressing the HPV1 E1∧E4 protein (Ad-1E4), the truncated protein E4-16K (Ad-1E4Δ2-15), and β-galactosidase (Ad-βGal) have been described previously (36). Cells grown in dishes or on glass slides were infected with the rAds at a multiplicity of infection of 50. After 2 h, the virus was removed and replaced with culture medium and incubated for the appropriate time period. For short-term growth assays, SCC-12F and SV-JD cells were infected with the rAds and the cells were harvested by trypsinization every 24 h for 6 days. The cells present were counted by using a glass hemocytometer. The efficiency of infection as judged by immunofluorescence staining was between 60 to 80% for the rAd-E4 viruses and 90 to 95% for Ad-βGal.
Flow cytometric analysis.
For DNA cell cycle analyses, cell pellets were resuspended in 70% ethanol and fixed at −20°C for 1 h. Cells were then pelleted by centrifugation and washed twice with phosphate-buffered saline (PBS). After the second wash, 1 ml of PBS containing 10 μg of RNase (Sigma Chemicals) per ml was added, and the samples were incubated at 37°C for 1 h. The cells were pelleted by centrifugation and resuspended in 500 μl of PBS containing 20 μg of propidium iodide (PI) per ml. The relative cellular DNA content of stained cells was measured by using an EPICS XL flow cytometer (Coulter Electronics, Luton, United Kingdom). PI fluorescence was collected by using a 488-nm long-pass filter, followed by a 635-nm band-pass filter. Single cells were selected for analysis by using the distribution of PI fluorescence signal integral against PI fluorescence signal peak to discriminate doublets and debris. The relative size of PI fluorescence signal integral (DNA content) in single cells was plotted as a frequency histogram. Equal numbers of cells (10,000) were analyzed by fluorescence-activated cell sorting. The proportions of cells in phases G0/G1, S, and G2/M were deconvoluted from the latter by using Multicycle dedicated cell cycle analysis software (Phoenix Flow Systems, San Diego, Calif.).
For the analysis of cell cycle profiles of cells labeled with 5-bromodeoxyuridine (BrdU), rAd-infected or plasmid-transfected cells were incubated at various times after infection or transfection with 33 μM of BrdU (Sigma Chemicals) for 2 h. The cells were harvested by trypsinization and pelleted by centrifugation. After fixation in 70% ethanol the samples were pelleted, washed twice in PBS, and treated with 1 ml of 2 M HCl at room temperature for 30 min. The samples were then washed twice in PBS and incubated at 37°C for 30 min in 500 μl of PBS containing 0.5% Tween 20, 0.5% bovine serum albumin, and 0.5 μg of fluorescein isothiocyanate-conjugated anti-BrdU monoclonal antibody (MAb; BD Biosciences, San Jose, Calif.). PI was added at a concentration of 5 μg ml−1 and incubated at room temperature for 15 min prior to dual-parameter flow cytometric analysis to detect PI and fluorescein isothiocyanate staining for nuclear DNA and BrdU content. Two-dimensional profiles were analyzed by using Win MDI 2.8 (version 1.0; Purdue University Cytometry Laboratories).
Nocodazole synchronization.
HeLa cells were infected with rAds, and after 48 h were treated with 10 μM nocodazole (Sigma Chemicals) for 16 h. The mitotic-arrested cells were removed from the cell monolayer by vigorous washing with culture medium and were pelleted and resuspended in 1 ml of culture medium. Aliquots of 100 μl were allowed to settle onto glass slides for approximately 3 h and then were processed for immunofluorescence analysis. Cells were incubated with appropriate primary and secondary antibodies, and DNA was visualized with 4,6-diamidino-2-phenylindole (DAPI; Sigma Chemicals).
Statistical analysis.
Each experiment was performed at least three times. Statistical analysis of data was performed by using single-factor analysis of variance against the control plasmid pcDNA3.1.
Immunofluorescence microscopy and Western blotting.
Cells grown on glass slides were fixed in 4% (wt/vol) paraformaldehyde in PBS for 5 min and permeabilized in acetone at −20°C for 10 min. Immunofluorescence staining was performed as described elsewhere (36). E4 proteins were detected by using the MAb 4.37 (1/250 dilution) (11). An MAb against β-galactosidase (1/400 dilution) was purchased from Promega (Madison, Wis.). MAbs specific for cyclin A and cyclin B1 were obtained from Santa Cruz Technologies and Upstate Technology, respectively. Both were used at a 1/50 dilution. Immune complexes were visualized by using AlexaFluor 488-labeled anti-mouse specific antibody conjugate (Molecular Probes), and nuclei were counterstained with DAPI. Immunofluorescence was observed by using a Nikon Eclipse E600 microscope with appropriate filter sets, and images were captured by using a Leica DC200 digital camera. Images were assembled in Adobe Photoshop 6.0.
For Western blotting, cells were lysed in 8 M urea, 25 mM Tris (pH 8.0), 0.15 M β-mercaptoethanol, 10 μg of leupeptin per ml, and 10 μg of pepstatin per ml. After sonication insoluble material was removed by high-speed centrifugation. Equal amounts of protein were electrophoresed on sodium dodecyl sulfate-polyacrylamide (10 to 12% acrylamide) gels. Separated proteins were transferred to a nitrocellulose membrane, and Western blotting was performed with the following primary antibodies: MAb 4.37 (HPV1 E4 proteins) and the anti-β-galactosidase MAb (1/1000 dilution). To control for loading, actin levels were determined by using an anti-β-actin MAb (1/40,000 dilution; Sigma Chemicals). Primary antibodies were detected with horseradish peroxidase-labeled anti-mouse immunoglobulins (Sigma Chemicals), and membranes were subjected to chemiluminescence detection (ECL; Amersham Pharmacia).
RESULTS
Loss of N-terminal residues from the HPV1 E1∧E4 protein is associated with suppression of keratinocyte growth.
In previous studies, we had noted that keratinocytes were unable to support the stable expression of HPV E4 proteins, suggesting that these viral proteins can interfere with normal cell growth (reference 34 and unpublished data). To investigate these observations in detail, we assessed the consequence of expression of the full-length HPV1 E1∧E4 polypeptide (17 kDa) on keratinocyte growth and also that of two N-terminal deletion proteins Δ2-15 and Δ2-58 (Fig. 2A). These two truncated proteins closely resemble the 16- and 11-kDa E4 polypeptides expressed in HPV1-induced warts, except that they have an N-terminal initiation methionine (33). We will refer to the full-length E1∧E4 protein as E4-17K, to Δ2-15 as E4-16K, and to Δ2-58 as E4-11K. Plasmids that express the different E4 polypeptides were transfected into SCC-12F keratinocytes. The SCC-12F cell line is an immortalized but nontumorigenic cell line derived from stratified squamous epithelium, and the cells behave like primary keratinocytes in vitro (26, 31). The E4 expression plasmids also express the antibiotic G418 resistance marker. After a 4-week selection by G418 sulfate, we observed that in comparison to the control (empty pcDNA3.1 vector), transfection of SCC-12F cells with the E4-16K-coding plasmid leads to a significant decrease in the number (38.5%) and size (55.5%) of colonies (Fig. 2B). There was no significant change, however, in the number or size of colonies generated following transfection with the E4-17K-expressing plasmid and only a slight reduction in colony number and size with the E4-11K expressing plasmid (Fig. 2B). No drug-resistant colonies were formed in mock-transfected cells. Similar results were obtained in HPV18-positive HeLa cervical tumor keratinocytes electroporated with the E4 expression plasmids (see Fig. 6A). Analysis of the clones for E4 expression at the end of the selection period showed that none had retained expression of any of the three transgenes, which is consistent with previous findings (reference 34 and our unpublished data).
