

Abstract
Fusion proteins of enveloped viruses categorized as class I are typified by two distinct heptad repeat domains within the transmembrane subunit. These repeats are important structural elements that assemble into the six-helix bundles characteristic of the fusion-activated envelope trimer. Peptides derived from these domains can be potent and specific inhibitors of membrane fusion and virus infection. To facilitate our understanding of retroviral entry, peptides corresponding to the two heptad repeat domains of the avian sarcoma and leukosis virus subgroup A (ASLV-A) TM subunit of the envelope protein were characterized. Two peptides corresponding to the C-terminal heptad repeat (HR2), offset from one another by three residues, were effective inhibitors of infection, while two overlapping peptides derived from the N-terminal heptad repeat (HR1) were not. Analysis of envelope mutants containing substitutions within the HR1 domain revealed that a single amino acid change, L62A, significantly reduced sensitivity to peptide inhibition. Virus bound to cells at 4°C became sensitive to peptide within the first 5 min of elevating the temperature to 37°C and lost sensitivity to peptide after 15 to 30 min, consistent with a transient intermediate in which the peptide binding site is exposed. In cell-cell fusion experiments, peptide inhibitor sensitivity occurred prior to a fusion-enhancing low-pH pulse. Soluble receptor for ASLV-A induces a lipophilic character in the envelope which can be measured by stable liposome binding, and this activation was found to be unaffected by inhibitory HR2 peptide. Finally, receptor-triggered conformational changes in the TM subunit were also found to be unaffected by inhibitory peptide. These changes are marked by a dramatic shift in mobility on sodium dodecyl sulfate-polyacrylamide gel electrophoresis, from a subunit of 37 kDa to a complex of about 80 kDa. Biotinylated HR2 peptide bound specifically to the 80-kDa complex, demonstrating a surprisingly stable envelope conformation in which the HR2 binding site is exposed. These experiments support a model in which receptor interaction promotes formation of an envelope conformation in which the TM subunit is stably associated with its target membrane and is able to bind a C-terminal peptide.
Viral envelope proteins with fusogenic capability can be classified into at least two distinct groups, based on common structural characteristics. Class I fusion proteins are found in many disparate virus families, including retroviruses, orthomyxoviruses, paramyxoviruses, arenaviruses (17), coronaviruses (4), and filoviruses (for a review, see references 6 and 13). Viral fusion proteins of alphaviruses and flaviviruses have been denoted as class II (20). Class I envelope proteins are synthesized as type I membrane proteins, which are generally cleaved into two functionally distinct domains. The N-terminal subunit (designated SU in the case of retroviruses) usually contains all of the receptor-binding capability and is therefore an important determinant of tropism. The C-terminal subunit (TM for retroviruses) is the membrane-anchored subunit, and it includes elements critical for mediating membrane fusion. Class I fusion proteins assemble into trimers, and the two subunits remain associated with one another after processing, generating a trimer of heterodimers.
The envelope element that interacts directly with the target lipid bilayer is referred to as the fusion peptide, a region of around 20 mostly hydrophobic amino acids at or near the amino terminus of TM. The TM subunit also has two regions predicted to display a helical secondary structure; these two regions are located between the fusion peptide and the membrane-spanning domain and are often referred to as HR1 and HR2 (for heptad repeats 1 and 2). In the absence of the SU domain, the heptad repeats form a highly stable coiled-coil known as the six-helix bundle, with three HR2 regions packed antiparallel against an internal HR1 trimer (6). The six-helix bundle is thought to represent the postfusion conformation of TM, and it is the formation of this stable structure that is thought to drive membrane fusion.
Synthetic peptides derived from the HR2 regions of envelopes from several viruses have proven to be specific and potent inhibitors of viral entry and cell-cell fusion mediated by their respective viral envelope proteins. This has been demonstrated for the retroviruses human immunodeficiency virus (HIV) (22, 48), simian immunodeficiency virus (33), human T-cell leukemia virus (34, 40), and feline immunodeficiency virus (27), two coronaviruses (4, 24), and several paramyxoviruses (23, 35, 51). Fuzeon (enfuvirtide) is a drug which has recently been approved for treating HIV infections based on this paradigm (5). Peptide inhibitors derived from the HR1 region, although less potent, have also been described (24, 47, 52).
The subgroup A avian sarcoma and leukosis virus (ASLV-A) is a prototypic alpharetrovirus that has been employed as a model for studying envelope triggering and viral entry. The receptor for ASLV-A, Tva, can be produced in a soluble form (2) which is sufficient for mediating virus entry into otherwise refractory receptor-deficient cell lines (7). The conversion of envelope to a fusion active state is correlated with a number of biochemical changes. A conformational transformation in the SU subunit results in increased protease sensitivity (8, 19), and changes in the TM subunit include exposure of the fusion peptide (19) and conversion to an oligomeric structure as observed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (1, 26, 29, 41). In addition, receptor binding induces envelope to bind liposomes, consistent with exposure of the fusion peptide (8, 9, 21). Unlike either the classical pH-dependent or pH-independent viruses, ASLV receptor binding can initiate envelope activation for subsequent further activation by acidic pH, revealing multiple triggering events (28). Here we describe the design of an effective HR2 inhibitor for ASLV-A. We used this inhibitor as a tool to probe conformational changes in TM, and we began to examine viral escape mechanisms for this important new type of fusion inhibitor.
MATERIALS AND METHODS
Viruses and cells.
QT6 cells, a quail myosarcoma line (American Type Culture Collection), were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS) and 2 mM l-glutamine. Turkey embryo fibroblasts (TEF) were maintained in M199 medium supplemented with 10% tryptose phosphate broth, 5% FCS, and 1% chick serum. Human 293T cells were maintained in DMEM with 10% bovine calf serum. DF-1 cells, a line derived from chicken embryo fibroblasts, were maintained in DMEM with 10% FCS. RCASBP(A)-GFP-infected DF-1 cells were kindly donated by Mark Federspiel (Mayo Clinic). HeLa cells containing an integrated HIV type 1 (HIV-1) long terminal repeat (LTR)-lacZ cassette (HeLa-MAGI) were obtained from the AIDS Reagent Program (National Institutes of Health) and were grown in DMEM with 10% FCS. Penicillin (100 U/ml) and streptomycin (100 μg/ml) were also added to all cell lines. Fusion target cells were generated by G418 selection of pCB6-Tva950-transfected (CaPO4) HeLa-MAGI cells. Independent clones were screened for high levels of Tva expression and preservation of HIV-1 Tat-inducible β-galactosidase expression; one clone with these properties was used for all of the cell-cell fusion assays.
Stocks of the replication-competent avian retroviral vector RCAS(A)AP (38) were produced on TEF. Murine leukemia virus (MLV) pseudotypes were produced by CaPO4 transfection of 293T cells as described previously (49). Virus-containing medium was filtered (0.45-μm pore size) and stored at −80°C. MLV(Ebola GP) pseudotypes were filtered and then concentrated by centrifugation through a cushion of 20% sucrose in Dulbecco's phosphate-buffered saline (D-PBS) at 200,000 × g for 45 min at 4°C. Pelleted virus was resuspended in HEPES-buffered saline (HBS; 20 mM HEPES [pH 7.7], 130 mM NaCl, 0.5 mM MgCl2) and stored at −80°C.
To purify ASLV-A for the liposome association and TM oligomerization assays, virus-containing supernatant was harvested from DF-1 cells chronically infected with a recombinant ASLV-A [RCASBP(A)-GFP], filtered (0.45-μm pore size), and layered onto a step gradient with steps of 20 and 60% sucrose in D-PBS. Following a 2-h centrifugation at 80,000 × g at 4°C, the 20-to-60% interface was isolated and diluted threefold with D-PBS. The diluted sample was layered above 20% sucrose in D-PBS and spun again, and the viral pellet was resuspended in HBS overnight at 4°C. For the TM oligomerization assay, this stock was used directly without freezing.
Plasmids.
Expression constructs pCB6-EnvA and pCB6-EboGP, encoding envelopes from ASLV-A (18) and the Zaire subtype of Ebola virus (49), respectively, have been described elsewhere. pCB6-myc-EnvA encodes a myc epitope tag near the amino terminus of SU and has also been described previously (3). Mutant plasmids were engineered with the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, Calif.) using pCB6-myc-EnvA as a template and the following mutagenic oligonucleotides (and their respective complements): OS676 (5′-CAGGCTAACTTGACAGCATCACTCCTCGGGG-3′) for pCB6-T55A; OS674 (5′-GACAACATCACTCGCCGGGGACTTATTGG-3′) for pCB6-L58A; OS592 (5′-CCTCGGGGACTTAGCGGATGATGTCACG-3′) for pCB6-L62A; and OS590 (5′-GATGTCACGAGTATTGAACACGCGGTCCTG-3′) for pCB6-R69E. Amino acid numbering begins after the polybasic cleavage site for both ASLV-A TM and Ebola GP2.