FIG. 2.

Expression of HPV1 E4 represses keratinocyte growth. (A) The structure of the full-length HPV1 E1∧E4 protein. E1, the small exon derived from the E1 ORF; PRR, the proline-rich region; C-terminal domain, a conserved domain necessary for E4 oligomerization (2, 33). The position of the leucine-rich motif 10LLGLL14 that is required for colocalization with the keratin intermediate filaments (33) is indicated by a bold line above the diagram, and the positions of the N termini of the proteolytically cleaved products of 16 kDa and 10 or 11 kDa (33) are indicated with arrows. The structure of the two mutant proteins Δ2-15 (E4-16K) and Δ 2-58 (E4-11K) is shown below. (B) Graphical representation of the relative number and size (diameter) of clones produced following transfection of SCC-12F cells with the different HPV1 E4-containing plasmids, relative to the empty pcDNA3.1 vector (left). Data are shown as means ± standard deviations, and the asterisk indicates a statistical significance of 99.9%. Examples of SCC-12F cells transfected with empty vector or the corresponding HPV1 E4-containing plasmids are shown on the right side of the panel. (C) Short-term growth profiles, performed in triplicate, of SCC-12F and SV-JD keratinocytes infected with the different rAds. MI, mock-infected.
FIG. 6.

Exogenous expression of cyclin B1 rescues E4-16K-induced repression of keratinocyte growth. (A) Graphical representation of the relative number of clones produced following transfection of HeLacells with the different HPV1 E4-containing plasmids, including the G2-defective plasmid E4-16KΔ13-16. Presented data are from three experiments, each with 10 dishes (6 cm) per plasmid. (B) Detection of cyclins A and B1 in SCC-12F cells expressing β-Gal, E4-17K, and E4-16K. Cells infected with the different rAds for 72 h were dual stained for β-galactosidase or E4 and the cyclins A and B1. (a) Histogram of the percentage of dual positive cells. A minimum of 400 cells were counted for each protein, and the results shown are from three independent experiments. (b) Immunofluorescence staining of cells showing examples of cyclin B1-negative E4-16K cells and cyclin B1-positive E4-17K cells. In merged images, E4 proteins are green, cyclin B1 is red, and nuclei are blue. Note that E4-17K is nuclear and cytoplasmic and associated with inclusion bodies, while E4-16K is predominantly in the nucleoplasm and nucleoli (33, 36).
We also examined the effect of the HPV1 E4 polypeptides on short-term keratinocyte growth. SCC-12F cells were infected with rAds that express E4-17K, E4-16K, or β-galactosidase, and cells were counted over a period of 5 days (Fig. 2C). Cell cultures expressing the E4-16K protein grew significantly more slowly than either mock-infected cells or cultures infected with the β-galactosidase-expressing virus. SCC-12F growth was also repressed by E4-17K protein, although not to the same extent as observed with the truncated polypeptide (Fig. 2C). Similar findings were obtained following infection of SV40-immortalized human epidermal keratinocytes (SV-JD) (Fig. 2C). Immunofluorescence microscopy and Western blotting confirmed that the frequency of infection with the different rAds was similar and that β-galactosidase and E4 protein levels were comparable (data not shown). We did note that in cells infected with the rAd expressing E4-17K, there was a progressive accumulation of the truncated 16-kDa polypeptide, an observation that is consistent with previous findings (34, 35). Therefore, repression of keratinocyte growth in E4-17K-expressing cells might reflect an accumulation of the truncated 16-kDa species. Collectively, these data suggest that a loss of extreme N-terminal amino acids from the full-length HPV1 E1∧E4 polypeptide is associated with suppression of the growth of immortalized human keratinocytes.
E4-16K induces an arrest in the G2 phase of the cell cycle.
Repression of cell growth by E4-16K, and also to some extent E4-17K, might reflect an arrest of the cells at a defined point in the cell cycle. To investigate this possibility, SCC-12F cells were infected with the different rAds, and cell cycle profiles were determined at different times postinfection (p. i.) by flow cytometry analysis (Fig. 3A). Infection with the virus expressing E4-17K or the control virus expressing β-galactosidase produced cell cycle profiles that were similar to those of mock-infected cells, and there was no significant change in the G2+M:G1 ratios, even at late times after infection. However, expression of the truncated E4-16K protein induced a marked increase in the G2/M population, which was evident between 48 and 72 h p. i. The ability of E4-16K to induce G2/M arrest was also evident in HeLa cells, occurring as early as 24 h after infection (Fig. 3A), and also in COS-1 cells transfected with E4 expression plasmids (Table 1). Western blot analysis showed that comparable levels of the three proteins were expressed in the infected cells and that failure of the full-length E1∧E4 protein to induce cell cycle arrest was not due to protein instability (Fig. 3B and data not shown).
FIG. 3.

Expression of E4-16K in keratinocytes induces a G2 arrest in the cell cycle. (A) Cell cycle analysis of SCC-12F and HeLa keratinocytes either mock infected or infected with rAds expressing the full-length E4-17K protein, the truncated E4-16K protein, or β-galactosidase (β-Gal). Cell cycle analysis was performed at 24, 48, 72, and 96 h p. i., but only the 24- and 72-h time points are shown. Arrows indicate the accumulation of E4-16K cells with a DNA content of greater than 4N. Results shown are representative of at least four independent experiments. (B) Western blot analysis of equivalent amounts of cell lysates prepared from HeLa cells expressing β-galactosidase and E4 proteins. Immunoblots were processed with E4 and β-galactosidase MAbs. (C) The percentage of mitotically arrested HeLa cells expressing β-galactosidase, E4-17K, and E4-16K, as determined by immunofluorescence staining. The data are derived from three experiments, and for each experiment a minimum of 200 cells were counted per transgene.
TABLE 1.
Cell cycle populations of COS-1 cells transfected with HPV1 E4 and cyclin B1-GFP expression plasmidsa
| Plasmid | G1 | S | G2/M | G2/M:G1 |
|---|---|---|---|---|
| pcDNA3.1 | 37.5 ± 2.1 | 33.5 ± 3.5 | 27.5 ± 2.1 | 0.73 |
| E4-17K | 36.0 ± 6.0 | 28.4 ± 4.8 | 24.8 ± 4.0 | 0.69 |
| E4-16K | 35.2 ± 6.1 | 25.8 ± 9.7 | 37.5 ± 1.2 | 1.07 |
| E4-16K plus cyclin B1 | 32.1 ± 6.2 | 41.7 ± 6.0 | 24.3 ± 1.8 | 0.76 |
| Cyclin B1 | 33.9 ± 5.8 | 34.0 ± 4.1 | 26.5 ± 3.5 | 0.78 |
Data shown represent the means ± standard deviations of three independent experiments.
Since single-parameter DNA histograms cannot discriminate G2 from M-phase cells, we used the microtubule disrupter nocodazole, which interferes with the formation of the mitotic spindle, to determine at what stage in the progression of cells through G2 and into mitosis the E4-16K-expressing cells were arrested. HeLa cells were infected with the different rAds and treated with nocodazole at 48 h p. i. to induce mitotic arrest. Isolated mitotic cells expressing β-galactosidase and E4 proteins were identified by immunofluorescence microscopy. As shown in Fig. 3C, 97.0 and 61.5% of mitotic cells were expressing β-galactosidase and the full-length E4-17K proteins, respectively. In contrast, only 3.4% of cells arrested in mitosis were E4-16K positive. The higher mitotic index for β-galactosidase cells (compared to E4-17K cells) was most likely because this virus had a greater efficiency of infection (as inferred from immunofluorescence staining) than the two E4 rAds, which were equivalent to each other. We therefore conclude that E4-16K induces cells to arrest in the G2 phase of the cell cycle (with a DNA content of 4N) and not in mitosis.
A proline-rich sequence in E4-16K is necessary for G2 cell cycle arrest.
Next, we wanted to identify which E4 sequences were mediating the G2 arrest. At this point in our investigation, two studies were published demonstrating that full-length E1∧E4 proteins from two oncogenic HPV types, 16 and 18, induced a G2 arrest of HeLa cervical keratinocytes (9, 23). The ability of these proteins to induce cell cycle arrest was mapped to a proline-rich sequence that lies close to the N terminus of the E1∧E4 protein (9). A similar region of the HPV1 E1∧E4 protein is also rich in proline amino acids (33) and is retained in the E4-16K protein (Fig. 2A). To test whether this region is required for the G2 arrest function of E4-16K, we generated a small set of E4-16K mutants containing deletions across part of the proline-rich region and across a neighboring region that is rich in charged residues (Fig. 4A). Because the efficiency of transfection into SCC-12F cells is very poor (<5%), we transfected the plasmids expressing the E4 mutants into HeLa cells (transfection efficiency of 30 to 35%). Cell cycle profiles were analyzed by flow cytometry, and the percentages of cells in G2/M are shown in Fig. 4B. E4-16K-transfected cells showed a significant increase in the G2/M population in comparison to E4-17K-transfected cells or to those transfected with the empty vector (Fig. 4B). Two of the three mutant 16K proteins containing deletions of proline-rich sequences, 16KΔ10-13 and 16KΔ13-16, did not induce an arrest. The third mutant, 16KΔ7-10, did induce a significant accumulation of cells in G2/M although the level of the cell cycle block was slightly less than observed with E4-16K. Deletion of charged residues (30 to 34 and 35 to 39) had no significant effect on E4-16K-induced cell cycle arrest. All E4 proteins with the exception of 16KΔ35-39 were expressed at equivalent levels (Fig. 4C). Similar G2/M profiles occurred following transfection of the different plasmids into COS-1 epithelial cells (data not shown). We conclude that a proline-rich sequence, 10PRVTPPS16, (equivalent to positions 24 to 30 in the full-length E1∧E4 sequence [33]) is necessary for E4-16K-mediated G2 arrest of human keratinocytes.
FIG. 4.

Deletion of amino acids in the proline-rich region abolishes E4-16K-mediated cell cycle arrest. (A) Structure of HPV1 E4-16K deletion mutants. The proline-rich region of E4-16K is underlined. (B) Percentages of HeLa cells that are in the G2/M phase of the cell cycle following transient transfection for 24 h with empty plasmid pcDNA3.1, pcDNA-1E4 (E4-17K), pcDNA-1Δ2-15 (E4-16K), and the E4 deletion mutant plasmids. The asterisk indicates a 95.0% significance. (C) Western blot analysis of equivalent amounts of HeLa cell lysates transfected with empty plasmid pcDNA3.1 and the various E4-expression plasmids. Blots were probed with an anti-E4 MAb.
The PRVTPPS sequence contains a threonine (Thr13) and serine residue (Ser16). Threonine and serine residues in the HPV1 E4 protein are known to be substrates for several different protein kinases (15), and phosphorylation of such residues might be necessary for HPV16 E1∧E4-induced cell cycle block (9). In order to determine whether Thr13 and Ser16 are required for the activity of the E4-16K protein, they were mutated to alanine residues by site-directed mutagenesis. Two out of the three proline residues (Pro14 and Pro15) were also replaced by alanines. With the exception of the mutant E4-16KT13A, expression of the other mutant proteins (E4-16KP14A, E4-16KP15A, and E4-16KS16A) induced an increase in the G2/M population in HeLa cells, comparable with that observed for the wild type E4-16K protein (Fig. 5A). In contrast, the cell cycle profile of E4-16KT13A-transfected cells was similar to the profile of cells expressing the full-length E4-17K protein (Fig. 5A). Equivalent steady-state expression levels of all the E4 proteins were noted following Western blotting (Fig. 5B). We therefore conclude that Thr13 in the proline-rich sequence is required for the G2 cell cycle arrest function of the E4-16K protein.
FIG. 5.
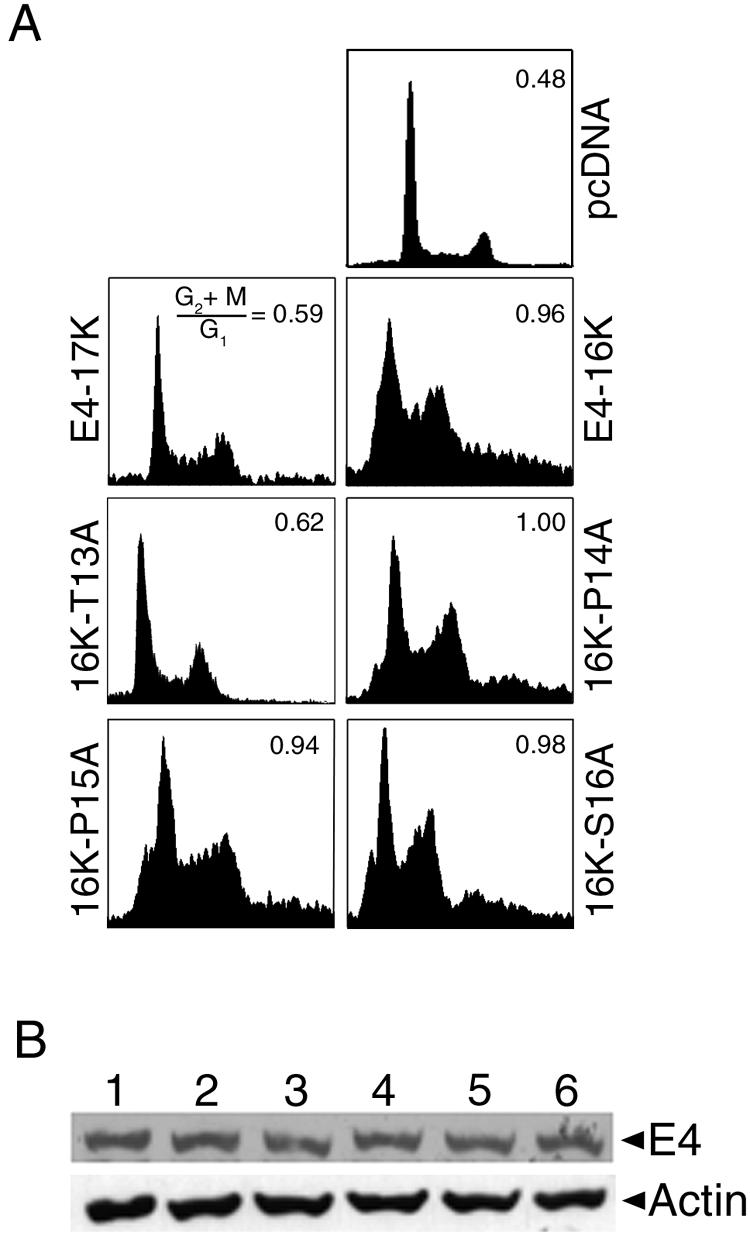
A threonine amino acid in the E4-16K proline-rich sequence is necessary for E4-16K-induced G2 arrest. (A) Cell cycle profiles of HeLa keratinocytes transiently transfected for 24 h with empty vector (pcDNA) and plasmids containing E4-17K, E4-16K, and alanine point substitutions of Thr13, Pro14, Pro15, and Ser16. Only the Thr13 → Ala mutant plasmid failed to induce a G2/M arrest in the cell cycle. (B) Western blot analysis of E4 protein expression in total protein lysates prepared from HeLa cells transfected with the E4-containing plasmids. Lane 1, E4-17K; lane 2, E4-16K; lane 3, 16K-T13A; lane 4, 16K-P14A; lane 5, 16K-P15A; and lane 6, 16K-S16A. As a protein loading control, the membrane was probed with an antibody against β-actin.