Proteins.
Peptides were synthesized by Alpha Diagnostic International, Inc., with N-terminal acetylation, C-terminal amidation, and purification to greater than 95%. The sequences (N to C termini) of the peptides are as follows: R42, RLASWSVKQANLTTSLLGDLLDDVT; R57, LLGDLLDDVTSIRHAVLQNRAAIDF; R99, FNLSDHSESIQKKFQLMKEHVNKIG; R102, SDHSESIQKKFQLMKEHVNKIGVDS; E109, IEPHDWTKNITDKIDQIIHDFVDKT;E112, HDWTKNITDKIDQIIHDFVDKTLPD. R99-Bio is identical to R99 except that it is biotinylated at its C terminus. All peptides were solubilized in water with the exception of R42, which was reconstituted in 25% dimethyl sulfoxide due to its insolubility in water. Aliquots of reconstituted peptide were stored at −20°C. Soluble ASLV-A receptor, sTva, was purified from the medium of Sf9 cells infected with a baculovirus recombinant as described elsewhere (2).
Viral infections.
Cells were seeded in 35-mm wells, 16 to 20 h prior to infection, at a density of approximately 4 × 105 cells per well. Viral stocks were diluted in medium (see above) and incubated with peptide in a total volume of 2 ml at room temperature for 2 min and then used to challenge the target cells. Infectivity was measured 48 h postinfection. For the MLV pseudotypes, titer was determined by staining for β-galactosidase activity and counting positive cells as described previously (49); titers were in the range of 2 × 105 to 5 × 105 infectious units per ml and were 3- to 10-fold higher when using centrifugal inoculation. For RCAS(A)AP infection of TEF cells, a histochemical stain for alkaline phosphatase (AP) activity was used to calculate viral titer (38).
To determine the incorporation of ASLV-A envelope into MLV pseudotypes, virus was concentrated and purified by centrifugation as described for MLV(Ebola GP) above, lysed in Triton lysis buffer (1% Triton X-100, 150 mM NaCl, 50 mM Tris [pH 8.0], 5 mM EDTA [pH 8.0]), separated by SDS-PAGE, and transferred to a nitrocellulose membrane. Envelope was detected by Western blotting as described previously (38) using an anti-ASLV-A TM antibody (50) at a 1:1,000 dilution.
Peptide addition and removal experiments.
QT6 cells were seeded and infected as described above using centrifugal inoculation at 4°C in a refrigerated, table-top centrifuge for 2.5 h at 1,700 × g. For the peptide addition experiments, unbound virus was washed away with ice-cold D-PBS, 37°C medium was added to the cells, and the plates were placed in a 37°C incubator. At various times after the temperature increase, R99 was added to a final concentration of either 10 μg/ml for wild-type MLV(EnvA) pseudotypes or 50 μg/ml for MLV(EnvA-L62A). Viral titers were determined as described above. The peptide inhibitor washout experiments were performed in much the same way. Following centrifugal inoculation, unbound virions were removed by washing with ice-cold D-PBS. Ice-cold medium containing 80 nM sTva and 10 μg of R99/ml was added to the cells for 1 h at 4°C. After this incubation, an equal volume (1 ml) of prewarmed medium (50°C) was added, and the cells were placed at 37°C. At the desired time points, the cells were placed on ice, washed three times with ice-cold medium, and then returned to the 37°C incubator. Viral titers were determined as described above. Incubation of cell-bound virus at 4°C with or without 10 μg of R99/ml, followed by three washes as above, yielded equivalent infectivity, indicating that the peptide was adequately removed and that the virus did not become peptide sensitive at this temperature.
Cell-cell fusion.
Effector cells were generated by CaPO4 transfection of 293T cells in 8.5-cm dishes with 0.5 μg of pCB6-EnvA and 10 μg of a plasmid encoding HIV-1 Tat (kindly provided by Michael Malim, King's College London). To enhance envelope expression, sodium butyrate was added at 20 h posttransfection to a final concentration of 10 mM and then removed 4 h later. At 48 h posttransfection, transfected cells and Tva-expressing HeLa-MAGI cells (see above) were released from their dishes by treatment with 5 mM EDTA in D-PBS without MgCl2 and CaCl2 and then coseeded in 96-well plates. The cells were allowed to adhere for 3 h and then washed once with D-PBS prior to incubation in fusion medium (RPMI medium 1640 with 0.2% bovine serum albumin and 10 mM HEPES adjusted to pH 5.25 or 7.0) for 10 min. The cells were washed again with D-PBS and returned to DMEM with 10% FCS. R99 was included at a final concentration of 5 μg/ml where indicated. Twelve hours later, β-galactosidase activity was measured using the Galacto-Star system (Applied Biosystems) as per the manufacturer's instructions. Luminescence was recorded with a Trilux 1450 MicroBeta luminescence counter (Wallac).
Peptide inhibition in the presence of bafilomycin.
QT6 cells were treated for 30 min at 37°C with or without 10 nM bafilomycin, cooled to 4°C, and infected with MLV(EnvA) pseudotype virus in the continued presence of bafilomycin with centrifugal inoculation for 2.5 h as described above. Soluble receptor (20 nM final sTva) and R99 (5 μg/ml final concentration) were added as indicated, and cells were warmed to 37°C and incubated at that temperature for 30 min. Medium containing receptor and peptide was removed, monolayers were washed with bafilomycin-containing medium, and fresh medium with 10 nM bafilomycin was added to the cells. After incubation at 37°C for another 1 h, cells were washed with D-PBS and drug-free medium was added. Viral titer was determined 48 h postinfection as described above.
Liposome association assays.
Liposomes were synthesized as a 2:1 ratio of egg phosphatidylcholine (13 mM) and cholesterol (6.5 mM) in chloroform with 40 mM n-octyl-β-d-glucopyranoside by using the Mini Lipoprep apparatus (Amika) as per the manufacturer's instructions and overnight dialysis against D-PBS without MgCl2 and CaCl2. Binding of soluble, oligomeric ASLV-A envelope to liposomes was performed as described previously (9). Proteins were separated by SDS-PAGE and analyzed by Western blotting with polyclonal antisera against the SU subunit of ASLV-A envelope. This rabbit antiserum was raised against an ASLV-A SU-immunoadhesin (53).
An “underlay” method was used to gauge association between virus and liposomes. Concentrated RCASBP(A)-GFP was incubated with sTva for 30 min at 4°C in a total volume of 40 μl of HBS. Liposomes (40 μl) were added, and the samples were incubated at 37°C for 30 min. Each sample was then mixed with 170 μl of 65% sucrose in D-PBS and used as the bottom layer of a three-step sucrose gradient, with a 150-μl middle layer of 25% and a 300-μl top step of 5%. The gradient was spun in 5- by 41-mm tubes (Beckman Ultra-Clear) at 150,000 × g for 2.5 h at 4°C. Following centrifugation, fractions of 300, 100, and 300 μl were collected, starting at the top. Proteins were precipitated with trichloroacetic acid as previously described (9), separated by SDS-PAGE, and analyzed by Western blotting with an anti-EnvA TM polyclonal antiserum.
TM oligomerization assay.
Purified RCASBP(A)-GFP was incubated on ice for 30 min along with, as specified, 250 ng of sTva, 20 μl of liposomes, 0.5 μg of R99-Bio, and 0, 0.5, or 5 μg of unmodified R99. Samples were then incubated at 37°C for 30 min and returned to ice, and the cross-linker bis(sulfosuccinimidyl)suberate was added to a final concentration of 4 mM for 2 h. The cross-linking reaction was stopped by addition of glycine to 80 mM on ice for 15 min. Low-SDS Laemmli buffer was added (final SDS concentration, 0.1%) and incubated at 37°C for 10 min prior to separation by electrophoresis on a Criterion Tris-HCl 4-to-15% polyacrylamide gradient gel (Bio-Rad). Following transfer to nitrocellulose, biotinylated peptides were detected with horseradish peroxidase-conjugated streptavidin and SuperSignal chemiluminescence (Pierce).
For immunoprecipitation of TM complexes, cross-linked samples were incubated in RIPA buffer (140 mM NaCl, 10 mM Tris-Cl [pH 8], 5 mM EDTA, 1% sodium deoxycholate, 1% Triton X-100, and 0.1% SDS) for 1 h at 4°C. Samples were precleared with protein A-Sepharose and Sepharose CL-4B (Sigma), and then 2 μl of polyclonal rabbit antiserum directed against a peptide derived from the cytoplasmic domain of TM (18) was added and incubated for 1 h at 4°C. To precipitate the immune complexes, a 1:1 mixture of protein A-Sepharose/Sepharose CL-4B was added and incubated for 1 h at 4°C. The precipitate pellet was washed once with RIPA buffer, eluted with Laemmli buffer at 100°C for 10 min, and then analyzed by SDS-PAGE as described above.