Exogenous expression of cyclin B1 abolishes E4-16K induced G2 arrest.
The progression of cells from interphase into mitosis is regulated by cyclin-cyclin-dependent kinase (cdk) activity (for a review, see reference 30). Commitment to cell division is mediated by activation of the cyclin B1-cdk1 complex, the core component of the mitosis-promoting factor. Cyclin B1-cdk1 activity is regulated, in part, by the accumulation of cyclin B1 during G2/M and its translocation from the cytoplasm to the nucleus (29). Failure of E4-16K cells to progress into mitosis might, therefore, be due to an aberration in the pathways that regulate cyclin B1-cdk1 activity. In the first instance we examined whether exogenous expression of cyclin B1 would enable E4-16K-expressing cells to progress through the cell cycle. In order to obtain sufficient levels of cotransfection of the expression plasmids, we used COS-1 epithelial cells. Cells were cotransfected with the E4-16K plasmid and a plasmid that expresses cyclin B1-GFP from the cytomegalovirus promoter (a kind gift from Jonathan Pines) and analyzed at 48 h posttransfection. Table 1 shows the mean percentage of cells in G1, S, and G2/M phases of the cell cycle after three independent experiments. E4-16K-expressing cells showed a significant accumulation of cells in G2/M (G2+M:G1, 1.07) in comparison to cells transfected with the E4-17K plasmid (G2+M:G1, 0.69) or the empty vector (G2+M:G1, 0.73), confirming that the truncated protein induces an arrest of COS-1 cells. However, cells cotransfected with E4-16K and cyclin B1-GFP expression plasmids did not accumulate in G2/M (G2+M:G1, 0.76) and had a similar number of cells in G2/M as cells expressing E4-17K, although we did note an increase in the number of cells in S phase in the cotransfected cells (Table 1). Expression of cyclin B1-GFP and E4 proteins was confirmed by immunofluorescence microscopy (data not shown). Our data suggest that exogenous expression of cyclin B1 can overcome E4-16K-induced G2 arrest of COS-1 epithelial cells.
In colony formation assays, E4-16K expression repressed the growth of SCC-12F keratinocytes (Fig. 2B). We would predict from our studies that the G2 arrest function of E4-16K mediates the suppression of colony growth. Furthermore, if cyclin B1 expression can rescue the cell cycle progression of E4-16K cells, then coexpression of the two proteins should restore colony formation. To test this hypothesis, we cotransfected HeLa cells with the cyclin B1-GFP plasmid, which contains the neomycin selectable marker and an E4-16K expression plasmid (pcDNA-1Δ2-15/Hyg) that carries the hygromycin B resistance gene, and selected for colonies by using G418 and hygromycin B. Also, cells were transfected with the cyclin B1 plasmid alone and the different E4 expression plasmids, including the G2 arrest-defective E4-16K mutant lacking the sequence TPPS (E4-16KΔ13-16). Expression of E4-16K caused a marked decrease in colony formation compared to cells transfected with E4-17K or the empty vector (Fig. 6A) and supports our findings derived from SCC-12F cells (Fig. 2B). In contrast, expression of E4-16KΔ13-16 that is unable to induce G2 arrest did not repress colony formation (Fig. 6A). Furthermore, the inhibitory effect of wild-type E4-16K was reversed upon cotransfection of cyclin B1 (Fig. 6A). Taken together, our results show that the G2 arrest function of E4-16K mediates the suppression of keratinocyte growth and that this function of E4-16K is overcome by exogenous expression of cyclin B1.
Immunofluorescence microscopy was used to assess whether E4-16K alters cyclin B1 protein expression or subcellular distribution. SCC-12F cells were infected with the different rAds, and at 96 h p. i. cells were stained for both cyclin B1 and E4 or β-galactosidase. As a control we also stained cells with an anti-cyclin A antibody. Flow cytometry of parallel cultures was used to confirm a G2/M arrest in the AdE4-16K-infected cells. Fifteen percent of E4-17K-positive cells were cyclin B1 positive, and this percentage was comparable to the proportion of β-galactosidase-expressing cells (Fig. 6B). In contrast, only 3.5% of E4-16K cells expressed cyclin B1 (Fig. 6B). This result was unexpected as cyclin B1 levels normally accumulate during G2/M (30), and, therefore, we would expect a majority of E4-16K cells to be expressing significant levels of cyclin B1. To exclude the possibility that the epitope recognized by the cyclin B1 MAb is masked in E4-16K cells, we used a cyclin B1 polyclonal antibody and obtained a similar decrease (∼70%) in cyclin B1-positive E4-16K cells (data not shown). In E4-16K cells that were cyclin B1 positive, cyclin B1 was predominantly nuclear, and there was no coincidence between the two proteins (data not shown). Cyclin A levels also increase in G2, and cyclin A is degraded by the anaphase promoting complex\cyclosome prior to cyclin B1. The number of cells expressing both E4-16K and cyclin A had increased compared to the controls, which is consistent with a G2 arrest (Fig. 6B, panel a), and indicates that the decrease in detection of cyclin B1 is specific.
E4-16K induces rereplication of the cellular genome.
Analysis of the cell cycle profiles of E4-expressing SCC-12F and HeLa cells showed that in AdE4-16K-infected cells there was, in addition to a G2 cell cycle block, also a consistent accumulation of cells with a DNA content of greater than 4N (8N and 16N populations are indicated by arrows in Fig. 3A). Although there was evidence that mock-infected cells and cells infected with rAds expressing E4-17K and β-galactosidase also had a population of >4N cells, these were at a level consistently lower than the levels observed in AdE4-16K-infected cultures (Fig. 3A). This result suggested that a portion of AdE4-16K-infected cells were continuing to replicate their DNA without completing cell division. Microscopical examination of rAd-infected SCC-12F and HeLa cells showed that a greater proportion of E4-16K-expressing cells contained large and often distorted nuclei than those expressing E4-17K or β-galactosidase (Fig. 7A, panels a and b, and data not shown). Notably, at later times p. i. (SCC-12F, >96 h; HeLa, >24 h) there was a marked induction of E4-16K cells with multiple nuclei (Fig. 7A, frames c and d, and B; also data not shown). We also noted that the E4-16K protein was both cytoplasmic and nuclear in cells with aberrant and multiple nuclei (Fig. 7A, a and c), while in cells with normal nuclear morphology, the protein is predominantly found in the nucleus (Fig. 6B, b) (see references 33 and 36).
FIG. 7.


E4-16K induces rereplication of the cellular genome. (A) Immunofluorescence microscopy of SCC-12F cells infected with Ad-E416K for 96 h. Cells were stained with an anti-E4 MAb (4.37; green), and nuclei were visualized with DAPI (frames a and c, blue). DAPI staining is also shown in gray scale (b and d). E4-16K-expressing cells contain large and sometimes distorted nuclei (a and b) and are also multinucleate (c and d). Bar, 5 μm. (B) Percentage of β-galactosidase-, E4-17K-, and E4-16K-positive SCC-12F keratinocytes that contain more than one nucleus (up to 5, multinucleate), as determined by immunofluorescence microscopy following infection with rAds. Data were collected every 24 h until 144 h p. i., but only the 96- and 144-h time points are shown. This experiment was repeated in triplicate, and the number of cells counted in each experiment was at least 400. (C) HeLa cells were transfected for 24 h with the empty vector, the E4-containing plasmids, and the panel of E4-16K deletion plasmids. Cells were stained with an E4 MAb, and nuclei were visualized by DAPI staining. The percentage of E4-positive multinucleate cells for each plasmid was recorded; the asterisk indicates a significant (99.9%) increase in multinucleate cells compared to cells transfected with pcDNA. (D) Incorporation of BrdU into DNA of HeLa cells transfected with the empty vector (pcDNA), E4-17K, and E4-16K plasmids (a) and plasmids expressing the alanine point substitutions within 13TPPS16 (b). The distributions of the G1, G2, and S phase populations are indicated. The percentage of BrdU-positive cells in S phase and with a >4N DNA content are as indicated. The G2/M:G1 ratios were 0.61 (pcDNA), 0.64 (E4-17K), 1.10 (E4-16K), 0.55 (16K-T13A), 1.10 (16K-P14A), 0.90 (16K-P15A), and 0.98 (16K-S16A).
To establish whether the G2 arrest function of E4-16K was associated with multinucleation, E4-16K mutant plasmids were transfected into HeLa cells, and the percentage of multinucleate E4-positive cells was determined at 48 h by immunofluorescence microscopy (Fig. 7C). While the numbers of multinucleate cells in cultures transfected with the empty vector control or E4-17 were equivalent, there was a 3.5-fold increase in E4-16K-positive multinucleate cells (Fig. 7C), findings that are in agreement with data from rAd-infected cells (Fig. 7B). The mutant proteins defective in G2 arrest induction (E4-16KΔ10-13 and E4-16KΔ13-16) failed to induce a significant increase in multinucleate cells (Fig. 7C), indicating that the sequence 10PRVTPPS16 is necessary for cell cycle arrest and aberrant cell division.
It was also possible to detect the population of rereplicating cells following incubation in the presence of BrdU, a thymidine analogue that is incorporated into cells undergoing DNA synthesis. HeLa cells were transiently transfected for 48 h with the empty vector or the plasmids encoding E4-17K and E4-16K and then pulse-labeled with BrdU for 2 h. Two-dimensional flow cytometry showed that BrdU incorporation into cells with a DNA content of >4N increased by between 2.1- and 4.0-fold in E4-16K-expressing cultures, compared to cells transfected with the empty vector and pcDNA1-E4 (E4-17K), respectively (Fig. 7D, a). Analysis of mutant proteins 16K-T13A, 16K-P14A, 16K-P15A, and 16K-S16A demonstrated that only the G2-defective mutant 16K-T13A failed to induce an increase in >4N cells (Fig. 7D, b). Because rereplication of the cellular genome does not occur in cells incapable of G2 arrest, this suggests that after arresting in G2, E4-16K cells eventually reenter the cycle and undergo additional rounds of DNA replication. This scenario would be consistent with the appearance of E4-16K-expressing cells with enlarged nuclei. The formation of cells containing multiple nuclei suggests that a population of E4-16K cells can undergo separation of sister chromatids and nuclear decondensation but fail to go through cytokinesis.
Full-length E1∧E4 protein does not block E4-16K-mediated G2 arrest but does inhibit an increase in cells of >4N. Our studies show that the full-length HPV1 E1∧E4 protein (E4-17K) does not induce an arrest in the cell cycle of human keratinocytes, and this is consistent with findings of HPV1 E1∧E4 expression in Saccharomyces pombe cells (9). Since both E1∧E4 and proteolysed E4 polypeptides are expressed in HPV1 infections (Fig. 1), we wanted to determine whether the E4-16K could still induce cell cycle arrest in the presence of the full-length protein. SCC-12F keratinocytes coinfected with rAds expressing E4-17K and E4-16K for 72 h showed a marked increase in the G2/M population, equivalent to the level observed in AdE4-16K-expressing cells (Fig. 8A). Western blot analysis of lysates prepared from these cultures demonstrated that in dual-infected cells, the E4-17K and E4-16K proteins were expressed in similar amounts as in cultures infected with the individual viruses (Fig. 8B). It would seem, therefore, that E4-16K is able to induce a G2 arrest in the presence of the E1∧E4 protein. In Ad-E4-17K-infected cells, a small amount of E4-17K is processed to 16 kDa (34), but the level is insufficient to interfere with cell cycle progression.
FIG. 8.


Coexpression of E4-17K and E4-16K does not prevent G2 arrest but does inhibit cellular DNA replication. (A) SCC-12F cells were infected with rAds for 72 h, and cell cycle profiles were analyzed by fluorescence-activated cell sorting. (B) Western blot analysis of E4 expression in SCC-12F cells infected with the different rAd E4 proteins. Note that a small amount of E4-16K is generated in cells infected with AdE4-17K (34). (C) SCC-12F cells were infected with rAds and pulse-labeled with BrdU. Coexpression of E4-17K and E4-16K inhibited BrdU incorporation into cells with a DNA content of greater than 4N (>4N) and also induced a decrease in BrdU incorporation into proliferating diploid cells. (D) COS-1 epithelial cells were transfected with expression plasmids and pulse-labeled with BrdU. The percentage of BrdU cells with a >4N DNA content (a) and the percentage of BrdU-positive S phase cells (b) are shown. Note that coexpression of E4-17K and E4-16K induces a significant decrease in BrdU incorporation into both populations of cells. The asterisk indicates that the increase in BrdU-positive cells of >4N and the decrease in BrdU-positive S-phase cells were significant (99.9%).
Next, we examined whether E4-17K influenced E4-16K-induced aberrant cell division. Immunofluorescence analysis showed that in contrast to cells infected with the AdE4-16K virus, there was no increase in the number of E4-positive multinucleate cells in dual-infected SCC-12F cells compared to controls (data not shown). A decrease in the >4N population was also apparent following BrdU-labeling of infected cultures (Fig. 8C). The level of BrdU-positive cells with a DNA content of >4N decreased from nearly 30.0% in AdE4-16K-infected cells to 19.0% in the dual-infected cultures, a level similar to that observed in the controls. The level of >4N BrdU-positive cells remained high (25.88%) following coinfection with AdE4-16K and Ad-βGal, indicating that a negative effect on E4-16K-induced chromosomal rereplication is mediated specifically by the E1∧E4 protein. Interestingly, further analysis of the flow cytometry profiles showed that in comparison to cells infected with the individual E4 viruses, coinfection also induced a marked reduction in BrdU incorporation into replicating diploid cells (Fig. 8C). The number of cells in S phase decreased from nearly 39.0% in Ad-E416K-infected cells to ∼13.0% in dual-infected cultures. A similar reduction was not apparent in cells expressing E4-17K alone (33.73%) or cultures coexpressing E4-16K and β-galactosidase (35.72%). Taken together, our data suggest that when expressed together, E4-17K and E4-16K can inhibit cellular proliferation. Consistent with these findings, we noted that in HeLa colony formation assays, compared to the empty vector control, E4-17K expression gave no significant reduction in colony formation, E4-16K expression induced an 83.6% reduction, and coexpression of E4-17K and E4-16K resulted in a 100% reduction of colony formation.