RESULTS
Synthetic peptide sequences.
The TM subunit of the ASLV envelope is similar to the membrane-anchored subunit of the envelope protein from the filovirus Ebola (GP2) in terms of primary sequence and predicted structural features (16, 44) (Fig. 1A). The crystal structure of the Ebola virus GP2 subunit demonstrated the existence of a six-helix bundle structure composed of the HR regions, similar to the structures of the HIV-1 and MLV TM subunits (25, 46). Using the Ebola virus GP2 structure as a guide, six different 25-residue-long peptides derived from the ASLV-A and the Ebola virus HR domains were designed and synthesized (Fig. 1A).
FIG. 1.

Comparison of the TM subunit of ASLV-A envelope with Ebola virus GP2. (A) Schematic diagram of the ASLV-A envelope. The fill patterns depict the predicted domains of the TM subunit as follows: diagonal lines, fusion peptide (FP); shaded, N- and C-terminal HRs (HR1 and HR2); horizontal lines, membrane-spanning domain (MSD). Shown below the schematic diagram is a sequence alignment of the heptad repeats and intervening sequences from the TM of ASLV-A and Ebola virus GP2 (Zaire subtype), with the one-letter amino acid code. For the Ebola sequence, dots indicate amino acids identical to ASLV-A TM and the asterisk in the ASLV-A sequence designates a space introduced in order to align the two sequences (16, 46). The cylinders mark regions predicted to be helical, and the brackets indicate the sequence of synthetic peptides. (B) Ribbon diagram showing the structure of Ebola virus GP2 (46) using RasMol modeling. The substitutions introduced into the HR1 domain of the TM subunit of ASLV-A are each in positions for which the sequence alignment displays identical residues between the two viruses; the side chains for these mutated residues are shown (ASLV numbering). The region corresponding to E109 and R99 is shown in dark.
Peptides R99 and R102 correspond to the predicted HR2 region of the ASLV-A TM, extending from residues 99 to 123 and 102 to 126, respectively. Ebola virus-specific peptides (E109 and E112, numbered according to the first residue of the peptide) analogous to R99 and R102 were also synthesized. In addition to the HR2 peptides, two overlapping peptides of 25 residues each, R42 and R57, were derived from the HR1 region of ASLV-A. The R42 sequence substitutes a serine in place of the cysteine found in ASLV-A TM position 45 to eliminate any potential complications imposed by disulfide bond formation.
Peptide inhibition of viral infection.
The ability of each peptide to inhibit viral entry was initially tested using viral pseudotypes. Pseudotypes based on an MLV core were produced by transient transfection of 293T cells with three plasmids encoding viral envelope, MLV gag/pol, and an MLV genome containing a β-galactosidase reporter gene (42). This system allows for envelope function to be tested independently of the virus from which it was derived. The four ASLV-specific peptides and one Ebola-specific peptide (E112) were tested for their ability to inhibit infection of avian QT6 cells by MLV(EnvA)-pseudotyped virus. Both of the ASLV HR2-derived peptides demonstrated significant inhibitory activity (Fig. 2A). The 50% inhibitory concentration (IC50) for R102 was approximately 15 μg/ml (5 μM), while R99 was more potent, with an IC50 of approximately 0.5 μg/ml (170 nM). The two HR1 peptides, R42 and R57, did not inhibit MLV(EnvA) infectivity at concentrations up to 100 μg/ml (data not shown). The Ebola virus-specific peptide, E112, had no effect on MLV(EnvA) (Fig. 2A). Interestingly, neither E112 nor E109 was able to inhibit the infectivity of pseudotyped virus carrying the Ebola virus glycoprotein at concentrations up to 200 μg/ml. R102 was unable to inhibit either MLV(Ebola GP) or MLV(MLV) infectivity (data not shown), thereby confirming the specificity of this peptide for ASLV envelope. To ensure that the inhibition observed was not limited to the pseudotype system, the two ASLV HR2 peptides were tested for their ability to block infection of recombinant ASLV-A that expresses an AP marker gene [RCAS(A)AP]. As seen in Fig. 2B, R99 and R102 inhibition of RCAS(A)AP infection of TEF cells correlated well with the inhibition seen with MLV(EnvA) infection of QT6 cells. The somewhat increased sensitivity to peptide exhibited by RCAS(A)AP might be due to differences in envelope density, cell type, or to the replication competence of RCAS(A)AP allowing secondary infections.
FIG. 2.
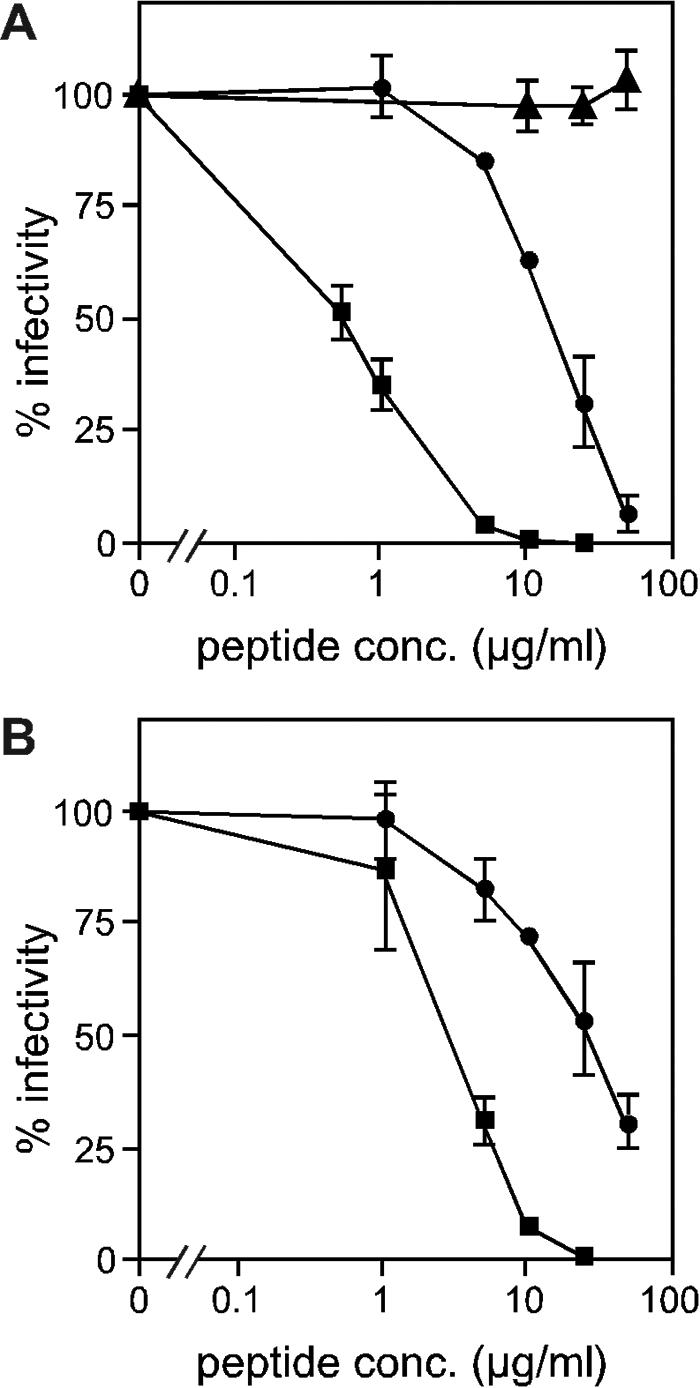
Peptide inhibition of infection. (A) MLV(EnvA) pseudotypes were used to infect QT6 cells in the presence of R99 (squares), R102 (circles), or Ebola virus-derived E112 (triangles). (B) ASLV-A [RCAS(A)AP] was used to infect TEF cells in the presence of R99 (squares) or R102 (circles). Percent infectivity is relative to the titer in the absence of peptide. Each data point is the average of three experiments, and error bars indicate standard deviations.
Identification of an HR1 mutant with diminished sensitivity to HR2 peptides.
Inhibitors derived from the HR2 region are believed to interact with HR1 domains exposed within a transient intermediate, thus blocking completion of the conformational changes necessary for fusion (14). Escape mutants of HIV-1, derived by passage of the virus in the presence of the HR2 peptide DP178, contain substitutions located within the HR1 region that decrease its affinity for the inhibitory peptide (37). To address whether ASLV HR1 residues can modulate peptide sensitivity, four amino acids predicted to interact with R102 by comparison to the Ebola virus GP2 structure were individually changed to either alanine or glutamic acid (Fig. 1B). MLV vectors pseudotyped with these mutant envelopes (T55A, L58A, L62A, and R69E) were assayed for glycoprotein function and peptide sensitivity. All four mutant envelopes were properly processed and incorporated into virions at levels comparable to wild type as assayed by Western blotting (data not shown). Mutants T55A, L62A, and R69E displayed normal levels of infectivity, while L58A was severely impaired and not tested further. The envelope proteins with mutations at positions 55 and 69 were sensitive to both HR2 peptides similar to wild type (Fig. 3). In contrast, MLV(EnvA-L62A) retained 95% of its infectivity at 50 μg of R102/ml, a concentration at which wild-type MLV(EnvA) was only 5% infectious (Fig. 3A). Similarly, MLV(EnvA-L62A) exhibited about a 20-fold increase in the R99 IC50, from 0.5 to 10 μg/ml (Fig. 3B).