To determine whether E4-17K-induced repression of cellular DNA synthesis was dependent on the G2 arrest function or a separate function of E4-16K, we proceeded to examine the effect of E4-17K on cellular DNA replication in the presence of the G2 arrest-defective E4-16K mutant E4-16KT13A. COS-1 epithelial cells were transfected with plasmids coding for the different HPV1 E4 proteins and labeled with BrdU 36 h after transfection. The G2 arrest function of E4-16K is active in COS-1 cells (Table 1). The viral protein also induced a significant increase in the rereplicating population, and coexpression of E4-17K and E4-16K induced a reduction in this cell population (Fig. 8D, a), consistent with the data obtained from rAd-infected SCC-12F keratinocytes. Furthermore, coexpression of the E4 proteins also mediated a marked decrease (2.6-fold) in BrdU incorporation into S phase nuclei compared to cells expressing E4-16K alone (Fig. 8D, b). However, the decrease in BrdU incorporation was not apparent when E4-17K was expressed in the presence of the E4-16KT13A (Fig. 8D, b), supporting the notion that the activity mediated by Thr13 in E4-16K is necessary for E4-17K/16K inhibition of cellular DNA synthesis.
We therefore conclude that following coexpression of HPV1 E1∧E4 and the truncated in vivo-like polypeptide E4-16K, the two proteins cooperate to negatively regulate the normal cell cycle progression of human keratinocytes.
DISCUSSION
This study presents the first evidence that a truncated in vivo-like protein of HPV1 E4 encodes biological functions that are distinct from those mediated by the full-length form. It supports the hypothesis that proteolytic processing of E4 modifies its biological function (33, 37). We have shown that expression of an HPV1 E4 deletion protein (E4-16K) whose primary structure closely resembles the 16-kDa polypeptide expressed in vivo induced a G2 arrest of human keratinocytes derived from squamous epithelia. The effect on the cell cycle was not dependent on the action of other HPV proteins. Continued high-level expression of the truncated protein gave rise to hyperploid cells, distinguished by large distorted nuclei, as well as cells containing multiple nuclei, indicating rereplication of the cellular genome and aberrant nuclear division. Intriguingly, the effect of E4-16K on cell division was partially altered upon coexpression of the full-length E1∧E4 protein. While the G2 arrest function was not affected, rereplication of the cellular genome and aberrant nuclear division were prevented. Moreover, coexpression of the two forms of E4 led to negative regulation of cellular proliferation.
While this study was in progress, two groups published evidence that the full-length E1∧E4 proteins of anogenital HPV types 16 and 18 induced a G2 arrest of cervical carcinoma cells (9, 23). Taken together with our findings, the data suggest that a G2 arrest function appears to be an E4 activity conserved between unrelated HPV types; HPV1 has a tropism for cutaneous palmar and plantar epithelium. In our study, there was no evidence that the full-length E1∧E4 protein had an effect on G2/M progression of keratinocytes. Thus, the G2 arrest function is specific to the truncated HPV1 polypeptide. However, we cannot rule out the possibility that in natural HPV1 infections, E1∧E4 does encode such a function and that it is dependent on a specific posttranslational event such as phosphorylation or interaction with cellular and/or viral factors that are not expressed in our culture systems.
The cell cycle arrest functions of HPV1 E4-16K and HPV 16 E1∧E4 are both dependent on a threonine residue contained within proline-rich domains. This suggests a common mechanism of action, perhaps one that involves phosphorylation of this residue (9). However, while there is limited homology between the immediate sequence neighboring the threonine residue of HPV1 and anogenital types (TPPSNRRP [HPV1], TPP-RPIP [HPV16], and TPP-HRIP [HPV18]; identical amino acids are in italics), this does not constitute a conserved motif between E4 proteins generally. For example, no such motif is present in the E1∧E4 protein of HPV2, a potent inducer of G2 arrest of epithelial cells (G. Knight and S. Roberts, unpublished data). Clearly, further experimentation is needed to clarify the role of the threonine residue in E4-mediated cell cycle arrest.
Normal G2/M transition is regulated by the activities of cyclin A-cdk1 and cyclin B1-cdk1 complexes (30). We found that the G2 arrest function of E4-16K correlated with its ability to suppress keratinocyte growth, and both growth suppression and G2 arrest were reversed by reconstitution of cyclin B1 expression (Fig. 6A and Table 1). Our results suggest that failure of E4-16K-expressing cells to transverse G2 to M might be via a mechanism that prevents the formation of active cyclin B1-cdk1 complexes. Immunofluorescence studies showed that cyclin B1 protein was not detected in a majority of E4-16K G2-arrested cells, and this finding was in contrast to results of cyclin A staining (Fig. 6B). Since the levels of both cyclin A and B1 proteins normally increase during G2/M, this result was somewhat surprising and therefore warrants further investigation. Because the decrease in cyclin B1 protein was apparent in E4-16K cells with single nuclei (Fig. 6B) and not in multinucleate cells (see below), the reduction in cyclin B1 cannot be a reflection of anaphase promoting complex\cyclosome-mediated degradation of cyclin B1 that would normally occur prior to sister chromatid separation (30). It is, therefore, tempting to speculate that the failure of E4-16K cells to undergo G2/M transition might involve inhibition of cyclin B1 protein accumulation and/or an inappropriate loss. Interestingly, an unscheduled decrease in cyclin B1 levels is associated with G2/M arrest induced by the human T-lymphotropic virus type 1 Tax oncoprotein and the Epstein-Barr virus BZLF1 protein (20-22).
Morphological changes (nuclear enlargement and multinucleation) and rereplication of the cellular genome were associated with the activity of Thr13 in E4-16K function (Fig. 7). These observations suggest that after arresting in G2, E4-16K cells eventually reenter the cell cycle and undergo additional rounds of DNA replication, a scenario consistent with nuclear enlargement. Multinucleation indicates that some of these cells enter mitosis but are unable to complete cell division. Perhaps the decrease in cyclin B1 levels in E4-16K cells contributes to the process of rereplication of the cellular genome, as it is known that cyclin B1 or cdk1 activity is required to prevent chromosomal reduplication in the fission yeast Schizosaccharomyces pombe and Drosophila (38, 41). Since aberrant nuclear morphology is also associated with expression of anogenital E1∧E4 proteins (reference 23 and our unpublished data), perhaps E4 proteins can also interfere with cellular pathways that regulate mitotic progression and execution of cytokinesis.