FIG. 3.
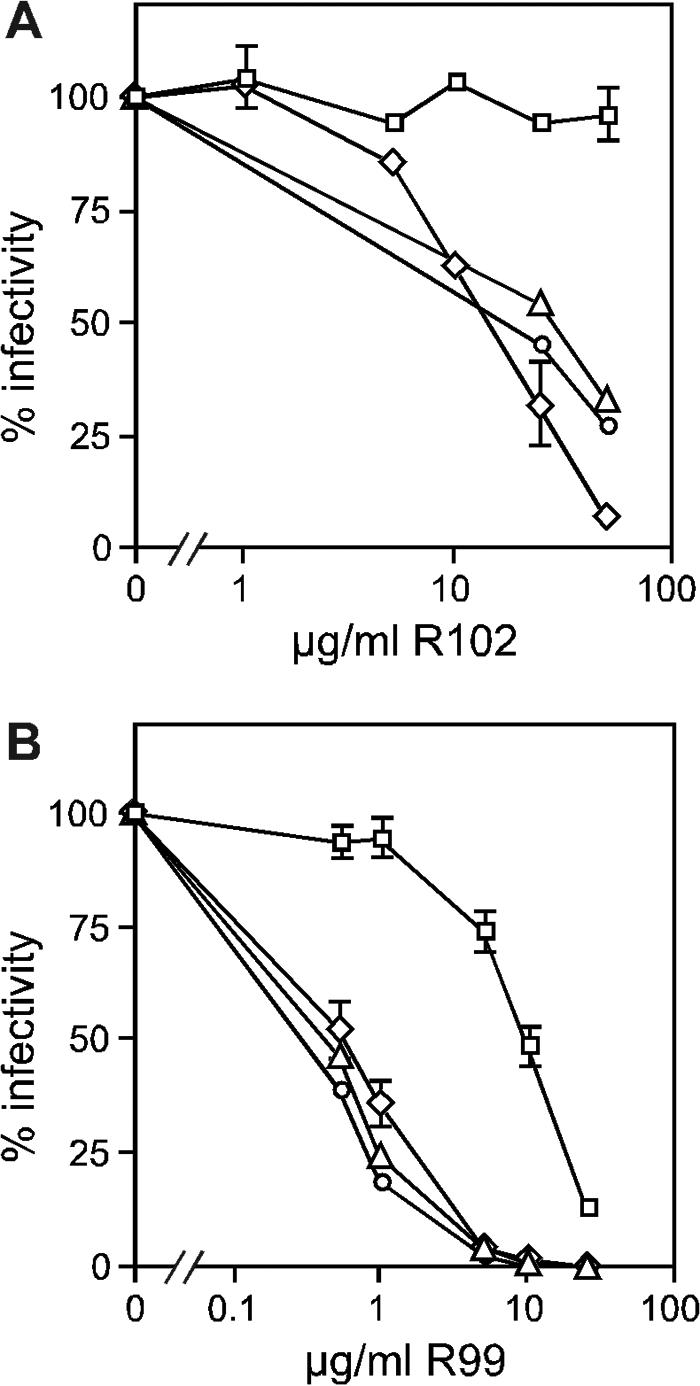
Effect of single amino acid substitutions in HR1 on sensitivity to HR2 peptides. MLV(EnvA) pseudotypes were generated with either wild-type ASLV-A envelope (diamonds) or HR1 mutants T55A (triangles), L62A (squares), or R69E (circles). Virus was incubated with either R102 (A) or R99 (B) for 2 min at room temperature prior to challenging QT6 cells. Percent infectivity is relative to the titer in the absence of peptide. Data points are the averages of three experiments (wild type and L62A) with error bars marking standard deviations, or they are from a single representative experiment (T55A and R69E).
Time dependence of susceptibility to HR2 peptide.
HR2 peptides are believed to exert their inhibition by targeting a transient intermediate. To examine the timing of the formation and disappearance of this intermediate, R99 was added to or removed from a viral infection at various times. Pseudotyped MLV(EnvA) virus was bound to QT6 cells at 4°C, cells were washed to remove unbound virions, and the temperature was raised to 37°C by the addition of warm medium.
For the R99 removal experiment, peptide was added to cells with bound virus after the 4°C wash and before elevating the temperature. At various times following the temperature shift, medium containing unbound peptide was removed, the cell monolayer was washed, and 37°C peptide-free medium was added. Initial experiments showed that it was difficult to achieve complete inhibition after peptide removal (data not shown). This observation suggested the presence of a reservoir of unexposed R99 binding sites, possibly representing envelope trimers on the virion that have not yet been triggered by engaging receptor on the host cell surface. To allow synchronous and complete activation of viral envelopes, soluble receptor (sTva) was added to cell-bound virus at 4°C. Addition of soluble receptor after binding virus to the cells did in fact now allow for the complete inhibition of infection in the R99 washout experiments (Fig. 4A). No inhibition occurred when the peptide was removed simultaneously with the temperature shift, indicating that the target of R99 action is not accessible at 4°C (Fig. 4A). Within the first 15 min at 37°C, virus became completely sensitive to R99, as removing the peptide after this time still caused full inhibition.
FIG. 4.
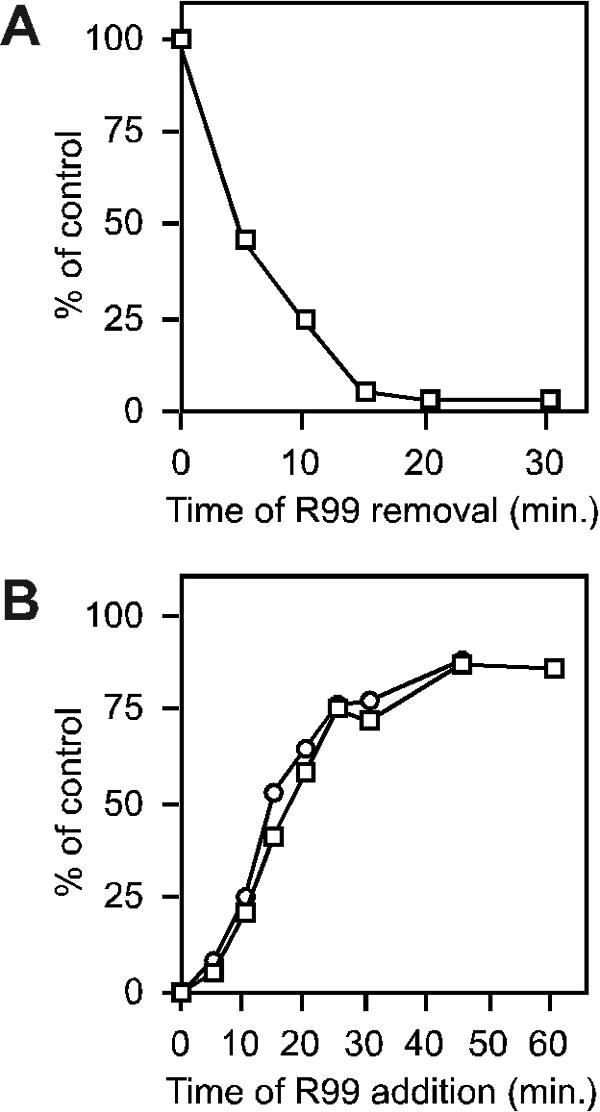
Sensitivity to HR2 peptide as a function of time at 37°C. Pseudotyped virus was allowed to bind QT6 cells at 4°C, and then unbound virus was washed away. (A) Peptide removal experiment to determine the time at which the R99 binding site is exposed. Cold medium containing 10 μg of R99/ml and 80 nM sTva was added to cells, and 1 h later cells were rapidly warmed to 37°C. For each time point, cells were washed to remove peptide and then returned to 37°C. Infectivity is normalized to a control in which the peptide was washed away prior to the warm-up step. Data points are averages of three replicates. (B) Peptide addition experiment to assess the longevity of its efficacy. Cells were warmed to 37°C, and R99 was added to a final concentration of 10 (wild-type) or 50 (L62A) μg/ml at the indicated time. MLV pseudotypes bore either a wild-type ASLV-A envelope (squares) or the L62A mutant (circles). For each pseudotype, infectivity was normalized to a control in which peptide was not added. Each data point is the average of three replicates.
The duration of the transiently accessible intermediate on which R99 acts was determined by adding the inhibitor at various times after raising the temperature. Inhibition was essentially complete when R99 was added within the first 5 min, but very little effect was seen when peptide was added after 45 min, demonstrating the disappearance of the targeted intermediate (Fig. 4B). To achieve more than a twofold reduction in infectivity, inhibitor had to be added within the first 15 min of elevating the temperature. EnvA(L62A) produced a very similar time-dependent curve (using five times as much peptide to compensate for its lower sensitivity), suggesting that the reduced peptide sensitivity of this envelope is not due to faster kinetics of envelope activation or fusion. These data together illustrate an envelope intermediate that emerges within 5 min and is no longer accessible after 30 to 45 min.
HR2 peptide inhibition does not require acid activation.
The activation of ASLV envelope has been proposed to be a multistep process involving receptor binding and acidic pH sequentially (29). Supporting this model, envelope-mediated cell-cell fusion is considerably enhanced by lowering the pH (12, 28, 29). In order to determine how this effect relates to exposure of the R99 peptide binding site, a quantitative cell-cell fusion assay was developed. Effector cells (293T cells transfected with ASLV-A envelope- and HIV-1 Tat-expressing plasmids) were mixed with target cells (a stable HeLa-MAGI cell line expressing the Tva receptor), exposed to pH 5.25 for 10 min, and then assayed 12 h later for β-galactosidase activity by chemiluminescence. The HeLa-MAGI cells contain an HIV-1 LTR-lacZ cassette such that Tat-induced β-galactosidase activity can be used to measure fusion of the two distinct cell types. When 5 μg of R99/ml was included throughout this assay, β-galactosidase activity was reduced more than 95% (Fig. 5, lane 2), demonstrating fusion inhibition. A similar level of inhibition was observed when unbound peptide was washed away immediately prior to the acidification step (Fig. 5, lane 3), showing that acid is not a requirement for exposure of the peptide binding site. Fusion was reduced approximately 50% when R99 was present only during the acidification step (Fig. 5, lane 4); however no inhibition was observed when R99 was added immediately after the acid pulse (Fig. 5, lane 5). These findings are consistent with an acid-induced envelope refolding event that eliminates the peptide binding site. The level of β-galactosidase activity detected when cell-cell fusion was done at neutral pH was about 10-fold lower than when done with an acid pulse, in agreement with a previous report (12). Cell-cell fusion performed at neutral pH was similarly inhibited by R99 (12) (data not shown).
FIG. 5.
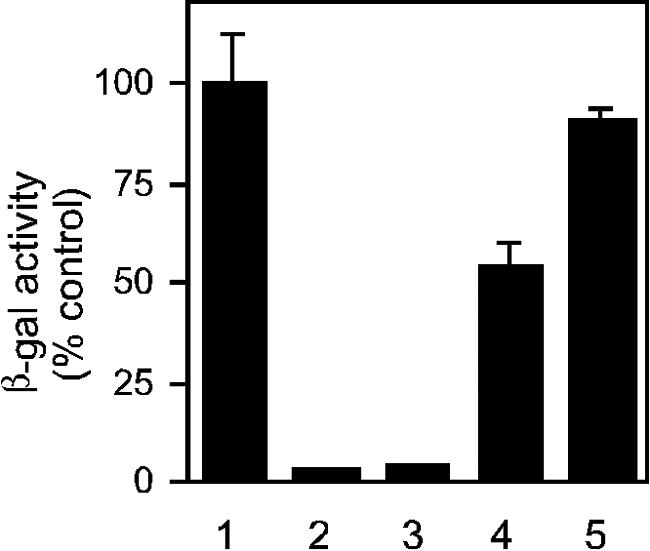
Inhibition of cell-cell fusion by HR2 peptide occurs prior to acid activation. Transfected 293T cells expressing ASLV-A envelope and HIV-1 Tat were seeded in an equimolar ratio with HeLa-MAGI cells stably expressing the Tva receptor. Cells were allowed to attach for 3 h, washed, pulsed with low-pH medium (pH 5.25), washed again, and assayed for β-galactosidase activity 12 h later. R99 was included at 5 μg/ml during the 3-h attachment period (lane 3), during the acid pulse (lane 4), during the 12-h recovery phase (lane 5), or throughout the entire assay (lane 2). Lane 1 is a control in which R99 was not used.
The observation that envelope-mediated cell-cell fusion can be inhibited by HR2 peptides without acid activation was extended to virus entry. Lysosomotropic agents that neutralize endocytic vesicles have been classically employed to examine the requirement for low pH in viral entry. In the presence of 10 nM bafilomycin, which is sufficient to neutralize avian cell endosomes (12; S. Matsuyama and J. M. White, personal communication), MLV(EnvA) pseudotypes are fully inhibitable by R99 (Table 1), again demonstrating that exposure of the HR2 peptide binding site does not require an acid trigger. As described previously, soluble receptor was added in this experiment to activate all envelopes and thus ensure that after removal of R99 and bafilomycin, infection would not be mediated by previously untriggered envelopes. In a separate control, QT6 cells treated with bafilomycin and R99 were found to be fully infectible if virus was added after removal of these agents (data not shown).
TABLE 1.
Neutralization of endosomes by bafilomycin does not reduce HR2 peptide efficacy
| Treatment | −Bafilomycin
|
+10 nM bafilomycin
|
||
|---|---|---|---|---|
| Titer | % Inhibition | Titer | % Inhibition | |
| sTvaa only | 4.4 × 106 | 6.0 × 106 | ||
| sTvaa + R99b | 1.8 × 105 | 96 | 1.4 × 105 | 98 |
20 nM.
5 μg/ml.
Effect of HR2 peptides on conversion of envelope to a fusogenic state.
Binding to the Tva receptor triggers several measurable changes in the ASLV-A envelope, including exposure of the fusion peptide (19) and conversion to a lipophilic, membrane-binding state (8, 9, 12, 21). This conversion can be demonstrated by liposome association of soluble envelope (an oligomer of the ectodomain), ASLV-A, or pseudotyped viral particles. Liposomes and associated protein or virus are separated from free virus or proteins by flotation on a sucrose gradient. When inhibitory peptide was added in excess prior to envelope triggering by soluble receptor, no significant effect was noted on the induction of liposome binding, either with soluble envelope (Fig. 6A) or viral particles (Fig. 6B). The more potent of the two HR2 peptides, R99, did not diminish envelope binding to liposomes when tested at concentrations as high as 300 μg/ml (data not shown). This result is consistent with the formation of an envelope intermediate upon soluble receptor triggering, in which the TM fusion peptide associates with the target lipid bilayer. Although this intermediate is thought to be the target of HR2 peptides, its formation would be predicted to be peptide insensitive.
FIG. 6.

Liposome binding is not blocked by HR2 peptides. Sucrose step gradients were used to separate liposomes by their buoyant densities. Soluble, oligomeric ASLV-A envelope (A) or ASLV-A virions (B) were incubated with or without soluble receptor (sTva) to activate envelope in the presence or absence of HR2 peptides. The R102 peptide was used at a concentration of 50 μg/ml (A), and R99 was used at 30 μg/ml (B). Each fraction (top, middle, bottom) was examined by SDS-PAGE and Western blotting with antisera against either the SU (A) or TM (B) subunit. Samples were loaded in the dense bottom fraction, so that liposome-associated material would float to the upper, less-dense fractions. The retarded migration of SU in the bottom fraction of the first gradient in panel A is an SDS-PAGE artifact.
HR2 peptide binds an activated form of TM.
Another assay for examining ASLV envelope activation is the detection of a stable, oligomeric TM complex (1, 26, 29, 41). The TM subunit from native virions migrates on SDS-polyacrylamide gels as a monomer with an apparent molecular mass of about 37 kDa. However, upon receptor engagement at 37°C, a large mobility shift occurs on SDS-PAGE (1, 26). Triggered envelope is characterized by a TM oligomer of about 70 to 80 kDa, denoted here as TM80, and to a variable extent by a 150-kDa species. A similar 90-kDa TM oligomer has recently been suggested to correspond to the fusion-active six-helix bundle conformation of ASLV envelope (41). Several lines of evidence suggest that the TM oligomer is significant to the fusogenic process (see Discussion). To address how the TM oligomer we observed relates to the six-helix bundle, the ability of HR2 peptide to affect oligomer formation was tested. Addition of fusion inhibitor peptide at concentrations up to 100 μg of R99/ml was unable to inhibit formation of this oligomeric form of TM (data not shown), casting doubt on the putative identity of TM80 and suggesting that the activated TM oligomer may instead be an intermediate conformer. To examine this possibility, biotinylated HR2 peptide R99 was used as a probe. Based on a predominant model, the HR2 binding site is exposed after receptor triggering and disappears upon formation of the six-helix bundle. To ensure that modification had not affected function, the biotinylated peptide was tested in an infection inhibition assay. Biotinylated R99 was fully functional for blocking virus infection (data not shown). Inclusion of biotinylated R99 with virus and sTva under conditions where the TM80 oligomer was produced, followed by treatment with a protein cross-linking agent, resulted in R99 peptide migration in a position similar to TM80 (Fig. 7). A slight shift in the migration of this band (designated TM90) to ∼90 kDa was noted, consistent with the additional mass provided by binding of three 3-kDa peptides per TM oligomer. Binding of biotinylated R99 to TM90 could be competed by addition of unlabeled ASLV-specific peptide (Fig. 7A). In contrast, the Ebola virus-specific HR2 peptide E109, even when included at a 10-fold molar excess, was unable to compete with biotinylated R99 (data not shown). Liposomes were included as shown, since they were found to increase the stability of the triggered oligomer or the efficiency of the conformational change (1).