An interesting finding of this study was that coexpression of the full-length E1∧E4 protein and the truncated E4-16K protein inhibited normal cellular proliferation and cellular DNA rereplication but did not prevent cells from arresting in G2 (Fig. 8). The inhibition of cellular DNA synthesis was not apparent upon expression of either protein independent of each other, suggesting that these two forms of the E4 protein are able to functionally cooperate to negatively influence cell proliferation. Moderate repression of short-term keratinocyte growth by E4-17K (Fig. 2C) probably reflects an accumulation of the truncated 16-kDa species, and subsequent cooperation between the two forms of E4. We are now in the process of elucidating the underlying molecular basis for the negative regulation of cellular proliferation by the HPV1 E4 protein. One possible explanation is that the two proteins interact directly with one another to form an active complex (2, 13, 35), mediated by sequences within the C termini of the proteins (2). This complex may be able to directly interfere with the process of cellular DNA synthesis. Alternatively, one of the E4 polypeptides might promote an interaction between the other E4 species and a cellular target by an indirect mechanism, perhaps one that is linked to the role of Thr13 in E4-16K function.
HPV1 E4 protein analysis in serial sections of warts taken parallel to the skin showed that the truncated E4 polypeptides appear in a progressive manner, with the 16-kDa species accumulating prior to the 10- and 11-kDa polypeptides (4) (Fig. 1). There was significant overlap between expression of the E1∧E4 and 16-kDa polypeptides, and in some sections the two proteins were present in equivalent amounts. Only in the deepest suprabasal layers was E1∧E4 protein detected by itself. In natural HPV1 infections, viral genome amplification initiates in cells that have moved up from the basal layer, and this correlates with the onset of E1∧E4 expression (4, 12, 28). These cells are stimulated to remain in an S-phase-like state through the activities of E7 (6), enabling the virus access to the host cell's replication machinery. The ability of HPV1 E1∧E4 protein to interact with cellular structures such as the keratin cytoskeleton and ND10 domains might facilitate the initial stage of the productive cycle, perhaps by organizing viral transcription and/or replication centers at E4-PML nuclear structures (34, 36). As genome amplification proceeds, the G2 arrest function of the truncated 16-kDa form might stimulate maintenance of the replication capacity of the differentiating keratinocyte by inhibition of further progression through the cell cycle. Continuous unscheduled host cell DNA synthesis (7), however, would present a drain on the nucleotide pool and on essential cellular replication factors that are also required by the virus for high-level replication of its own genome; HPV1 is a hugely efficient virus, producing as many as 1012 virus particles in cutaneous verrucae (3). Inhibition of host chromosome replication by E4 would enable cellular replication enzymes and nucleotides to be sequestered to viral DNA replication centers. Thus, the overall effect of E4 activity would be to keep infected keratinocytes in a metabolically active state without competing host DNA synthesis in order to maximize virus genome amplification. Such a scenario is not without precedent. Recent studies of herpesviruses (cytomegalovirus and Epstein-Barr virus) have shown that they have evolved mechanisms that allow activation of S-phase promoting cdk activity while cellular DNA synthesis is inhibited, presumably to support high-level viral DNA replication (19, 40).
Evidence to support a role for HPV E4 as a major regulator of the productive phase of the virus life cycle has yet to be presented. However, loss of E4 protein in papillomas induced in rabbits following inoculation with mutant E4 knockout CRPV genomes has been shown to severely compromise efficient viral genome amplification in suprabasal keratinocytes of papillomas (27). It was also noted that the E4 knockout genome was far more efficient at producing papillomas than the wild-type DNA, suggesting that CRPV E4 might negatively control CRPV DNA-containing keratinocyte proliferation. Thus, perhaps the ability to modify cell proliferation is a function conserved between E4 proteins of different virus types. This novel biological activity might be dependent on cooperation between different posttranslationally modified forms of the protein, as we have shown for HPV1 E4.
Acknowledgments
We are grateful to Andrew Turnell for critical reading of the manuscript and for his advice and encouragement during the course of this study. We thank Jonathan Pines for his kind gift of the cyclin B1-GFP expression vector. We acknowledge Michele McNally for excellent technical assistance, Gabriele Wölfler for help in making the proline deletion mutants, and David Lloyd for his advice and help with the flow cytometry studies.
This work was supported by a Cancer Research UK (CRUK) program grant (C427/2303) to S.R. and P.H.G. (CRUK Gibb Fellow).
REFERENCES
- 1.Almeida, J. D., A. F. Howatson, and M. G. Williams. 1962. Electron microscope study of human warts; sites of virus production and nature of the inclusion bodies. J. Invest. Dermatol. 38:337-345. [PubMed] [Google Scholar]
- 2.Ashmole, I., P. H. Gallimore, and S. Roberts. 1998. Identification of conserved hydrophobic C-terminal residues of the human papillomavirus type 1 E1∧E4 protein necessary for E4 oligomerisation in vivo. Virology 240:221-231. [DOI] [PubMed] [Google Scholar]
- 3.Barrera-Oro, J. G., K. O. Smith, and J. L. Melnick. 1962. Quantitation of papova virus particles in human warts. J. Natl. Cancer Inst. 29:583-595. [PubMed] [Google Scholar]
- 4.Breitburd, F., O. Croissant, and G. Orth. 1987. Expression of human papillomavirus type-1 E4 gene products in warts, In B. M. Steinberg, J. L. Brandsma, and L. B. Taichman (ed.), Papillomaviruses. Cancer Cells 5:115-122. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. [Google Scholar]
- 5.Brown, D. R., L. Pratt, J. T. Bryan, K. H. Fife, and K. Jansen. 1996. Virus-like particles and E1∧E4 protein expressed from the human papillomavirus type 11 bicistronic E1∧E4L̂1 transcript. Virology 222:43-50. [PubMed] [Google Scholar]
- 6.Cheng, S., D.-C. Schmidt-Grimminger, T. Murrant, T. R. Broker, and L. T. Chow. 1995. Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes Dev. 9:2335-2349. [DOI] [PubMed] [Google Scholar]
- 7.Chien, W.-M., F. Noya, H. M. Benedict-Hamilton, T. R. Broker, and L. T. Chow. 2002. Alternative fates of keratinocytes transduced by human papillomavirus type 18 E7 during squamous differentiation. J. Virol. 76:2964-2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chow, L. T., S. S. Reilly, T. R. Broker, and L. B. Taichman. 1987. Identification and mapping of human papillomavirus type 1 RNA transcripts recovered from plantar warts and infected epithelial cell cultures. J. Virol. 61:2581-2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davy, C. E., D. J. Jackson, Q. Wang, K. Raj, P. J. Masterson, N. F. Fenner, S. Southern, S. Cuthill, J. B. A. Millar, and J. Doorbar. 2002. Identification of a G2 arrest domain in the E1∧E4 protein of human papillomavirus type 16. J. Virol. 76:9806-9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doorbar, J., D. Campbell, R. J. A. Grand, and P. H. Gallimore. 1986. Identification of the human papillomavirus-1a E4 gene products. EMBO J. 5:355-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doorbar, J., H. S. Evans, I. Coneron, L. V. Crawford, and P. H. Gallimore. 1988. Analysis of HPV1 E4 gene expression using epitope-defined antibodies. EMBO J. 7:825-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doorbar, J., C. Foo, N. Coleman, L. Medcalf, O. Hartley, T. Prospero, S. Napthine, J. Sterling, G. Winter, and H. Griffin. 1997. Characterization of events during the late stages of HPV16 infection in vivo using high affinity synthetic Fabs to E4. Virology 238:40-52. [DOI] [PubMed] [Google Scholar]