FIG. 7.

HR2 peptide binds to a triggered form of TM. ASLV-A was incubated on ice with combinations of soluble receptor (sTva), liposomes, and/or the HR2 peptide R99, triggered at 37°C, and then cross-linked on ice. Samples were examined by SDS-PAGE and Western blotting and probed with horseradish peroxidase-conjugated streptavidin to detect biotinylated R99 (R99-Bio). Locations of molecular mass standards are shown on the left of each blot. (A) R99-Bio is detected in a position consistent with binding to triggered, oligomeric TM and can be competed by unlabeled R99. The band labeled TM90 migrates somewhat slower than triggered TM in the absence of HR2 peptide (see Results). Soluble receptor (sTva) and/or liposomes were added to the reaction mixture as indicated. Unlabeled R99 was included either in an equimolar amount (1×) or as a 10-fold excess (10×) to R99-Bio, as shown. (B) Appearance of R99-Bio in the TM90 position is dependent upon incubation time at 37°C. (C) TM90 is efficiently immunoprecipitated by antiserum directed against the TM subunit.
The protein species identified by biotinylated R99 is dependent upon the presence of soluble receptor, sTva, which is consistent with its identity as triggered TM. The correlation between activated TM and the R99 binding partner was strengthened by demonstrating that this species was predominant after incubation at 37°C for 10 to 20 min (Fig. 7B). This time course is similar to that observed for liposome binding by envelope upon sTva activation (9, 21). To confirm that the band recognized by biotinylated R99 was indeed TM, an immunoprecipitation experiment was performed. A predominant band corresponding to TM90 was observed upon immunoprecipitation with TM-specific antiserum and probing for biotinylated peptide (Fig. 7C). In addition, some of the 150-kDa species was seen, as well as monomeric gp37. The monomeric form might result from dissociation induced by boiling these complexes for several minutes prior to SDS-PAGE. As above, incorporation of peptide into the TM oligomer complex was dependent on receptor (Fig. 7C). Taken together, these data demonstrate that receptor activation of ASLV envelope produces an oligomeric TM species that interacts physically with HR2 peptides.
DISCUSSION
Here we report the design and characterization of synthetic peptides corresponding to the predicted HR domains of the TM subunit of the ASLV-A envelope. ASLV has been employed as a model retrovirus to study receptor-triggered conformational changes in envelope, so a TM-derived peptide inhibitor that targets and permits analysis of fusion intermediates would be a useful tool. Both HR2-derived peptides inhibited virus entry, although R99 was more effective than R102. The structure of the Ebola virus GP2 ectodomain, the sequence of which is similar to ASLV TM, was applied to the design of these peptides. Based on this model, R102 is predicted to include the entire C-terminal HR, while R99 is N-terminally shifted three residues. Thus, R99, while the more potent of the two inhibitors, includes less of HR2 and more of the loop between HR1 and HR2. The basis for the difference in potency is unclear. A separate preparation of R99 has been shown to be slightly more potent than that discussed here, with an IC50 of about 0.075 μg/ml, or 25 nM (12) and, in fact, subsequent experiments with newer peptide stocks have supported this lower value.
Ebola virus-specific peptides analogous to the ASLV HR2 peptides were ineffective at inhibiting infection of MLV(Ebola GP)-pseudotyped virus. In contrast, a peptide identical to E109 has been reported to block infection of VSV(Ebola GP)-pseudotyped virus (45), although the apparent IC50 (2 mg/ml) was 20-fold higher than the maximum concentration tested in this work. A possible reason for the remarkable difference in HR2 peptide efficacy between the Ebola viral and retroviral envelopes is the mode of virus entry. To date, all of the reported HR2 inhibitors have been directed against pH-independent viruses, with the exception of the Ebola virus peptide mentioned above. The location of the virus at the time that the HR2 peptide-sensitive, transient intermediate is formed might play a significant role in sensitivity. The peptide concentration in an endosomal compartment, where a pH-dependent envelope such as the Ebola virus GP (43, 49) is triggered, would be expected to be lower than at the cell surface, where a pH-independent envelope would be triggered. Although acidic pH appears to play a role in ASLV entry, the initial triggering of envelope is dependent on receptor and not pH (9, 12, 19, 21, 26, 28, 29). Given the analogous peptides E109 and R99 and the similarity between Ebola GP2 and ASLV TM, this might be a demonstration of the important role that mode of entry plays in susceptibility to HR2 peptides. Alternatively, the basis for the difference between E109 and R99 may be a sequence-specific structure. In order to preserve the sequence alignment, ASLV TM has an insertion of one residue within its HR2 in comparison to Ebola GP2 (16, 46), and this distinction might contribute to the difference in peptide potency. Finally, a general kinetic difference between acid- and receptor-activated conformational changes might impart significant disparity in susceptibility to peptide inhibitors. For HIV-1, sensitivity to an HR2 peptide correlates with fusion kinetics (36).
Only one of the three viable HR1 mutations, L62A, displayed a change in sensitivity to HR2 peptides. This mutation increased the IC50 for R99 about 20-fold for infectivity of pseudotyped MLV(EnvA) particles (Fig. 3B) and about 10-fold in a quantitative cell-cell fusion assay (data not shown). Since these substitutions were selected based on the likelihood of involvement in HR1-HR2 interactions, these results might expose the limitations of the Ebola GP2 structure as a surrogate for ASLV TM. Alternatively, perhaps HR2 binding to HR1 is not easily perturbed by single amino acid substitutions. We are currently examining ASLV-A mutants that were selected by passage in the presence of R99, which should provide useful information on the array of envelope residues that can influence HR2 peptide sensitivity.
The putative target for HR2 peptides is the coiled-coil formed by a trimer of the N-terminal HR. Current models for class I viral fusion proteins suggest that this target becomes accessible following envelope activation and becomes inaccessible after formation of the stable six-helix bundle, or the homotrimer of hairpins. Consistent with this model, R99 has no effect on virus binding (12), but instead acts upon a transiently accessible intermediate that appears within several minutes at 37°C (Fig. 4A). The ability of R99 to inhibit infection is then lost after 15 to 30 min (Fig. 4B), which is somewhat more rapid than for inhibition of HIV-1 envelope-mediated cell-cell fusion by an HR2 peptide (30). An important distinction of the viral infectivity experiment, however, is that it evaluates only loss of inhibition and does not directly measure six-helix bundle formation. Endocytosis of viral particles might be expected to decrease the effectiveness of HR2 peptide as well. Thus, our inhibitor addition experiments do not distinguish between clearance of a linear intermediate from the cell surface or progression to a peptide-resistant conformation of TM. In fact, inhibition appears to be lost slower than the reported rate of ASLV internalization from the cell surface (31). What is clear from both the infection and cell-cell fusion studies is that the peptide inhibition of membrane fusion is independent of a low-pH environment. The most straightforward explanation of these results is that receptor engagement alone, most likely at the cell surface, induces structural rearrangements in TM that expose the peptide inhibitor binding sites.
Several assays have been developed to study ASLV-A envelope triggering, and an HR2 inhibitor is a useful tool for investigating the nature of the events these assays are measuring. Conformational changes in SU, manifested by increased protease sensitivity (8, 19), are unaffected by R99 (data not shown). Similarly, ASLV-A envelope achieves a lipophilic state in the presence of HR2 peptides (Fig. 6). These peptide-insensitive transformations most likely characterize the conversion of envelope from its native, metastable structure to an elongated, peptide-sensitive, prehairpin intermediate. Lipid mixing between virus and host cell has been shown to be inhibited by R99, albeit at higher concentrations than those needed to inhibit infection (12). Cell-cell fusion, which occurs more readily after exposure to acidic medium, is also blocked by peptide (Fig. 5) at concentrations similar to those needed to block infectivity (data not shown). Inhibition of cell-cell fusion by R99 is observed to a similar extent whether it is performed at neutral or acidic pH (reference 12 and data not shown).
The TM subunit undergoes a significant conformational change upon receptor activation, such that its migration by SDS-PAGE shifts from a monomeric to oligomeric, possibly trimeric species that we have designated as TM80 (1, 29, 41). It has been proposed that receptor binding and low pH operate synergistically to induce TM refolding to a stable fusogenic conformation (41); other parameters which have been shown to influence the efficiency with which this structure forms include time, temperature, presence of target lipid, Ca2+, virus preparation, and possibly glycosylation (1, 10, 29, 41). Several observations have suggested the idea that the triggered, oligomeric form of TM90 is the stable six-helix bundle. First, the receptor, time, and temperature requirements for this rearrangement all mirror the conditions necessary for viral entry (1). Second, triggered TM migration on SDS-PAGE has been reported to be 70 to 90 kDa depending upon the conditions, which could be a dense trimer of the native, 37-kDa monomer. Third, its stability is reminiscent of six-helix bundles from other class I viral envelopes (14), with reports that it has a melting temperature in the 65 to 80°C range (1, 29), or even that it is stable at 100°C in low concentrations of SDS (41). Finally, mutations in the SU subunit that impair viral entry at a postbinding step also impair the TM refolding event; TM mutations which suppress the entry defect also restore the ability of TM to undergo conformational change (1). Acidic pH has been shown to be necessary for efficient TM refolding (29, 41), consistent with a proposed role for endocytosis and acidification in viral entry (11, 29). However, our results and those of Matsuyama et al. (26) demonstrate that conformational change can be induced without an acidification step. Furthermore, in the experiments described here no noticeable difference was detected when the pH of the assay mixture was lowered. One reason for the discrepancy of our results and those of others may be that the assay was performed near enough to the reported pH threshold for activation of 6.7 to 7.0 (41) that further acidification had no effect. Another possibility is that the protein species detected by Western blotting may be heterogeneous, with a mix of intermediate and stable end product. This possibility might explain why oligomeric TM appears characteristic of a stable six-helix bundle and yet, as we demonstrated, still has the ability to bind HR2 peptide. Thus, differences in the manner that the assay is performed could dictate the distribution of detectable species. Use of a relatively low concentration of SDS as in this study (0.1%) might favor detection of less stable intermediates; under neutral pH conditions, the activated form of TM is not observed as reliably with SDS concentrations above 0.5% (data not shown).
An intermediate in the fusogenic pathway might not be expected to display the stability of triggered TM. Possibly this species is heterogeneous as mentioned or, alternatively, the HR2 peptide is binding a precursor to triggered TM, and thus some amount is trapped upon conversion to the stable fusogenic conformation. Arguing against this idea is the observation that the TM conversion is not blocked by the peptide, which is the expected result if it corresponds to a postfusogenic six-helix bundle. This result might be seen if the peptide were used at concentrations too low to inhibit, although it was tested at concentrations several times higher than that which completely blocks viral entry. Similar observations have been made by Matsuyama et al. (26). In addition, the mobility shift observed for TM90 suggests that the majority of triggered TM is associated with R99-Bio. A final possibility is that this species is neither a true intermediate nor an authentic postfusion structure, but instead is a dead-end artifact that has refolded nonproductively. However, as outlined above, the biological relevance of the TM oligomer is supported by several lines of evidence. Thus, it remains likely that R99-Bio is binding a species that is an intermediate. Since acidic pH appears to play an important role in ASLV-A entry, perhaps the absence of an acid pulse in these experiments has allowed the capture of this stage. It is unclear if the final, six-helix bundle conformation is formed under these assay conditions. Alternatively, it may form but not be distinguishable from the intermediate form when resolved by SDS-PAGE. R99 can be seen bound to the 150-kDa form of TM (Fig. 7C), although generally R99 decoration of TM90 appears to be more robust. This, coupled with the observation that formation of the 150-kDa species is not blocked by HR2 peptide, suggests that the larger species is an oligomeric variant of TM90 and not a final conformation. It has been recently suggested that the final structure migrates at about 100 kDa (26), a form that is not readily apparent in the data reported here. Generation of this 100-kDa form requires a low-pH pulse and is inhibited by R99 (26).
These results support the existence of a receptor-induced, HR2 peptide-sensitive intermediate. Similarly, HIV-1 is able to bind HR2 peptide only after CD4 triggering of its envelope (15), and the paramyxovirus SV5 becomes sensitive to HR2 peptide only after host cell binding and subsequent thermal activation (39). The surprisingly stable ASLV intermediate is reached in the absence of acid exposure but is quickly transcended during acidification (Fig. 5). This interpretation is consistent with the observation that endocytosed virions can remain infectious for several hours in the presence of ammonium chloride (31). Envelope-mediated liposome binding occurs in the presence of HR2 peptide (Fig. 6) and does not require acidification (9, 21, 28), suggesting that the stable intermediate is a linear, prehairpin structure, with its fusion peptide anchored in the target membrane. The role of acidification is thus likely to be downstream of this intermediate, possibly in formation of six-helix bundles. However, the observation that envelope-mediated lipid mixing occurs at neutral pH and yet is blocked by HR2 peptide (12) may mean that transition of the stable peptide-sensitive envelope to a final conformation involves more than a single step, analogous to the multiple stages suggested for activation of influenza virus hemagglutinin (32).
Acknowledgments
We thank Shutoku Matsuyama and Sue Ellen Delos for sharing their data, John Young for providing the SUA-rIgG expression plasmid used for generating antisera, and Andy Piefer, Jackie Reeves, and Graham Simmons for critical reading of the manuscript.
This work was funded by National Institutes of Health grants AI43455 (P.B.), CA76256 (P.B.), F32-AI050341 (S.M.A.), T32-GM07229 (R.C.N.), T32-AI07324 (J.W.B.), and AI22470 (J.M.W.).
REFERENCES
- 1.Balliet, J. W. 1998. Early events in subgroup A avian sarcoma and leukosis virus entry. Ph.D. thesis. University of Pennsylvania, Philadelphia.
- 2.Balliet, J. W., J. Berson, C. M. D'Cruz, J. Huang, J. Crane, J. M. Gilbert, and P. Bates. 1999. Production and characterization of a soluble, active form of Tva, the subgroup A avian sarcoma and leukosis virus receptor. J. Virol. 73:3054-3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balliet, J. W., K. Gendron, and P. Bates. 2000. Mutational analysis of the subgroup A avian sarcoma and leukosis virus putative fusion peptide domain. J. Virol. 74:3731-3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bosch, B. J., R. van der Zee, C. A. M. de Haan, and P. J. M. Rottier. 2003. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 77:8801-8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cervia, J. S., and M. A. Smith. 2003. Enfuvirtide (T-20): a novel human immunodeficiency virus type 1 fusion inhibitor. Clin. Infect. Dis. 37:1102-1106. [DOI] [PubMed] [Google Scholar]
- 6.Colman, P. M., and M. C. Lawrence. 2003. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 4:309-319. [DOI] [PubMed] [Google Scholar]
- 7.Damico, R., and P. Bates. 2000. Soluble receptor-induced retroviral infection of receptor-deficient cells. J. Virol. 74:6469-6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Damico, R., L. Rong, and P. Bates. 1999. Substitutions in the receptor-binding domain of the avian sarcoma and leukosis virus envelope uncouple receptor-triggered structural rearrangements in the surface and transmembrane subunits. J. Virol. 73:3087-3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Damico, R. L., J. Crane, and P. Bates. 1998. Receptor-triggered membrane association of a model retroviral glycoprotein. Proc. Natl. Acad. Sci. USA 95:2580-2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delos, S. E., M. J. Burdick, and J. M. White. 2002. A single glycosylation site within the receptor-binding domain of the avian sarcoma/leukosis virus glycoprotein is critical for receptor binding. Virology 294:354-363. [DOI] [PubMed] [Google Scholar]
- 11.Diaz-Griffero, F., S. A. Hoschander, and J. Brojatsch. 2002. Endocytosis is a critical step in entry of subgroup B avian leukosis viruses. J. Virol. 76:12866-12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Earp, L. J., S. E. Delos, R. C. Netter, P. Bates, and J. M. White. 2003. The avian retrovirus avian sarcoma/leukosis virus subtype A reaches the lipid mixing stage of fusion at neutral pH. J. Virol. 77:3058-3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Earp, L. J., S. E. Delos, H. E. Park, and J. M. White. 2004. The many mechanisms of viral membrane fusion proteins. Curr. Top. Microbiol. Immunol. 285:25-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eckert, D. M., and P. S. Kim. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70:777-810. [DOI] [PubMed] [Google Scholar]