- 13.Doorbar, J., E. Medcalf, and S. Napthine. 1996. Analysis of HPV1 E4 complexes and their association with keratins in vivo. Virology 218:114-126. [DOI] [PubMed] [Google Scholar]
- 14.Fehrmann, F., and L. A. Laimins. 2003. Human papillomaviruses: targeting differentiating epithelial cells for malignant transformation. Oncogene 22:5201-5207. [DOI] [PubMed] [Google Scholar]
- 15.Grand, R. J. A., J. Doorbar, K. J. Smith, I. Coneron, and P. H. Gallimore. 1989. Phosphorylation of the human papillomavirus type 1 E4 proteins in vivo and in vitro. Virology 170:201-213. [DOI] [PubMed] [Google Scholar]
- 16.Grassman, K., B. Rapp, H. Maschek, K. U. Petry, and T. Iftner. 1996. Identification of a differentiation-inducible promoter in the E7 open reading frame of human papillomavirus type 16 (HPV-16) in raft cultures of a new cell line containing high copy numbers of episomal HPV-16 DNA. J. Virol. 70:2339-2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hummel, M., J. B. Hudson, and L. A. Laimins. 1992. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J. Virol. 66:6070-6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hummel, M., H. B. Lim, and L. A. Laimins. 1995. Human papillomavirus type 31b late gene expression is regulated through protein kinase C-mediated changes in RNA processing. J. Virol. 69:3381-3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kudoh, A., M. Fujita, T. Kiyono, K. Kuzushima, Y. Sugaya, S. Izuta, Y. Nishiyama, and T. Tsurumi. 2003. Reactivation of lytic replication from B cells latently infected with Epstein-Barr virus occurs with high S-phase cyclin-dependent kinase activity while inhibiting cellular DNA replication. J. Virol. 77:851-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang, M-H., T. Geisbert, Y. Yao, S. H. Hinrichs, and C.-Z. Giam. 2002. Human T-lymphotropic virus type 1 oncoprotein Tax promotes S-phase entry but blocks mitosis. J. Virol. 76:4022-4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu, B., M.-H. Liang, Y. Kuo, W. Boros, T. Lkeinberger, J. Blancato, and C-Z Giam. 2003. Human T-lymphotropic virus type 1 oncoprotein tax promotes unscheduled degradation of Pds1p/securin and Clb2p/cyclin B1 and causes chromosomal instability. Mol. Cell. Biol. 23:5269-5281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mauser, A., E. Holley-Guthrie, D. Simpson, W. Kaufmann, and S. Kenney. 2002. The Epstein-Barr virus immediate early protein BZLF1 induces both a G2 and a mitotic block. J. Virol. 76:10030-10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakahara, T., A. Nishimura, M. Tanaka, T. Ueno, A. Ishimoto, and H. Sakai. 2002. Modulation of the cell division cycle by human papillomavirus type 18 E4. J. Virol. 76:10914-10920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nasseri, M., R. Hirochika, T. R. Broker, and L. T. Chow. 1987. A human papilloma virus type 11 transcript encoding an E1∧E4 protein. Virology 159:433-439. [DOI] [PubMed] [Google Scholar]
- 25.Palermo-Dilts, D. A., T. R. Broker, and L. T. Chow. 1990. Human papillomavirus type 1 produces redundant as well as polycistronic mRNAs in plantar warts. J. Virol. 64:3144-3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parkinson, E. K., P. Grabham, and A. Emmerson. 1983. A subpopulation of cultured human keratinocytes which is resistant to the induction of terminal differentiation-related changes by phorbol, 12-myristate, 13-acetate: evidence for an increase in the resistant population following transformation. Carcinogenesis 4:857-861. [DOI] [PubMed] [Google Scholar]
- 27.Peh, W. L., J. L. Brandsma, N. D. Christensen, N. M. Cladel, X. Wu, and J. Doorbar. 2004. The viral E4 protein is required for the completion of the cottontail rabbit papillomavirus productive cycle in vivo. J. Virol. 78:2142-2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peh, W. L., K. Middleton, N. Christensen, P. Nicholls, K. Egawa, K. Soltar, J. Brandsma, A. Percival, J. Lewis, W. J. Liu, and J. Doorbar. 2002. Life cycle heterogeneity in animal models of human papillomavirus-associated disease. J. Virol. 76:10401-10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pines, J., and T. Hunter. 1991. Human cyclins A and B1 are differentially located in the cell and undergo cell cycle-dependent nuclear transport. J. Cell Biol. 115:1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pines, J., and C. L. Rieder. 2001. Re-staging mitosis: a contemporary view of mitotic progression. Nat. Cell Biol. 3:E3-E6. [DOI] [PubMed] [Google Scholar]
- 31.Rheinwald, J. G., and M. A. Beckett. 1981. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultures from human squamous cell carcinomas. Cancer Res. 41:1657-1663. [PubMed] [Google Scholar]
- 32.Roberts, S. 2002. Biology of the E4 protein, 119-142. In D. J. McCance (ed.), Human papillomaviruses, vol. 8. Elsevier Science B. V., Amsterdam, The Netherlands.
- 33.Roberts, S., I. Ashmole, L. J. Gibson, S. M. Rookes, G. J. Barton, and P. H. Gallimore. 1994. Mutational analysis of human papillomavirus E4 proteins: identification of structural features important in the formation of cytoplasmic E4/cytokeratin networks in epithelial cells. J. Virol. 68:6432-6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts, S., I. Ashmole, G. D. Johnson, J. W. Kreider, and P. H. Gallimore. 1993. Cutaneous and mucosal papillomavirus E4 proteins form intermediate filament-like structures in epithelial cells. Virology 197:176-187. [DOI] [PubMed] [Google Scholar]
- 35.Roberts, S., I. Ashmole, T. M. T. Sheehan, A. H. Davies, and P. H. Gallimore. 1994. Human papillomavirus type 1 E4 protein is a zinc-binding protein. Virology 202:865-874. [DOI] [PubMed] [Google Scholar]
- 36.Roberts, S., M. L. Hillman, G. L. Knight, and P. H. Gallimore. 2003. The ND10 component promyelocytic leukemia protein relocates to human papillomavirus type 1 E4 intranuclear inclusion bodies in cultured keratinocytes and in warts. J. Virol. 77:673-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rogel-Gaillard, C., F. Breitburd, and G. Orth. 1992. Human papillomavirus type 1 E4 proteins differing by their N-terminal ends have distinct cellular localizations when transiently expressed in vitro. J. Virol. 66:816-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sigrist, S. J., and C. F. Lehner. 1997. Drosophila fizzy-related down-regulates mitotic cyclins and is required for cell proliferation arrest and entry into endocycles. Cell 90:671-681. [DOI] [PubMed] [Google Scholar]
- 39.Stoler, M. H., S. M. Wolinsky, A. Whitbeck, T. R. Broker, and L. T. Chow. 1989. Differentiation-linked human papillomavirus types 6 and 11 transcription in genital condylomata revealed by in situ hybridisation with message-specific RNA probes. Virology 172:331-340. [DOI] [PubMed] [Google Scholar]
- 40.Wiesbusch, L., and C. Hagemeier. 2001. The human cytomegalovirus immediate early 2 protein dissociates cellular DNA synthesis from cyclin-dependent kinase activation. EMBO J. 20:1086-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wuarin, J., V. Buck, P. Nurse, and J. B. A. Millar. 2002. Stable association of mitotic cyclin B/Cdc2 to replication origins prevents endoreduplication. Cell 111:419-431. [DOI] [PubMed] [Google Scholar]