- 15.Furuta, R. A., C. T. Wild, Y. Weng, and C. D. Weiss. 1998. Capture of an early fusion-active conformation of HIV-1 gp41. Nat. Struct. Biol. 5:276-279. [DOI] [PubMed] [Google Scholar]
- 16.Gallaher, W. R. 1996. Similar structural models of the transmembrane proteins of Ebola and avian sarcoma viruses. Cell 85:477-478. [DOI] [PubMed] [Google Scholar]
- 17.Gallaher, W. R., C. DiSimone, and M. J. Buchmeier. 2001. The viral transmembrane superfamily: possible divergence of Arenavirus and Filovirus glycoproteins from a common RNA virus ancestor. BMC Microbiol. [Online.] 10.1186/1471-2180-1-1. [DOI] [PMC free article] [PubMed]
- 18.Gilbert, J. M., P. Bates, H. E. Varmus, and J. M. White. 1994. The receptor for the subgroup A avian leukosis-sarcoma viruses binds to subgroup A but not to subgroup C envelope glycoprotein. J. Virol. 68:5623-5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilbert, J. M., L. D. Hernandez, J. W. Balliet, P. Bates, and J. M. White. 1995. Receptor-induced conformational changes in the subgroup A avian leukosis and sarcoma virus envelope glycoprotein. J. Virol. 69:7410-7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heinz, F. X., and S. L. Allison. 2001. The machinery for flavivirus fusion with host cell membranes. Curr. Opin. Microbiol. 4:450-455. [DOI] [PubMed] [Google Scholar]
- 21.Hernandez, L. D., R. J. Peters, S. E. Delos, J. A. T. Young, D. A. Agard, and J. M. White. 1997. Activation of a retroviral membrane fusion protein: soluble receptor-induced liposome binding of the ALSV envelope glycoprotein. J. Cell Biol. 139:1455-1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang, S., K. Lin, N. Strick, and A. R. Neurath. 1993. HIV-1 inhibition by a peptide. Nature 365:113. [DOI] [PubMed] [Google Scholar]
- 23.Lambert, D. M., S. Barney, A. L. Lambert, K. Guthrie, R. Medinas, D. E. Davis, T. Bucy, J. Erickson, G. Merutka, and S. R. Petteway, Jr. 1996. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc. Natl. Acad. Sci. USA 93:2186-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu, S., G. Xiao, Y. Chen, Y. He, J. Niu, C. R. Escalante, H. Xiong, J. Farmar, A. K. Debnath, P. Tien, and S. Jiang. 2004. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet 363:938-947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malashkevich, V. N., B. J. Schneider, M. L. McNally, M. A. Milhollen, J. X. Pang, and P. S. Kim. 1999. Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9-Å resolution. Proc. Natl. Acad. Sci. USA 96:2662-2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsuyama, S., S. E. Delos, and J. M. White. 2004. Sequential roles of receptor binding and low pH in forming prehairpin and hairpin conformations of a retroviral envelope glycoprotein. J. Virol. 78:8201-8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Medinas, R. J., D. M. Lambert, and W. A. Tompkins. 2002. C-terminal gp40 peptide analogs inhibit feline immunodeficiency virus: cell fusion and virus spread. J. Virol. 76:9079-9086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melikyan, G. B., R. J. O. Barnard, R. M. Markosyan, J. A. T. Young, and F. S. Cohen. 2004. Low pH is required for avian sarcoma and leukosis virus Env-induced hemifusion and fusion pore formation but not for pore growth. J. Virol. 78:3753-3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mothes, W., A. L. Boerger, S. Narayan, J. M. Cunningham, and J. A. T. Young. 2000. Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell 103:679-689. [DOI] [PubMed] [Google Scholar]
- 30.Muñoz-Barroso, I., S. Durell, K. Sakaguchi, E. Appella, and R. Blumenthal. 1998. Dilation of the human immunodeficiency virus-1 envelope glycoprotein fusion pore revealed by the inhibitory action of a synthetic peptide from gp41. J. Cell Biol. 140:315-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Narayan, S., R. J. O. Barnard, and J. A. T. Young. 2003. Two retroviral entry pathways distinguished by lipid raft association of the viral receptor and differences in viral infectivity. J. Virol. 77:1977-1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park, H. E., J. A. Gruenke, and J. M. White. 2003. Leash in the groove mechanism of membrane fusion. Nat. Struct. Biol. 10:1048-1053. [DOI] [PubMed] [Google Scholar]
- 33.Peisajovich, S. G., S. A. Gallo, R. Blumenthal, and Y. Shai. 2003. C-terminal octylation rescues an inactive T20 mutant: implications for the mechanism of HIV/simian immunodeficiency virus-induced membrane fusion. J. Biol. Chem. 278:21012-21017. [DOI] [PubMed] [Google Scholar]
- 34.Piñón, J. D., S. M. Kelly, N. C. Price, J. U. Flanagan, and D. W. Brighty. 2003. An antiviral peptide targets a coiled-coil domain of the human T-cell leukemia virus envelope glycoprotein. J. Virol. 77:3281-3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rapaport, D., M. Ovadia, and Y. Shai. 1995. A synthetic peptide corresponding to a conserved heptad repeat domain is a potent inhibitor of Sendai virus-cell fusion: an emerging similarity with functional domains of other viruses. EMBO J. 14:5524-5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reeves, J. D., S. A. Gallo, N. Ahmad, J. L. Miamidian, P. E. Harvey, M. Sharron, S. Pöhlmann, J. N. Sfakianos, C. A. Derdeyn, R. Blumenthal, E. Hunter, and R. W. Doms. 2002. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc. Natl. Acad. Sci. USA 99:16249-16254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rimsky, L. T., D. C. Shugars, and T. J. Matthews. 1998. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J. Virol. 72:986-993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rong, L., and P. Bates. 1995. Analysis of the subgroup A avian sarcoma and leukosis virus receptor: the 40-residue, cysteine-rich, low-density lipoprotein receptor repeat motif of Tva is sufficient to mediate viral entry. J. Virol. 69:4847-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Russell, C. J., T. S. Jardetzky, and R. A. Lamb. 2001. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 20:4024-4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sagara, Y., Y. Inoue, H. Shiraki, A. Jinno, H. Hoshino, and Y. Maeda. 1996. Identification and mapping of functional domains on human T-cell lymphotropic virus type 1 envelope proteins by using synthetic peptides. J. Virol. 70:1564-1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith, J. G., W. Mothes, S. C. Blacklow, and J. M. Cunningham. 2004. The mature avian leukosis virus subgroup A envelope glycoprotein is metastable, and refolding induced by the synergistic effects of receptor binding and low pH is coupled to infection. J. Virol. 78:1403-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soneoka, Y., P. M. Cannon, E. E. Ramsdale, J. C. Griffiths, G. Romano, S. M. Kingsman, and A. J. Kingsman. 1995. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucleic Acids Res. 23:628-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takada, A., C. Robison, H. Goto, A. Sanchez, K. G. Murti, M. A. Whitt, and Y. Kawaoka. 1997. A system for functional analysis of Ebola virus glycoprotein. Proc. Natl. Acad. Sci. USA 94:14764-14769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Volchkov, V. E., V. M. Blinov, and S. V. Netesov. 1992. The envelope glycoprotein of Ebola virus contains an immunosuppressive-like domain similar to oncogenic retroviruses. FEBS Lett. 305:181-184. [DOI] [PubMed] [Google Scholar]
- 45.Watanabe, S., A. Takada, T. Watanabe, H. Ito, H. Kida, and Y. Kawaoka. 2000. Functional importance of the coiled-coil of the Ebola virus glycoprotein. J. Virol. 74:10194-10201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weissenhorn, W., A. Carfí, K.-H. Lee, J. J. Skehel, and D. C. Wiley. 1998. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol. Cell 2:605-616. [DOI] [PubMed] [Google Scholar]
- 47.Wild, C., T. Oas, C. McDanal, D. Bolognesi, and T. Matthews. 1992. A synthetic peptide inhibitor of human immunodeficiency virus replication: correlation between solution structure and viral inhibition. Proc. Natl. Acad. Sci. USA 89:10537-10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wild, C. T., D. C. Shugars, T. K. Greenwell, C. B. McDanal, and T. J. Matthews. 1994. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 91:9770-9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wool-Lewis, R. J., and P. Bates. 1998. Characterization of Ebola virus entry by using pseudotyped viruses: identification of receptor-deficient cell lines. J. Virol. 72:3155-3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wool-Lewis, R. J., and P. Bates. 1999. Endoproteolytic processing of the Ebola virus envelope glycoprotein: cleavage is not required for function. J. Virol. 73:1419-1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Young, J. K., R. P. Hicks, G. E. Wright, and T. G. Morrison. 1997. Analysis of a peptide inhibitor of paramyxovirus (NDV) fusion using biological assays, NMR, and molecular modeling. Virology 238:291-304. [DOI] [PubMed] [Google Scholar]
- 52.Young, J. K., D. Li, M. C. Abramowitz, and T. G. Morrison. 1999. Interaction of peptides with sequences from the Newcastle disease virus fusion protein heptad repeat regions. J. Virol. 73:5945-5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zingler, K., and J. A. T. Young. 1996. Residue Trp-48 of Tva is critical for viral entry but not for high-affinity binding to the SU glycoprotein of subgroup A avian leukosis and sarcoma viruses. J. Virol. 70:7510-7516. [DOI] [PMC free article] [PubMed] [Google Scholar]